Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery



Abstract
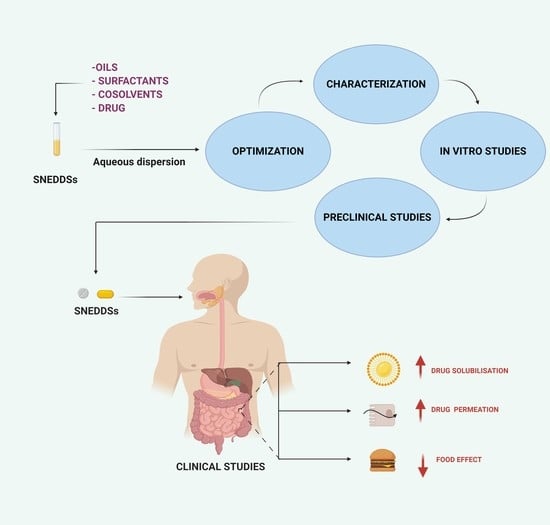
:
1. Introduction
2. General Components of SNEDDSs and Their Role in Formulation Performance
2.1. Oil Phase
2.2. Surfactants
2.3. Cosurfactants/Cosolvents
3. Optimization of SNEDDSs Formulations
4. Physico-Chemical Characterization of SNEDDSs Formulation
4.1. Particle Size
4.2. Zeta Potential
4.3. Emulsification Time Measurement
4.4. Transmittance Percentage Measurement
4.5. Morphology
4.6. Viscosity Measurement
4.7. Cloud Point Measurement
4.8. Thermodynamic Stability Studies
5. In Vitro Assessment of SNEDDSs Formulations
5.1. In Vitro Dissolution
5.2. In Vitro Lipolysis
5.3. In Vitro Permeation Studies
6. Ex Vivo Permeation Studies
7. In Vivo Pharmacokinetics Studies
8. Advancements in SNEDDSs
8.1. Supersaturated SNEDDSs
8.2. Mucus-Permeating SNEDDSs
8.3. Solid SNEDDSs
8.3.1. Methods of Production
8.3.2. Solid-State Characterization of Solid SNEDDSs
8.4. SNEDDSs for the Oral Delivery of Hydrophilic Macromolecules
8.4.1. Ion Pairing
8.4.2. Double Emulsification Technique
8.4.3. The Use of Hydrophilic Cosolvent
8.4.4. Chemical Modification
8.5. Targeted SNEDDSs
8.6. SNEDDSs for the Oral Delivery of Herbal Drugs
9. Challenges
10. SNEDDSs from an Industrial Perspective
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shah, M.K.; Khatri, P.; Vora, N.; Patel, N.K.; Jain, S.; Lin, S. Lipid Nanocarriers: Preparation, Characterization and Absorption Mechanism and Applications to Improve Oral Bioavailability of Poorly Water-Soluble Drugs. In Biomedical Applications of Nanoparticles; William Andrew Publisher: Norwich, NY, USA, 2019; pp. 117–147. ISBN 9780128165065. [Google Scholar]
- Boyd, B.J.; Bergström, C.A.S.; Vinarov, Z.; Kuentz, M.; Brouwers, J.; Augustijns, P.; Brandl, M.; Bernkop-Schnürch, A.; Shrestha, N.; Préat, V.; et al. Successful Oral Delivery of Poorly Water-Soluble Drugs Both Depends on the Intraluminal Behavior of Drugs and of Appropriate Advanced Drug Delivery Systems. Eur. J. Pharm. Sci. 2019, 137, 104967. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Morozowich, W. Development of Supersaturatable Self-Emulsifying Drug Delivery System Formulations for Improving the Oral Absorption of Poorly Soluble Drugs. Expert Opin. Drug Deliv. 2006, 3, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Charman, W.N. Transport and Absorption of Drugs via the Lymphatic System. Adv. Drug Deliv. Rev. 2001, 50, 1–2. [Google Scholar] [CrossRef]
- Williams, H.D.; Ford, L.; Igonin, A.; Shan, Z.; Botti, P.; Morgen, M.M.; Hu, G.; Pouton, C.W.; Scammells, P.J.; Porter, C.J.H.; et al. Unlocking the Full Potential of Lipid-Based Formulations Using Lipophilic Salt/ionic Liquid Forms. Adv. Drug Deliv. Rev. 2019, 142, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Siqueira Jørgensen, S.D.; Al Sawaf, M.; Graeser, K.; Mu, H.; Müllertz, A.; Rades, T. The Ability of Two in vitro Lipolysis Models Reflecting the Human and Rat Gastro-Intestinal Conditions to Predict the in vivo Performance of SNEDDS Dosing Regimens. Eur. J. Pharm. Biopharm. 2018, 124, 116–124. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid Formulations for Oral Administration of Drugs: Non-Emulsifying, Self-Emulsifying and “Self-Microemulsifying” Drug Delivery Systems. Eur. J. Pharm. Sci. 2000, 11, 93–98. [Google Scholar] [CrossRef]
- Ujhelyi, Z.; Vecsernyés, M.; Fehér, P.; Kósa, D.; Arany, P.; Nemes, D.; Sinka, D.; Vasvári, G.; Fenyvesi, F.; Váradi, J.; et al. Physico-Chemical Characterization of Self-Emulsifying Drug Delivery Systems. Drug Discov. Today Technol. 2018, 27, 81–86. [Google Scholar] [CrossRef]
- Li, Z.; Xu, D.; Yuan, Y.; Wu, H.; Hou, J.; Kang, W.; Bai, B. Advances of Spontaneous Emulsification and Its Important Applications in Enhanced Oil Recovery Process. Adv. Colloid Interface Sci. 2020, 277, 102119. [Google Scholar] [CrossRef]
- Rachmawati, H.; Rasaputri, D.H.; Susilowidodo, R.A.; Darijanto, S.T.; Sumirtapura, Y.C. The Influence of Oils and Surfactants on the Formation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) Containing Therapeutic Protein. Proc. Int. Conf. Mater. Sci. Technol. 2011, 247, 3–9. [Google Scholar]
- Ding, W.; Hou, X.; Cong, S.; Zhang, Y.; Chen, M.; Lei, J.; Meng, Y.; Li, X.; Li, G. Co-Delivery of Honokiol, a Constituent of Magnolia Species, in a Self-Microemulsifying Drug Delivery System for Improved Oral Transport of Lipophilic Sirolimus. Drug Deliv. 2016, 23, 2513–2523. [Google Scholar] [CrossRef]
- Hetényi, G.; Griesser, J.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Comparison of the Protective Effect of Self-Emulsifying Peptide Drug Delivery Systems towards Intestinal Proteases and Glutathione. Int. J. Pharm. 2017, 523, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Bernkop-Schnürch, A. SEDDS: A Game Changing Approach for the Oral Administration of Hydrophilic Macromolecular Drugs. Adv. Drug Deliv. Rev. 2019, 142, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, R.N.; Benita, S. Self-Emulsifying Drug Delivery Systems (SEDDS) for Improved Oral Delivery of Lipophilic Drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.T.; Åkesson, P.; Juréus, A.; Saaby, L.; Abu-Rmaileh, R.; Abrahamsson, B.; Østergaard, J.; Müllertz, A. Bioavailability of Cinnarizine in Dogs: Effect of SNEDDS Loading Level and Correlation with Cinnarizine Solubilization during in Vitro Lipolysis. Pharm. Res. 2013, 30, 3101–3113. [Google Scholar] [CrossRef]
- Pouton, C.W.; Porter, C.J.H. Formulation of Lipid-Based Delivery Systems for Oral Administration: Materials, Methods and Strategies. Adv. Drug Deliv. Rev. 2008, 60, 625–637. [Google Scholar] [CrossRef]
- Date, A.A.; Desai, N.; Dixit, R.; Nagarsenker, M. Self-Nanoemulsifying Drug Delivery Systems: Formulation Insights, Applications and Advances. Nanomedicine 2010, 1595–1616. [Google Scholar] [CrossRef]
- Singh, B.; Bandopadhyay, S.; Kapil, R.; Singh, R.; Katare, O.P. Self-Emulsifying Drug Delivery Systems (SEDDS): Formulation Development, Characterization, and Applications. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 427–521. [Google Scholar] [CrossRef]
- Kollipara, S.; Gandhi, R.K. Pharmacokinetic Aspects and in Vitro–in Vivo Correlation Potential for Lipid-Based Formulations. Acta Pharm. Sin. B 2014, 4, 333–349. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-Based Formulations for Oral Administration of Poorly Water-Soluble Drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Rajpoot, K.; Tekade, M.; Pandey, V.; Nagaraja, S.H.; Youngren-Ortiz, S.R.; Tekade, R.K. Self-Microemulsifying Drug-Delivery System: Ongoing Challenges and Future Ahead; Tekade, R.K., Ed.; Drug Delivery Systems; Academic Press: Cambridge, MA, USA, 2020; pp. 393–454. [Google Scholar] [CrossRef]
- Qian, J.; Meng, H.; Xin, L.; Xia, M.; Shen, H.; Li, G.; Xie, Y. Self-Nanoemulsifying Drug Delivery Systems of Myricetin: Formulation Development, Characterization, and in Vitro and in Vivo Evaluation. Colloids Surfaces B Biointerfaces 2017, 160, 101–109. [Google Scholar] [CrossRef]
- Maher, S.; Brayden, D.J. Overcoming Poor Permeability: Translating Permeation Enhancers for Oral Peptide Delivery. Drug Discov. Today Technol. 2012, 9, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with Self-Emulsifying Drug Delivery System from Pharmacokinetic Point of View. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef] [Green Version]
- Izgelov, D.; Shmoeli, E.; Domb, A.J.; Hoffman, A. The Effect of Medium Chain and Long Chain Triglycerides Incorporated in Self-Nano Emulsifying Drug Delivery Systems on Oral Absorption of Cannabinoids in Rats. Int. J. Pharm. 2020, 580, 119201. [Google Scholar] [CrossRef]
- Gracia, G.; Cao, E.; Johnston, A.P.R.; Porter, C.J.H.; Trevaskis, N.L. Organ-Specific Lymphatics Play Distinct Roles in Regulating HDL Trafficking and Composition. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G725–G735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L. Lipid Excipients and Delivery Systems for Pharmaceutical Development: A Regulatory Perspective. Adv. Drug Deliv. Rev. 2008, 60, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, C.H.; Xu, Z.P. Self-Nanoemulsifying Drug-Delivery System. In Nanocarriers for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2019; pp. 421–449. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupančič, O.; Grieβinger, J.A.; Rohrer, J.; Pereira de Sousa, I.; Danninger, L.; Partenhauser, A.; Sündermann, N.E.; Laffleur, F.; Bernkop-Schnürch, A. Development, in Vitro and in Vivo Evaluation of a Self-Emulsifying Drug Delivery System (SEDDS) for Oral Enoxaparin Administration. Eur. J. Pharm. Biopharm. 2016, 109, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.M.; Cui, F.D.; Mu, C.F.; Choi, M.K.; Kim, J.S.; Chung, S.J.; Shim, C.K.; Kim, D.D. Docetaxel Microemulsion for Enhanced Oral Bioavailability: Preparation and in Vitro and in Vivo Evaluation. J. Control. Release 2009, 140, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Mountfield, R.J.; Senepin, S.; Schleimer, M.; Walter, I.; Bittner, B. Potential Inhibitory Effects of Formulation Ingredients on Intestinal Cytochrome P450. Int. J. Pharm. 2000, 211, 89–92. [Google Scholar] [CrossRef]
- Rege, B.D.; Kao, J.P.Y.; Polli, J.E. Effects of Nonionic Surfactants on Membrane Transporters in Caco-2 Cell Monolayers. Eur. J. Pharm. Sci. 2002, 16, 237–246. [Google Scholar] [CrossRef]
- Čerpnjak, K.; Zvonar, A.; Gašperlin, M.; Vrečer, F. Lipid-Based Systems as a Promising Approach for Enhancing the Bioavailability of Poorly Water-Soluble Drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouton, C.W. Formulation of Poorly Water-Soluble Drugs for Oral Administration: Physicochemical and Physiological Issues and the Lipid Formulation Classification System. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Weisan, P.; Jiayu, L.; Ying, Z. Preparation, Evaluation, and NMR Characterization of Vinpocetine Microemulsion for Transdermal Delivery. Drug Dev. Ind. Pharm. 2004, 30, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Borhade, V.; Pathak, S.; Sharma, S.; Patravale, V. Clotrimazole Nanoemulsion for Malaria Chemotherapy. Part I: Preformulation Studies, Formulation Design and Physicochemical Evaluation. Int. J. Pharm. 2012, 431, 138–148. [Google Scholar] [CrossRef]
- Kale, A.A.; Patravale, V.B. Design and Evaluation of Self-Emulsifying Drug Delivery Systems (SEDDS) of Nimodipine. AAPS Pharmscitech 2008, 9, 191–196. [Google Scholar] [CrossRef]
- Nepal, P.R.; Han, H.K.; Choi, H.K. Preparation and in Vitro-in Vivo Evaluation of Witepsol®® H35 Based Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) of Coenzyme Q10. Eur. J. Pharm. Sci. 2010, 39, 224–232. [Google Scholar] [CrossRef]
- Memvanga, P.B.; Coco, R.; Préat, V. An Oral Malaria Therapy: Curcumin-Loaded Lipid-Based Drug Delivery Systems Combined with β-Arteether. J. Control. Release 2013, 172, 904–913. [Google Scholar] [CrossRef]
- Memvanga, P.B.; Préat, V. Formulation Design and in Vivo Antimalarial Evaluation of Lipid-Based Drug Delivery Systems for Oral Delivery of β-Arteether. Eur. J. Pharm. Biopharm. 2012, 82, 112–119. [Google Scholar] [CrossRef]
- Jain, S.; Garg, T.; Kushwah, V.; Thanki, K.; Agrawal, A.K.; Dora, C.P. α-Tocopherol as Functional Excipient for Resveratrol and Coenzyme Q10-Loaded SNEDDS for Improved Bioavailability and Prophylaxis of Breast Cancer. J. Drug Target. 2017, 25, 554–565. [Google Scholar] [CrossRef]
- Yanfei, M.; Guoguang, C.; Lili, R.; Pingkai, O. Controlled Release of Glaucocalyxin—A Self-Nanoemulsifying System from Osmotic Pump Tablets with Enhanced Bioavailability. Pharm. Dev. Technol. 2017, 22, 148–155. [Google Scholar] [CrossRef]
- Hosny, K.M.; Aldawsari, H.M.; Bahmdan, R.H.; Sindi, A.M.; Kurakula, M.; Alrobaian, M.M.; Aldryhim, A.Y.; Alkhalidi, H.M.; Bahmdan, H.H.; Khallaf, R.A.; et al. Preparation, Optimization, and Evaluation of Hyaluronic Acid-Based Hydrogel Loaded with Miconazole Self-Nanoemulsion for the Treatment of Oral Thrush. AAPS Pharmscitech 2019, 20, 297. [Google Scholar] [CrossRef] [PubMed]
- Batool, A.; Arshad, R.; Razzaq, S.; Nousheen, K.; Kiani, M.H.; Shahnaz, G. Formulation and Evaluation of Hyaluronic Acid-Based Mucoadhesive Self Nanoemulsifying Drug Delivery System (SNEDDS) of Tamoxifen for Targeting Breast Cancer. Int. J. Biol. Macromol. 2020, 152, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Zhang, M.; Pang, Y.; Li, Z.; Zhao, A.; Feng, J. Self-Emulsifying Drug Delivery System and the Applications in Herbal Drugs. Drug Deliv. 2015, 22, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.; Baskaran, R.; Yoo, B.K. Self-Nanoemulsifying Drug Delivery System of Lutein: Physicochemical Properties and Effect on Bioavailability of Warfarin. Biomol. Ther. 2013, 21, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Date, A.A.; Nagarsenker, M.S. Design and Evaluation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Cefpodoxime Proxetil. Int. J. Pharm. 2007, 329, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Khattab, A.; Mohamed, M.; Basalious, E.B. Design of Self-Nanoemulsifying System to Enhance Absorption and Bioavailability of Poorly Permeable Aliskiren Hemi-Fumarate. J. Drug Deliv. Sci. Technol. 2020, 57, 101646. [Google Scholar] [CrossRef]
- Shah, M.K.; Madan, P.; Lin, S. Elucidation of Intestinal Absorption Mechanism of Carvedilol-Loaded Solid Lipid Nanoparticles Using Caco-2 Cell Line as an in-Vitro Model. Pharm. Dev. Technol. 2015, 20, 877–885. [Google Scholar] [CrossRef]
- Shukla, M.; Jaiswal, S.; Sharma, A.; Srivastava, P.K.; Arya, A.; Dwivedi, A.K.; Lal, J. A Combination of Complexation and Self-Nanoemulsifying Drug Delivery System for Enhancing Oral Bioavailability and Anticancer Efficacy of Curcumin. Drug Dev. Ind. Pharm. 2017, 43, 847–861. [Google Scholar] [CrossRef]
- Yadav, P.; Rastogi, V.; Verma, A. Application of Box–Behnken Design and Desirability Function in the Development and Optimization of Self-Nanoemulsifying Drug Delivery System for Enhanced Dissolution of Ezetimibe. Futur. J. Pharm. Sci. 2020, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, K.C.; Jena, J.; Jena, G.K.; Patra, C.N.; Rao, M.E.B. QBD-Based Systematic Development of BosentanSNEDDS: Formulation, Characterization and Pharmacokinetic Assessment. J. Drug Deliv. Sci. Technol. 2018, 47, 31–42. [Google Scholar] [CrossRef]
- Sanka, K.; Suda, D.; Bakshi, V. Optimization of Solid-Self Nanoemulsifying Drug Delivery System for Solubility and Release Profile of Clonazepam Using Simplex Lattice Design. J. Drug Deliv. Sci. Technol. 2016, 33, 114–124. [Google Scholar] [CrossRef]
- El-Zahaby, S.A.; AbouGhaly, M.H.H.; Abdelbary, G.A.; El-Gazayerly, O.N. Zero-Order Release and Bioavailability Enhancement of Poorly Water Soluble Vinpocetine from Self-Nanoemulsifying Osmotic Pump Tablet. Pharm. Dev. Technol. 2018, 23, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Ujilestari, T.; Martien, R.; Ariyadi, B.; Dono, N.D. Zuprizal. Self-Nanoemulsifying Drug Delivery System (SNEDDS) of Amomum Compactum Essential Oil: Design, Formulation, and Characterization. J. Appl. Pharm. Sci. 2018, 8, 14–21. [Google Scholar] [CrossRef]
- Das, S.S.; Singh, A.; Kar, S.; Ghosh, R.; Pal, M.; Fatima, M.; Singh, N.; Singh, S.K. Application of QbD Framework for Development of Self-Emulsifying Drug Delivery Systems. In Pharmaceutical Quality by Design; Academic Press: Cambridge, MA, USA, 2019; pp. 297–350. [Google Scholar] [CrossRef]
- Garg, V.; Kaur, P.; Singh, S.K.; Kumar, B.; Bawa, P.; Gulati, M.; Yadav, A.K. Solid Self-Nanoemulsifying Drug Delivery Systems for Oral Delivery of Polypeptide-K: Formulation, Optimization, in-Vitro and in-Vivo Antidiabetic Evaluation. Eur. J. Pharm. Sci. 2017, 109, 297–315. [Google Scholar] [CrossRef]
- Gündoğdu, T.K.; Deniz, I.; Çalişkan, G.; Şahin, E.S.; Azbar, N. Experimental Design Methods for Bioengineering Applications. Crit. Rev. Biotechnol. 2016, 36, 368–388. [Google Scholar] [CrossRef]
- Rad, A.H.; Pirouzian, H.R.; Toker, O.S.; Konar, N. Application of Simplex Lattice Mixture Design for Optimization of Sucrose-Free Milk Chocolate Produced in a Ball Mill. LWT 2019, 115, 108435. [Google Scholar] [CrossRef]
- Astuti, I.Y.; Marchaban, M.; Martien, R.; Nugroho, A.E. Design and Optimization of Self Nano-Emulsifying Drug Delivery System Containing a New Anti-Inflammatory Agent Pentagamavunon-0. Indones. J. Chem. 2017, 17, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Antony, J. Full Factorial Designs. In Design of Experiments for Engineers and Scientists, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 63–85. ISBN 9780080994178. [Google Scholar] [CrossRef]
- Karamanidou, T.; Karidi, K.; Bourganis, V.; Kontonikola, K.; Kammona, O.; Kiparissides, C. Effective Incorporation of Insulin in Mucus Permeating Self-Nanoemulsifying Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2015, 97, 223–229. [Google Scholar] [CrossRef]
- De Aguiar, P.F.; Bourguignon, B.; Khots, M.S.; Massart, D.L.; Phan-Than-Luu, R. D-Optimal Designs. Chemom. Intell. Lab. Syst. 1995, 30, 199–210. [Google Scholar] [CrossRef]
- Talekar, S.D.; Haware, R.V.; Dave, R.H. Evaluation of Self-Nanoemulsifying Drug Delivery Systems Using Multivariate Methods to Optimize Permeability of Captopril Oral Films. Eur. J. Pharm. Sci. 2019, 130, 215–224. [Google Scholar] [CrossRef]
- Cunha, S.; Costa, C.P.; Moreira, J.N.; Lobo, J.M.S.; Silva, A.C. Using the Quality by Design (QbD) Approach to Optimize Formulations of Lipid Nanoparticles and Nanoemulsions: A Review. Nanomed. Nanotechnol. Biol. Med. 2020, 102206. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.; Salentinig, S.; Prestidge, C.A.; Boyd, B.J. Self-Assembled Structures Formed during Lipid Digestion: Characterization and Implications for Oral Lipid-Based Drug Delivery Systems. Drug Deliv. Transl. Res. 2014, 4, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Franzen, U.; Vermehren, C.; Jensen, H.; Østergaard, J. Physicochemical Characterization of a PEGylated Liposomal Drug Formulation Using Capillary Electrophoresis. Electrophoresis 2011, 32, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Schmidt, M. Pitfalls and Novel Applications of Particle Sizing by Dynamic Light Scattering. Biomaterials 2016, 98, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Phillies, G.D.J. Interpretation of Fluorescence Correlation Spectra of Biopolymer Solutions. Biopolymers 2016, 105, 260–266. [Google Scholar] [CrossRef]
- Piñeiro, L.; Novo, M.; Al-Soufi, W. Fluorescence Emission of Pyrene in Surfactant Solutions. Adv. Colloid Interface Sci. 2015, 215, 1–12. [Google Scholar] [CrossRef]
- Khan, M.F.; Singh, M.K.; Sen, S. Measuring Size, Size Distribution, and Polydispersity of Water-in-Oil Microemulsion Droplets Using Fluorescence Correlation Spectroscopy: Comparison to Dynamic Light Scattering. J. Phys. Chem. B 2016, 120, 1008–1020. [Google Scholar] [CrossRef]
- Chamieh, J.; Merdassi, H.; Rossi, J.C.; Jannin, V.; Demarne, F.; Cottet, H. Size Characterization of Lipid-Based Self-Emulsifying Pharmaceutical Excipients during Lipolysis Using Taylor Dispersion Analysis with Fluorescence Detection. Int. J. Pharm. 2018, 537, 94–101. [Google Scholar] [CrossRef]
- Vithani, K.; Jannin, V.; Pouton, C.W.; Boyd, B.J. Colloidal Aspects of Dispersion and Digestion of Self-Dispersing Lipid-Based Formulations for Poorly Water-Soluble Drugs. Adv. Drug Deliv. Rev. 2019, 142, 16–34. [Google Scholar] [CrossRef]
- Chamieh, J.; Jannin, V.; Demarne, F.; Cottet, H. Hydrodynamic Size Characterization of a Self-Emulsifying Lipid Pharmaceutical Excipient by Taylor Dispersion Analysis with Fluorescent Detection. Int. J. Pharm. 2016, 513, 262–269. [Google Scholar] [CrossRef]
- Tominaga, T.; Nishinaka, M. Tracer Diffusion of Ionic Micelles: Effects of Size and Interactions. J. Chem. Soc. Faraday Trans. 1993, 89, 3459–3464. [Google Scholar] [CrossRef]
- Chamieh, J.; Davanier, F.; Jannin, V.; Demarne, F.; Cottet, H. Size Characterization of Commercial Micelles and Microemulsions by Taylor Dispersion Analysis. Int. J. Pharm. 2015, 492, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Wan, J.; Xu, H.; Yang, X. A New Solid Self-Microemulsifying Formulation Prepared by Spray-Drying to Improve the Oral Bioavailability of Poorly Water Soluble Drugs. Eur. J. Pharm. Biopharm. 2008, 70, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.M.; Nazzal, S. Novel Sulforaphane-Enabled Self-Microemulsifying Delivery Systems (SFN-SMEDDS) of Taxanes: Formulation Development and in Vitro Cytotoxicity against Breast Cancer Cells. Int. J. Pharm. 2018, 536, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.X.; Tang, D.; Feng, L.; Zheng, Z.G.; Wang, R.S.; Wu, A.G.; Duan, T.T.; He, B.; Zhu, Q. Development of Self-Microemulsifying Drug Delivery System for Oral Bioavailability Enhancement of Berberine Hydrochloride. Drug Dev. Ind. Pharm. 2013, 39, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, S.; Guo, Y.; Zhao, Q.; Zhang, X.; Pan, W.; Yang, X. Preparation and Pharmacokinetics Evaluation of Oral Self-Emulsifying System for Poorly Water-Soluble Drug Lornoxicam. Drug Deliv. 2015, 22, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, S.P.; Yuen, K.H. Influence of Lipolysis and Droplet Size on Tocotrienol Absorption from Self-Emulsifying Formulations. Int. J. Pharm. 2004, 281, 67–78. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Mueller, E.A.; van Bree, J.B.; Tetzloff, W.; Kutz, K. Reduced Inter- and Intraindividual Variability in Cyclosporine Pharmacokinetics from a Microemulsion Formulation. J. Pharm. Sci. 1994, 83, 444–446. [Google Scholar] [CrossRef]
- Nielsen, F.S.; Petersen, K.B.; Müllertz, A. Bioavailability of Probucol from Lipid and Surfactant Based Formulations in Minipigs: Influence of Droplet Size and Dietary State. Eur. J. Pharm. Biopharm. 2008, 69, 553–562. [Google Scholar] [CrossRef]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.S.; Porter, C.J.H. 50 Years of Oral Lipid-Based Formulations: Provenance, Progress and Future Perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-Nano-Emulsifying Drug Delivery Systems: An Update of the Biopharmaceutical Aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, I.P.; Steiner, C.; Schmutzler, M.; Wilcox, M.D.; Veldhuis, G.J.; Pearson, J.P.; Huck, C.W.; Salvenmoser, W.; Bernkop-Schnürch, A. Mucus Permeating Carriers: Formulation and Characterization of Highly Densely Charged Nanoparticles. Eur. J. Pharm. Biopharm. 2015, 97, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Netsomboon, K.; Bernkop-Schnürch, A. Mucoadhesive vs. Mucopenetrating Particulate Drug Delivery. Eur. J. Pharm. Biopharm. 2016, 98, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Corbo, D.C.; Liu, J.-C.; Chienx, Y.W. Characterization of the Barrier Properties of Mucosal Membranes. J. Pharm. Sci. 1990, 79, 202–206. [Google Scholar] [CrossRef]
- Gershanik, T.; Benita, S. Positively Charged Self-Emulsifying Oil Formulation for Improving Oral Bioavailability of Progesterone. Pharm. Dev. Technol. 1996, 1, 147–157. [Google Scholar] [CrossRef]
- Salimi, E.; Le-Vinh, B.; Zahir-Jouzdani, F.; Matuszczak, B.; Ghaee, A.; Bernkop-Schnürch, A. Self-Emulsifying Drug Delivery Systems Changing Their Zeta Potential via a Flip-Flop Mechanism. Int. J. Pharm. 2018, 550, 200–206. [Google Scholar] [CrossRef]
- Basalious, E.B.; Shawky, N.; Badr-Eldin, S.M. SNEDDS Containing Bioenhancers for Improvement of Dissolution and Oral Absorption of Lacidipine. I: Development and Optimization. Int. J. Pharm. 2010, 391, 203–211. [Google Scholar] [CrossRef]
- Wang, L.; Dong, J.; Chen, J.; Eastoe, J.; Li, X. Design and Optimization of a New Self-Nanoemulsifying Drug Delivery System. J. Colloid Interface Sci. 2009, 330, 443–448. [Google Scholar] [CrossRef]
- Czajkowska-Kośnik, A.; Szekalska, M.; Amelian, A.; Szymańska, E.; Winnicka, K. Development and Evaluation of Liquid and Solid Self-Emulsifying Drug Delivery Systems for Atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Mandal, S.S. Microemulsion Drug Delivery System: A Platform for Improving Dissolution Rate of Poorly Water Soluble Drug. Int. J. Pharm. Sci. Nanotechnol. 2011, 3, 1214–1219. [Google Scholar] [CrossRef]
- Patel, J.; Dhingani, A.; Garala, K.; Raval, M.; Sheth, N. Quality by Design Approach for Oral Bioavailability Enhancement of Irbesartan by Self-Nanoemulsifying Tablets. Drug Deliv. 2014, 21, 412–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazzal, S.; Smalyukh, I.I.; Lavrentovich, O.D.; Khan, M.A. Preparation and in Vitro Characterization of a Eutectic Based Semisolid Self-Nanoemulsified Drug Delivery System (SNEDDS) of Ubiquinone: Mechanism and Progress of Emulsion Formation. Int. J. Pharm. 2002, 235, 247–265. [Google Scholar] [CrossRef]
- Bali, V.; Ali, M.; Ali, J. Study of Surfactant Combinations and Development of a Novel Nanoemulsion for Minimising Variations in Bioavailability of Ezetimibe. Colloids Surfaces B Biointerfaces 2010, 76, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Parikh, K.J.; Sawant, K.K. Solubilization of Vardenafil HCl in Lipid-Based Formulations Enhances Its Oral Bioavailability in Vivo: A Comparative Study Using Tween®—20 and Cremophor®—EL. J. Mol. Liq. 2019, 277, 189–199. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Katare, O.P.; Singh, B. Optimized Self Nano-Emulsifying Systems of Ezetimibe with Enhanced Bioavailability Potential Using Long Chain and Medium Chain Triglycerides. Colloids Surfaces B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef]
- Gupta, S.; Chavhan, S.; Sawant, K.K. Self-Nanoemulsifying Drug Delivery System for Adefovir Dipivoxil: Design, Characterization, in Vitro and Ex Vivo Evaluation. Colloids Surfaces A Physicochem. Eng. Asp. 2011, 392, 145–155. [Google Scholar] [CrossRef]
- Elsheikh, M.A.; Elnaggar, Y.S.R.; Gohar, E.Y.; Abdallah, O.Y. Nanoemulsion Liquid Preconcentrates for Raloxifene Hydrochloride: Optimization and in Vivo Appraisal. Int. J. Nanomed. 2012, 7, 3787–3802. [Google Scholar] [CrossRef] [Green Version]
- Elnaggar, Y.S.R.; El-Massik, M.A.; Abdallah, O.Y. Self-Nanoemulsifying Drug Delivery Systems of Tamoxifen Citrate: Design and Optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef]
- Shafiq, S.; Shakeel, F.; Talegaonkar, S.; Ahmad, F.J.; Khar, R.K.; Ali, M. Development and Bioavailability Assessment of Ramipril Nanoemulsion Formulation. Eur. J. Pharm. Biopharm. 2007, 66, 227–243. [Google Scholar] [CrossRef]
- Zhuang, X.; Tian, X.; Zheng, Y.; Lan, N.; Liu, L.; Zhang, R.; Liu, Y. Formulation and Physicochemical Characterisation of a Novel Self-Microemulsifying Delivery System as Hydrotropic and Solubilising Agent for Penfluridol. Procedia Eng. 2011, 18, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Hedge, O.J.; Bergström, C.A.S. Suitability of Artificial Membranes in Lipolysis-Permeation Assays of Oral Lipid-Based Formulations. Pharm. Res. 2020, 37, 9. [Google Scholar] [CrossRef] [PubMed]
- Alvebratt, C.; Keemink, J.; Edueng, K.; Cheung, O.; Strømme, M.; Bergström, C.A.S. An in Vitro Dissolution-digestion-permeation Assay for the Study of Advanced Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2020, 149, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-Emulsifying Drug Delivery Systems: An Approach to Enhance Oral Bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Ramesh, D. Design, Optimization and Evaluation of Glipizide Solid Self-Nanoemulsifying Drug Delivery for Enhanced Solubility and Dissolution. Saudi Pharm. J. 2015, 23, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, G.K.; Mantelou, P.; Karavasili, C.; Chatzopoulou, P.; Katsantonis, D.; Irakli, M.; Mygdalia, A.; Vizirianakis, I.S.; Fatouros, D.G. Development and Characterization of a Self-Nanoemulsifying Drug Delivery System Comprised of Rice Bran Oil for Poorly Soluble Drugs. AAPS Pharmscitech 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Abouhussein, D.M.N.; Bahaa El Din Mahmoud, D.; Mohammad, F.E. Design of a Liquid Nano-Sized Drug Delivery System with Enhanced Solubility of Rivaroxaban for Venous Thromboembolism Management in Paediatric Patients and Emergency Cases. J. Liposome Res. 2019, 29, 399–412. [Google Scholar] [CrossRef]
- Vertzoni, M.; Dressman, J.; Butler, J.; Hempenstall, J.; Reppas, C. Simulation of Fasting Gastric Conditions and Its Importance for the in Vivo Dissolution of Lipophilic Compounds. Eur. J. Pharm. Biopharm. 2005, 60, 413–417. [Google Scholar] [CrossRef]
- Kamboj, S.; Rana, V. Quality-by-Design Based Development of a Self-Microemulsifying Drug Delivery System to Reduce the Effect of Food on Nelfinavir Mesylate. Int. J. Pharm. 2016, 501, 311–325. [Google Scholar] [CrossRef]
- Prajapat, M.D.; Patel, N.J.; Bariya, A.; Patel, S.S.; Butani, S.B. Formulation and Evaluation of Self-Emulsifying Drug Delivery System for Nimodipine, a BCS Class II Drug. J. Drug Deliv. Sci. Technol. 2017, 39, 59–68. [Google Scholar] [CrossRef]
- Barba, A.A.; Dalmoro, A.; Bochicchio, S.; de Simone, V.; Caccavo, D.; Iannone, M.; Lamberti, G. Engineering Approaches for Drug Delivery Systems Production and Characterization. Int. J. Pharm. 2020, 581, 119267. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution Media Simulating Conditions in the Proximal Human Gastrointestinal Tract: An Update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, F.; Zarmpi, P.; Kuiper, J.; Fotaki, N. Investigation of Drug Partition Kinetics to Fat in Simulated Fed State Gastric Conditions Based on Drug Properties. Eur. J. Pharm. Sci. 2020, 146, 105263. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Reppas, C. In Vitro-in Vivo Correlations for Lipophilic, Poorly Water-Soluble Drugs. Eur. J. Pharm. Sci. 2000, 11, 73–80. [Google Scholar] [CrossRef]
- Nicolaides, E.; Symillides, M.; Dressman, J.B.; Reppas, C. Biorelevant Dissolution Testing to Predict the Plasma Profile of Lipophilic Drugs after Oral Administration. Pharm. Res. 2001, 18, 380–388. [Google Scholar] [CrossRef]
- van der Meer, J.W.M.; Keuning, J.J.; Scheijgrond, H.W.; Heykants, J.; van Cutsem, J.; Brugmans, J. The Influence of Gastric Acidity on the Bio-Availability of Ketoconazole. J. Antimicrob. Chemother. 1980, 6, 552–554. [Google Scholar] [CrossRef]
- Galia, E.; Nicolaides, E.; Hörter, D.; Löbenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of Various Dissolution Media for Predicting In Vivo Performance of Class I and II Drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef]
- Mendes, C.; Buttchevitz, A.; Kruger, J.H.; Caon, T.; de Oliveira Benedet, P.; Lemos-Senna, E.; Silva, M.A.S. Self-Nanoemulsified Drug Delivery System of Hydrochlorothiazide for Increasing Dissolution Rate and Diuretic Activity. AAPS Pharmscitech 2017, 18, 2494–2504. [Google Scholar] [CrossRef]
- Khan, J.; Hawley, A.; Rades, T.; Boyd, B.J. In Situ Lipolysis and Synchrotron Small-Angle X-Ray Scattering for the Direct Determination of the Precipitation and Solid-State Form of a Poorly Water-Soluble Drug during Digestion of a Lipid-Based Formulation. J. Pharm. Sci. 2016, 105, 2631–2639. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of in Vitro Digestion Profiles across a Range of Representative Formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Bakala-N’Goma, J.C.; Williams, H.D.; Sassene, P.J.; Kleberg, K.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the Establishment of Standardized in Vitro Tests for Lipid-Based Formulations. 5. Lipolysis of Representative Formulations by Gastric Lipase. Pharm. Res. 2015, 32, 1279–1287. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. Use of a Dynamic in Vitro Lipolysis Model to Rationalize Oral Formulation Development for Poor Water Soluble Drugs: Correlation with in Vivo Data and the Relationship to Intra-Enterocyte Processes in Rats. Pharm. Res. 2006, 23, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Hoffman, A. The Effect of Different Lipid Based Formulations on the Oral Absorption of Lipophilic Drugs: The Ability of in Vitro Lipolysis and Consecutive Ex Vivo Intestinal Permeability Data to Predict in Vivo Bioavailability in Rats. Eur. J. Pharm. Biopharm. 2007, 67, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Crum, M.F.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Porter, C.J.H. A New in Vitro Lipid Digestion—In Vivo Absorption Model to Evaluate the Mechanisms of Drug Absorption from Lipid-Based Formulations. Pharm. Res. 2016, 33, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Reis, P.; Miller, R.; Leser, M.; Watzke, H. Lipase-Catalyzed Reactions at Interfaces of Two-Phase Systems and Microemulsions. Appl. Biochem. Biotechnol. 2009, 158, 706–721. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.J.H.; Kaukonen, A.M.; Taillardat-Bertschinger, A.; Boyd, B.J.; O’Connor, J.M.; Edwards, G.A.; Charman, W.N. Use of in Vitro Lipid Digestion Data to Explain the in Vivo Performance of Triglyceride-Based Oral Lipid Formulations of Poorly Water-Soluble Drugs: Studies with Halofantrine. J. Pharm. Sci. 2004, 93, 1110–1121. [Google Scholar] [CrossRef]
- Fatouros, D.G.; Mullertz, A. In Vitro Lipid Digestion Models in Design of Drug Delivery Systems for Enhancing Oral Bioavailability. Expert Opin. Drug Metab. Toxicol. 2008, 4, 65–76. [Google Scholar] [CrossRef]
- Larsen, A.; Holm, R.; Pedersen, M.L.; Müllertz, A. Lipid-Based Formulations for Danazol Containing a Digestible Surfactant, Labrafil® M2125CS: In Vivo Bioavailability and Dynamic in Vitro Lipolysis. Pharm. Res. 2008, 25, 2769–2777. [Google Scholar] [CrossRef]
- Christophersen, P.C.; Christiansen, M.L.; Holm, R.; Kristensen, J.; Jacobsen, J.; Abrahamsson, B.; Müllertz, A. Fed and Fasted State Gastro-Intestinal in Vitro Lipolysis: In Vitro in Vivo Relations of a Conventional Tablet, a SNEDDS and a Solidified SNEDDS. Eur. J. Pharm. Sci. 2014, 57, 232–239. [Google Scholar] [CrossRef]
- Fei, Y.; Kostewicz, E.S.; Sheu, M.T.; Dressman, J.B. Analysis of the Enhanced Oral Bioavailability of Fenofibrate Lipid Formulations in Fasted Humans Using an in Vitro-in Silico-in Vivo Approach. Eur. J. Pharm. Biopharm. 2013, 85, 1274–1284. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Müllertz, A.; Rades, T. In Vitro and in Vivo Performance of Novel Supersaturated Self-Nanoemulsifying Drug Delivery Systems (super-SNEDDS). J. Control. Release 2012, 160, 25–32. [Google Scholar] [CrossRef]
- Bibi, H.A.; Holm, R.; Bauer-Brandl, A. Simultaneous Lipolysis/permeation in Vitro Model, for the Estimation of Bioavailability of Lipid Based Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2017, 117, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.T.; Tran, C.S.; Pham, T.M.H.; Nguyen, H.A.; Nguyen, T.L.; Chi, S.C.; Nguyen, D.D.; Bui, T.B.H. Development of Solidified Self-Microemulsifying Drug Delivery Systems Containing L-Tetrahydropalmatine: Design of Experiment Approach and Bioavailability Comparison. Int. J. Pharm. 2018, 537, 9–21. [Google Scholar] [CrossRef]
- Stillhart, C.; Kuentz, M. Comparison of High-Resolution Ultrasonic Resonator Technology and Raman Spectroscopy as Novel Process Analytical Tools for Drug Quantification in Self-Emulsifying Drug Delivery Systems. J. Pharm. Biomed. Anal. 2012, 59, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Holm, R.; Rades, T.; Müllertz, A. Characterising Lipid Lipolysis and Its Implication in Lipid-Based Formulation Development. AAPS J. 2012, 14, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Stillhart, C.; Cavegn, M.; Kuentz, M. Study of Drug Supersaturation for Rational Early Formulation Screening of Surfactant/co-Solvent Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Stillhart, C.; Imanidis, G.; Kuentz, M. Insights into Drug Precipitation Kinetics during in Vitro Digestion of a Lipid-Based Drug Delivery System Using in-Line Raman Spectroscopy and Mathematical Modeling. Pharm. Res. 2013, 30, 3114–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, D.B.; Anby, M.U.; Hawley, A.; Boyd, B.J. Real Time Evolution of Liquid Crystalline Nanostructure during the Digestion of Formulation Lipids Using Synchrotron Small-Angle X-ray Scattering. Langmuir 2011, 27, 9528–9534. [Google Scholar] [CrossRef]
- Phan, S.; Hawley, A.; Mulet, X.; Waddington, L.; Prestidge, C.A.; Boyd, B.J. Structural Aspects of Digestion of Medium Chain Triglycerides Studied in Real Time Using sSAXS and Cryo-TEM. Pharm. Res. 2013, 30, 3088–3100. [Google Scholar] [CrossRef]
- Vithani, K.; Hawley, A.; Jannin, V.; Pouton, C.; Boyd, B.J. Solubilisation Behaviour of Poorly Water-Soluble Drugs during Digestion of Solid SMEDDS. Eur. J. Pharm. Biopharm. 2018, 130, 236–246. [Google Scholar] [CrossRef]
- Psimadas, D.; Georgoulias, P.; Valotassiou, V.; Loudos, G. Molecular Nanomedicine towards Cancer. J. Pharm. Sci. 2012, 101, 2271–2280. [Google Scholar] [CrossRef]
- Heshmati, N.; Cheng, X.; Dapat, E.; Sassene, P.; Eisenbrand, G.; Fricker, G.; Müllertz, A. In Vitro and in Vivo Evaluations of the Performance of an Indirubin Derivative, Formulated in Four Different Self-Emulsifying Drug Delivery Systems. J. Pharm. Pharmacol. 2014, 66, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Berthelsen, R.; Holm, R.; Jacobsen, J.; Kristensen, J.; Abrahamsson, B.; Müllertz, A. Kolliphor Surfactants Affect Solubilization and Bioavailability of Fenofibrate. Studies of in Vitro Digestion and Absorption in Rats. Mol. Pharm. 2015, 12, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Do, T.T.; Van Speybroeck, M.; Mols, R.; Annaert, P.; Martens, J.; Van Humbeeck, J.; Vermant, J.; Augustijns, P.; van den Mooter, G. The Conflict between in Vitro Release Studies in Human Biorelevant Media and the in Vivo Exposure in Rats of the Lipophilic Compound Fenofibrate. Int. J. Pharm. 2011, 414, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.T.; Kuentz, M.; Vertzoni, M.; Kostewicz, E.S.; Fei, Y.; Faisal, W.; Stillhart, C.; O’Driscoll, C.M.; Reppas, C.; Dressman, J.B. Comparison of in Vitro Tests at Various Levels of Complexity for the Prediction of in Vivo Performance of Lipid-Based Formulations: Case Studies with Fenofibrate. Eur. J. Pharm. Biopharm. 2014, 86, 427–437. [Google Scholar] [CrossRef]
- Mosgaard, M.D.; Sassene, P.; Mu, H.; Rades, T.; Müllertz, A. Development of a High-Throughput in Vitro Intestinal Lipolysis Model for Rapid Screening of Lipid-Based Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2015, 94, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Mosgaard, M.D.; Sassene, P.J.; Mu, H.; Rades, T.; Müllertz, A. High-Throughput Lipolysis in 96-Well Plates for Rapid Screening of Lipid-Based Drug Delivery Systems. J. Pharm. Sci. 2017, 106, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Keemink, J.; Mårtensson, E.; Bergström, C.A.S. Lipolysis-permeation setup for simultaneous study of digestion and absorption in vitro. Mol. Pharm. 2019, 16, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Alskär, L.C.; Parrow, A.; Keemink, J.; Johansson, P.; Abrahamsson, B.; Bergström, C.A.S. Effect of Lipids on Absorption of Carvedilol in Dogs: Is Coadministration of Lipids as Efficient as a Lipid-Based Formulation? J. Control. Release 2019, 304, 90–100. [Google Scholar] [CrossRef]
- Ciappellano, S.G.; Tedesco, E.; Venturini, M.; Benetti, F. In Vitro Toxicity Assessment of Oral Nanocarriers. Adv. Drug Deliv. Rev. 2016, 106, 381–401. [Google Scholar] [CrossRef]
- Avdeef, A. The Rise of PAMPA. Expert Opin. Drug Metab. Toxicol. 2005, 1, 325–342. [Google Scholar] [CrossRef]
- Cabrera-Pérez, M.Á.; Sanz, M.B.; Sanjuan, V.M.; González-Álvarez, M.; Álvarez, I.G. Importance and Applications of Cell-and Tissue-Based in Vitro Models for Drug Permeability Screening in Early Stages of Drug Development. In Concepts and Models for Drug Permeability Studies: Cell and Tissue based In Vitro Culture Models; Woodhead Publishing: Cambridge, UK, 2016; pp. 3–29. [Google Scholar] [CrossRef]
- Hiremath, P.S.; Soppimath, K.S.; Betageri, G.V. Proliposomes of Exemestane for Improved Oral Delivery: Formulation and in Vitro Evaluation Using PAMPA, Caco-2 and Rat Intestine. Int. J. Pharm. 2009, 380, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Berben, P.; Bauer-Brandl, A.; Brandl, M.; Faller, B.; Flaten, G.E.; Jacobsen, A.C.; Brouwers, J.; Augustijns, P. Drug Permeability Profiling Using Cell-Free Permeation Tools: Overview and Applications. Eur. J. Pharm. Sci. 2018, 119, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Artificial Membrane Technologies to Assess Transfer and Permeation of Drugs in Drug Discovery. Compr. Med. Chem. II 2006, 5, 453–487. [Google Scholar] [CrossRef]
- Diukendjieva, A.; Tsakovska, I.; Alov, P.; Pencheva, T.; Pajeva, I.; Worth, A.P.; Madden, J.C.; Cronin, M.T.D. Advances in the Prediction of Gastrointestinal Absorption: Quantitative Structure-Activity Relationship (QSAR) Modelling of PAMPA Permeability. Comput. Toxicol. 2019, 10, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Nekkanti, V.; Wang, Z.; Betageri, G.V. Pharmacokinetic Evaluation of Improved Oral Bioavailability of Valsartan: Proliposomes versus Self-Nanoemulsifying Drug Delivery System. AAPS Pharmscitech 2016, 17, 851–862. [Google Scholar] [CrossRef]
- Dumont, C.; Bourgeois, S.; Fessi, H.; Jannin, V. Lipid-Based Nanosuspensions for Oral Delivery of Peptides, a Critical Review. Int. J. Pharm. 2018, 541, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Sambuy, Y.; de Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 Cell Line as a Model of the Intestinal Barrier: Influence of Cell and Culture-Related Factors on Caco-2 Cell Functional Characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Föger, F.; Kopf, A.; Loretz, B.; Albrecht, K.; Bernkop-Schnürch, A. Correlation of in Vitro and in Vivo Models for the Oral Absorption of Peptide Drugs. Amino Acids 2008, 35, 233–241. [Google Scholar] [CrossRef]
- Artursson, P.; Palm, K.; Luthman, K. Caco-2 Monolayers in Experimental and Theoretical Predictions of Drug Transport. Adv. Drug Deliv. Rev. 2012, 64, 280–289. [Google Scholar] [CrossRef]
- Buya, A.B.; Ucakar, B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Design and Evaluation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDSs) for Senicapoc. Int. J. Pharm. 2020, 580, 119180. [Google Scholar] [CrossRef]
- Dantzig, A.H.; Bergin, L. Uptake of the Cephalosporin, Cephalexin, by a Dipeptide Transport Carrier in the Human Intestinal Cell Line, Caco-2. BBA Biomembr. 1990, 1027, 211–217. [Google Scholar] [CrossRef]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized Conditions for Prediction of Intestinal Drug Permeability Using Caco-2 Cells. Eur. J. Pharm. Sci. 2000, 10, 195–204. [Google Scholar] [CrossRef]
- Obringer, C.; Manwaring, J.; Goebel, C.; Hewitt, N.J.; Rothe, H. Suitability of the in Vitro Caco-2 Assay to Predict the Oral Absorption of Aromatic Amine Hair Dyes. Toxicol. Vitr. 2016, 32, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Love, S.A.; Maurer-Jones, M.A.; Thompson, J.W.; Lin, Y.-S.; Haynes, C.L. Assessing Nanoparticle Toxicity. Annu. Rev. Anal. Chem. 2012, 5, 181–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a Novel Therapeutic Approach for the Treatment of Vemurafenib Resistant Melanoma: Preformulation Studies, Formulation Development and in Vitro Evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, L.; Shen, R.; Gong, L.; Tian, Z.; Qiu, H.; Shi, Z.; Gao, L.; Sun, H.; Zhang, G. Self-Nanoemulsifying System Improves Oral Absorption and Enhances Anti-Acute Myeloid Leukemia Activity of Berberine. J. Nanobiotechnol. 2018, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Alam, M.N.; Ahmad, F.J.; Singh, B. Chylomicron Mimicking Nanocolloidal Carriers of Rosuvastatin Calcium for Lymphatic Drug Targeting and Management of Hyperlipidemia. Colloids Surfaces B Biointerfaces 2019, 177, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhang, Q.; Shi, W.; Wang, J. Mechanisms of Oral Absorption Improvement for Insoluble Drugs by the Combination of Phospholipid Complex and SNEDDS. Drug Deliv. 2019, 26, 1155–1166. [Google Scholar] [CrossRef]
- Aktas, Y.; Celik Tekeli, M.; Celebi, N. Development and Characterization of Exendin-4 Loaded Self-Nanoemulsifying System and in Vitro Evaluation on Caco-2 Cell Line. J. Microencapsul. 2020, 37, 41–51. [Google Scholar] [CrossRef]
- Kontogiannidou, E.; Meikopoulos, T.; Virgiliou, C.; Bouropoulos, N.; Gika, H.; Vizirianakis, I.S.; Müllertz, A.; Fatouros, D.G. Towards the Development of Self-Nano-Emulsifying Drug Delivery Systems (SNEDDS) Containing Trimethyl Chitosan for the Oral Delivery of Amphotericin B: In Vitro Assessment and Cytocompatibility Studies. J. Drug Deliv. Sci. Technol. 2020, 56, 101524. [Google Scholar] [CrossRef]
- Jones, C.F.; Grainger, D.W. In Vitro Assessments of Nanomaterial Toxicity. Adv. Drug Deliv. Rev. 2009, 61, 438–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquis, B.J.; Love, S.A.; Braun, K.L.; Haynes, C.L. Analytical Methods to Assess Nanoparticle Toxicity. Analyst 2009, 134, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Riviere, N.A.; Inman, A.O.; Zhang, L.W. Limitations and Relative Utility of Screening Assays to Assess Engineered Nanoparticle Toxicity in a Human Cell Line. Toxicol. Appl. Pharmacol. 2009, 234, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Botha, N.; Gehringer, M.M.; Downing, T.G.; van de Venter, M.; Shephard, E.G. The Role of Microcystin-LR in the Induction of Apoptosis and Oxidative Stress in CaCo2 Cells. Toxicon 2004, 43, 85–92. [Google Scholar] [CrossRef]
- Fisichella, M.; Bérenguer, F.; Steinmetz, G.; Auffan, M.; Rose, J.; Prat, O. Reply to Comment on Fisichella et al. (2012), “Intestinal Toxicity Evaluation of TiO2 Degraded Surface-Treated Nanoparticles: A Combined Physico-Chemical and Toxicogenomics Approach in Caco-2 Cells” by Faust et al. Part. Fibre Toxicol. 2012, 9, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Holder, A.L.; Goth-Goldstein, R.; Lucas, D.; Koshland, C.P. Particle-Induced Artifacts in the MTT and LDH Viability Assays. Chem. Res. Toxicol. 2012, 25, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- Alvi, M.M.; Chatterjee, P. A Prospective Analysis of Co-Processed Non-Ionic Surfactants in Enhancing Permeability of a Model Hydrophilic Drug. AAPS Pharmscitech 2014, 15, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004; pp. 1–31. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/23805433 (accessed on 17 April 2020). [PubMed]
- Desai, H.H.; Bu, P.; Shah, A.V.; Cheng, X.; Serajuddin, A.T.M. Evaluation of Cytotoxicity of Self-Emulsifying Formulations Containing Long-Chain Lipids Using Caco-2 Cell Model: Superior Safety Profile Compared to Medium-Chain Lipids. J. Pharm. Sci. 2020, 109, 1752–1764. [Google Scholar] [CrossRef]
- Grassi, M.; Cadelli, G. Theoretical Considerations on the in Vivo Intestinal Permeability Determination by Means of the Single Pass and Recirculating Techniques. Int. J. Pharm. 2001, 229, 95–105. [Google Scholar] [CrossRef]
- Prajapati, S.T.; Joshi, H.A.; Patel, C.N. Preparation and Characterization of Self-Microemulsifying Drug Delivery System of Olmesartan Medoxomil for Bioavailability Improvement. J. Pharm. 2013, 2013, 728425. [Google Scholar] [CrossRef]
- Dezani, T.M.; Dezani, A.B.; Junior, J.B.D.S.; Serra, C.H.D.R. Single-Pass Intestinal Perfusion (SPIP) and Prediction of Fraction Absorbed and Permeability in Humans: A Study with Antiretroviral Drugs. Eur. J. Pharm. Biopharm. 2016, 104, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Liu, Y.; Zhao, B.; Tang, M.; Dong, H.; Zhang, L.; Lv, B.; Wei, L. Ex Vivo and in Situ Approaches Used to Study Intestinal Absorption. J. Pharmacol. Toxicol. Methods 2013, 68, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, L.P.; Luo, S.Q.; Jiang, H.D.; Zeng, S. Intestinal Absorption of Luteolin from Peanut Hull Extract Is More Efficient than That from Individual Pure Luteolin. J. Agric. Food Chem. 2008, 56, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, R.; Bandyopadhyay, S.; Kapil, R.; Garg, B. Optimized Nanoemulsifying Systems with Enhanced Bioavailability of Carvedilol. Colloids Surfaces B Biointerfaces 2013, 101, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kazi, M.; Al-Swairi, M.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Badran, M.M.; Khan, A.A.; Alanazi, A.M.; Hussain, M.D. Evaluation of Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Poorly Water-Soluble Talinolol: Preparation, in Vitroand in vivo Assessment. Front. Pharmacol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Kaur, R.; Khurana, R.K.; Rana, V.; Sharma, T.; Singh, B. QbD-Based Development of Cationic Self-Nanoemulsifying Drug Delivery Systems of Paclitaxel with Improved Biopharmaceutical Attributes. AAPS Pharmscitech 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Sandhu, P.S.; Beg, S.; Mehta, F.; Singh, B.; Trivedi, P. Novel Dietary Lipid-Based Self-Nanoemulsifying Drug Delivery Systems of Paclitaxel with P-Gp Inhibitor: Implications on Cytotoxicity and Biopharmaceutical Performance. Expert Opin. Drug Deliv. 2015, 12, 1809–1822. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, M.; Morrison, R.A.; Chong, S. Commonly Used Surfactant, Tween® 80, Improves Absorption of P-Glycoprotein Substrate, Digoxin, in Rats. Arch. Pharm. Res. 2003, 26, 768–772. [Google Scholar] [CrossRef]
- Seo, Y.G.; Kim, D.H.; Ramasamy, T.; Kim, J.H.; Marasini, N.; Oh, Y.K.; Kim, D.W.; Kim, J.K.; Yong, C.S.; Kim, J.O.; et al. Development of Docetaxel-Loaded Solid Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Enhanced Chemotherapeutic Effect. Int. J. Pharm. 2013, 452, 412–420. [Google Scholar] [CrossRef]
- Truong, D.H.; Tran, T.H.; Ramasamy, T.; Choi, J.Y.; Lee, H.H.; Moon, C.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of Solid Self-Emulsifying Formulation for Improving the Oral Bioavailability of Erlotinib. AAPS Pharmscitech 2016, 17, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Faisal, W.; Ruane-O’Hora, T.; O’Driscoll, C.M.; Griffin, B.T. A Novel Lipid-Based Solid Dispersion for Enhancing Oral Bioavailability of Lycopene—In Vivo Evaluation Using a Pig Model. Int. J. Pharm. 2013, 453, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Bourkaib, N.; Zhou, J.; Yao, J.; Fang, Z.; Mezghrani, O. Combination of β-Cyclodextrin Inclusion Complex and Self-Microemulsifying Drug Delivery System for Photostability and Enhanced Oral Bioavailability of Methotrexate: Novel Technique. Drug Dev. Ind. Pharm. 2013, 39, 918–927. [Google Scholar] [CrossRef]
- Negi, L.M.; Tariq, M.; Talegaonkar, S. Nano Scale Self-Emulsifying Oil Based Carrier System for Improved Oral Bioavailability of Camptothecin Derivative by P-Glycoprotein Modulation. Colloids Surfaces B Biointerfaces 2013, 111, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, P.; Nie, S.; Pan, W. Preparation and Evaluation of SEDDS and SMEDDS Containing Carvedilol. Drug Dev. Ind. Pharm. 2005, 31, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Jing, B.; Wang, Z.; Yang, R.; Zheng, X.; Zhao, J.; Tang, S.; He, Z. Enhanced Oral Bioavailability of Felodipine by Novel Solid Self-Microemulsifying Tablets. Drug Dev. Ind. Pharm. 2016, 42, 506–512. [Google Scholar] [CrossRef]
- Bakhle, S.S.; Avari, J.G. Development and Characterization of Solid Self-Emulsifying Drug Delivery System of Cilnidipine. Chem. Pharm. Bull. 2015, 63, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Chopra, M.; Nayak, U.Y.; Kumar Gurram, A.; Sreenivasa Reddy, M.; Koteshwara, K.B. Formulation, Characterization and In Vivo Evaluation of Self-Nanoemulsifying Drug Delivery System for Oral Delivery of Valsartan. Curr. Nanosci. 2013, 10, 263–270. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Verma, S.; Verma, P.R.P.; Singh, S.K.; Chakraborty, A. Fabrication of Liquid and Solid Self-Double Emulsifying Drug Delivery System of Atenolol by Response Surface Methodology. J. Drug Deliv. Sci. Technol. 2017, 41, 45–57. [Google Scholar] [CrossRef]
- Li, P.; Tan, A.; Prestidge, C.A.; Nielsen, H.M.; Müllertz, A. Self-Nanoemulsifying Drug Delivery Systems for Oral Insulin Delivery: In Vitro and in Vivo Evaluations of Enteric Coating and Drug Loading. Int. J. Pharm. 2014, 477, 390–398. [Google Scholar] [CrossRef]
- Bari, A.; Chella, N.; Sanka, K.; Shastri, N.R.; Diwan, P.V. Improved Anti-Diabetic Activity of Glibenclamide Using Oral Self Nano Emulsifying Powder. J. Microencapsul. 2015, 32, 54–60. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Deng, W.; Omari-Siaw, E.; Wang, Q.; Wang, S.; Wang, S.; Cao, X.; Xu, X.; Yu, J. Self-Nanoemulsifying Drug Delivery System of Trans-Cinnamic Acid: Formulation Development and Pharmacodynamic Evaluation in Alloxan-Induced Type 2 Diabetic Rat Model. Drug Dev. Res. 2015, 76, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wankhade, V.P.; Atram, S.C.; Bobade, N.N.; Pande, S.D.; Tapar, K.K. Formulation and Optimization of SNEDDS of Gliclazide Using Response Surface Methodology. Asian J. Pharm. 2012, 6, 289–294. [Google Scholar] [CrossRef]
- Menzel, C.; Holzeisen, T.; Laffleur, F.; Zaichik, S.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnürch, A. In Vivo Evaluation of an Oral Self-Emulsifying Drug Delivery System (SEDDS) for Exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Jain, A.K.; Pohekar, M.; Thanki, K. Novel Self-Emulsifying Formulation of Quercetin for Improved in Vivo Antioxidant Potential: Implications for Drug-Induced Cardiotoxicity and Nephrotoxicity. Free Radic. Biol. Med. 2013, 65, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Mamadou, G.; Charrueau, C.; Dairou, J.; Limas Nzouzi, N.; Eto, B.; Ponchel, G. Increased Intestinal Permeation and Modulation of Presystemic Metabolism of Resveratrol Formulated into Self-Emulsifying Drug Delivery Systems. Int. J. Pharm. 2017, 521, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Hong, M.; Liu, C.; Pei, Y. Application of Box-Behnken Design in Understanding the Quality of Genistein Self-Nanoemulsified Drug Delivery Systems and Optimizing Its Formulation. Pharm. Dev. Technol. 2009, 14, 642–649. [Google Scholar] [CrossRef]
- Taha, E.I.; Al-Saidan, S.; Samy, A.M.; Khan, M.A. Preparation and in Vitro Characterization of Self-Nanoemulsified Drug Delivery System (SNEDDS) of All-Trans-Retinol Acetate. Int. J. Pharm. 2004, 285, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Garg, B.; Beg, S.; Kaur, R.; Kumar, R.; Katare, O.P.; Singh, B. Long-Chain Triglycerides-Based Self-Nanoemulsifying Oily Formulations (SNEOFs) of Darunavir with Improved Lymphatic Targeting Potential. J. Drug Target. 2018, 26, 252–266. [Google Scholar] [CrossRef]
- Garg, B.; Katare, O.P.; Beg, S.; Lohan, S.; Singh, B. Systematic Development of Solid Self-Nanoemulsifying Oily Formulations (S-SNEOFs) for Enhancing the Oral Bioavailability and Intestinal Lymphatic Uptake of Lopinavir. Colloids Surfaces B Biointerfaces 2016, 141, 611–622. [Google Scholar] [CrossRef]
- Patel, D.; Sawant, K.K. Oral Bioavailability Enhancement of Acyclovir by Self-Microemulsifying Drug Delivery Systems (SMEDDS). Drug Dev. Ind. Pharm. 2007, 33, 1318–1326. [Google Scholar] [CrossRef]
- Hussain, A.; Kumar Singh, S.; Ranjan Prasad Verma, P.; Singh, N.; Jalees Ahmad, F. Experimental Design-Based Optimization of Lipid Nanocarrier as Delivery System against Mycobacterium Species: In Vitro and in Vivo Evaluation. Pharm. Dev. Technol. 2017, 22, 910–927. [Google Scholar] [CrossRef] [PubMed]
- Wasan, E.K.; Bartlett, K.; Gershkovich, P.; Sivak, O.; Banno, B.; Wong, Z.; Gagnon, J.; Gates, B.; Leon, C.G.; Wasan, K.M. Development and Characterization of Oral Lipid-Based Amphotericin B Formulations with Enhanced Drug Solubility, Stability and Antifungal Activity in Rats Infected with Aspergillus Fumigatus or Candida Albicans. Int. J. Pharm. 2009, 372, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Gurav, N.P.; Dandagi, P.M.; Gadad, A.P.; Masthiholimath, V.S. Solubility Enhancement of Satranidazole Using Self Emulsified Drug Delivery Systems. Indian J. Pharm. Educ. Res. 2016, 50, S68–S75. [Google Scholar] [CrossRef]
- Perlman, M.E.; Murdande, S.B.; Gumkowski, M.J.; Shah, T.S.; Rodricks, C.M.; Thornton-Manning, J.; Freel, D.; Erhart, L.C. Development of a Self-Emulsifying Formulation That Reduces the Food Effect for Torcetrapib. Int. J. Pharm. 2008, 351, 15–22. [Google Scholar] [CrossRef] [PubMed]
- El-Say, K.M.; Ahmed, T.A.; Ahmed, O.A.A.; Hosny, K.M.; Abd-Allah, F.I. Self-Nanoemulsifying Lyophilized Tablets for Flash Oral Transmucosal Delivery of Vitamin K: Development and Clinical Evaluation. J. Pharm. Sci. 2017, 106, 2447–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julianto, T.; Yuen, K.H.; Noor, A.M. Improved Bioavailability of Vitamin E with a Self Emulsifying Formulation. Int. J. Pharm. 2000, 200, 53–57. [Google Scholar] [CrossRef]
- Postolache, P.; Petrescu, O.; Dorneanu, V.; Zanini, A.C. Cyclosporine Bioavailability of Two Physically Different Oral Formulations. Eur. Rev. Med. Pharmacol. Sci. 2002, 6, 127–131. [Google Scholar] [PubMed]
- Abdelbary, G.; Amin, M.; Salah, S. Self Nano-Emulsifying Simvastatin Based Tablets: Design and in Vitro/in Vivo Evaluation. Pharm. Dev. Technol. 2013, 18, 1294–1304. [Google Scholar] [CrossRef]
- Roche Laboratoiries Home Page. Available online: http://www.pharmatimes.com/news/roche_to_pull_Fortovase®_hiv_drug_998246 (accessed on 18 May 2020).
- Mohsin, K.; Alamri, R.; Ahmad, A.; Raish, M.; Alanazi, F.K.; Hussain, M.D. Development of Self-Nanoemulsifying Drug Delivery Systems for the Enhancement of Solubility and Oral Bioavailability of Fenofibrate, A Poorly Water-Soluble Drug. Int. J. Nanomed. 2016, 11, 2829–2838. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Ha, E.; Kim, M. Current Status of Supersaturable Self-Emulsifying Drug Delivery Systems. Pharmaceutics 2020, 12, 365. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and Lipid-Based Formulations: Optimizing the Oral Delivery of Lipophilic Drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.Ø.; Schultz, K.; Mollgaard, B.; Kristensen, H.G.; Mullertz, A. Solubilisation of Poorly Water-Soluble Drugs during in Vitro Lipolysis of Medium- and Long-Chain Triacylglycerols. Eur. J. Pharm. Sci. 2004, 23, 287–296. [Google Scholar] [CrossRef]
- Kaukonen, A.M.; Boyd, B.J.; Porter, C.J.H.; Charman, W.N. Drug Solubilization Behavior during in Vitro Digestion of Simple Triglyceride Lipid Solution Formulations. Pharm. Res. 2004, 21, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Holm, R.; Garmer, M.; Karlsson, J.J.; Müllertz, A.; Rades, T. Supersaturated Self-Nanoemulsifying Drug Delivery Systems (Super-SNEDDS) Enhance the Bioavailability of the Poorly Water-Soluble Drug Simvastatin in Dogs. AAPS J. 2013, 15, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, T.; Plakogiannis, F.M. Development and Oral Bioavailability Assessment of a Supersaturated Self-Microemulsifying Drug Delivery System (SMEDDS) of Albendazole. J. Pharm. Pharmacol. 2010, 62, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Katare, O.; Singh, B. Development of Optimized Supersaturable Self-Nanoemulsifying Systems of Ezetimibe: Effect of Polymers and Efflux Transporters. Expert Opin. Drug Deliv. 2014, 11, 479–492. [Google Scholar] [CrossRef]
- Strindberg, S.; Plum, J.; Stie, M.B.; Christiansen, M.L.; Hagner Nielsen, L.; Rades, T.; Müllertz, A. Effect of Supersaturation on Absorption of Indomethacin and Tadalafil in a Single Pass Intestinal Perfusion Rat Model, in the Absence and Presence of a Precipitation Inhibitor. Eur. J. Pharm. Biopharm. 2020, 151, 108–115. [Google Scholar] [CrossRef]
- van Speybroeck, M.; Mellaerts, R.; Mols, R.; Do Thi, T.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; van den Mooter, G.; Augustijns, P. Enhanced Absorption of the Poorly Soluble Drug Fenofibrate by Tuning Its Release Rate from Ordered Mesoporous Silica. Eur. J. Pharm. Sci. 2010, 41, 623–630. [Google Scholar] [CrossRef]
- Bannow, J.; Yorulmaz, Y.; Löbmann, K.; Müllertz, A.; Rades, T. Improving the Drug Load and in Vitro Performance of Supersaturated Self-Nanoemulsifying Drug Delivery Systems (super-SNEDDS) Using Polymeric Precipitation Inhibitors. Int. J. Pharm. 2020, 575, 118960. [Google Scholar] [CrossRef]
- Quan, G.; Niu, B.; Singh, V.; Zhou, Y.; Wu, C.Y.; Pan, X.; Wu, C. Supersaturable Solid Self-Microemulsifying Drug Delivery System: Precipitation Inhibition and Bioavailability Enhancement. Int. J. Nanomed. 2017, 12, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.C. Role of Mucus Layers in Gut Infection and Inflammation. Curr. Opin. Microbiol. 2012, 15, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaichik, S.; Steinbring, C.; Jelkmann, M.; Bernkop-Schnürch, A. Zeta Potential Changing Nanoemulsions: Impact of PEG-Corona on Phosphate Cleavage. Int. J. Pharm. 2020, 581, 119299. [Google Scholar] [CrossRef] [PubMed]
- Hintzen, F.; Perera, G.; Hauptstein, S.; Müller, C.; Laffleur, F.; Bernkop-Schnürch, A. In Vivo Evaluation of an Oral Self-Microemulsifying Drug Delivery System (SMEDDS) for Leuprorelin. Int. J. Pharm. 2014, 472, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Dünnhaupt, S.; Kammona, O.; Waldner, C.; Kiparissides, C.; Bernkop-Schnürch, A. Nano-Carrier Systems: Strategies to Overcome the Mucus Gel Barrier. Eur. J. Pharm. Biopharm. 2015, 96, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Griesser, J.; Hetényi, G.; Kadas, H.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Self-Emulsifying Peptide Drug Delivery Systems: How to Make Them Highly Mucus Permeating. Int. J. Pharm. 2018, 538, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Friedl, H.; Dünnhaupt, S.; Hintzen, F.; Waldner, C.; Parikh, S.; Pearson, J.P.; Wilcox, M.D.; Bernkop-Schnürch, A. Development and Evaluation of a Novel Mucus Diffusion Test System Approved by Self-Nanoemulsifying Drug Delivery Systems. J. Pharm. Sci. 2013, 102, 4406–4413. [Google Scholar] [CrossRef]
- Suchaoin, W.; Pereira de Sousa, I.; Netsomboon, K.; Lam, H.T.; Laffleur, F.; Bernkop-Schnürch, A. Development and in Vitro Evaluation of Zeta Potential Changing Self-Emulsifying Drug Delivery Systems for Enhanced Mucus Permeation. Int. J. Pharm. 2016, 510, 255–262. [Google Scholar] [CrossRef]
- Griesser, J.; Hetényi, G.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Hydrophobic Ion Pairing: Key to Highly Payloaded Self-Emulsifying Peptide Drug Delivery Systems. Int. J. Pharm. 2017, 520, 267–274. [Google Scholar] [CrossRef]
- Nazir, I.; Fürst, A.; Lupo, N.; Hupfauf, A.; Gust, R.; Bernkop-Schnürch, A. Zeta Potential Changing Self-Emulsifying Drug Delivery Systems: A Promising Strategy to Sequentially Overcome Mucus and Epithelial Barrier. Eur. J. Pharm. Biopharm. 2019, 144, 40–49. [Google Scholar] [CrossRef]
- Prüfert, F.; Fischer, F.; Leichner, C.; Zaichik, S.; Bernkop-Schnürch, A. Development and In Vitro Evaluation of Stearic Acid Phosphotyrosine Amide as New Excipient for Zeta Potential Changing Self-Emulsifying Drug Delivery Systems. Pharm. Res. 2020, 37, 79. [Google Scholar] [CrossRef] [Green Version]
- Bernkop-Schnürch, A. Thiomers: A New Generation of Mucoadhesive Polymers. Adv. Drug Deliv. Rev. 2005, 57, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, J.; Dnnhaupt, S.; Hombach, J.; Bernkop-Schnrch, A. Thiomer Nanoparticles: Stabilization via Covalent Cross-Linking. Drug Deliv. 2011, 18, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.; Partenhauser, A.; Hauptstein, S.; Gallati, C.M.; Matuszczak, B.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnürch, A. Mucus Permeating Thiolated Self-Emulsifying Drug Delivery Systems. Eur. J. Pharm. Biopharm. 2016, 98, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Efiana, N.A.; Phan, T.N.Q.; Wicaksono, A.J.; Bernkop-Schnürch, A. Mucus Permeating Self-Emulsifying Drug Delivery Systems (SEDDS): About the Impact of Mucolytic Enzymes. Colloids Surfaces B Biointerfaces 2018, 161, 228–235. [Google Scholar] [CrossRef]
- Bonengel, S.; Haupstein, S.; Perera, G.; Bernkop-Schnürch, A. Thiolated and S-Protected Hydrophobically Modified Cross-Linked Poly(acrylic Acid)—A New Generation of Multifunctional Polymers. Eur. J. Pharm. Biopharm. 2014, 88, 390–396. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Adamovic, N.T.; Lam, H.T.; Rohrer, J.; Partenhauser, A.; Bernkop-Schnürch, A. Self-Emulsifying Drug Delivery Systems (SEDDS): Proof-of-Concept How to Make Them Mucoadhesive. Eur. J. Pharm. Biopharm. 2017, 112, 51–57. [Google Scholar] [CrossRef]
- Abdulkarim, M.; Sharma, P.K.; Gumbleton, M. Self-Emulsifying Drug Delivery System: Mucus Permeation and Innovative Quantification Technologies. Adv. Drug Deliv. Rev. 2019, 142, 62–74. [Google Scholar] [CrossRef]
- Leichner, C.; Menzel, C.; Laffleur, F.; Bernkop-Schnürch, A. Development and in Vitro Characterization of a Papain Loaded Mucolytic Self-Emulsifying Drug Delivery System (SEDDS). Int. J. Pharm. 2017, 530, 346–353. [Google Scholar] [CrossRef]
- Balakrishnan, P.; Lee, B.J.; Oh, D.H.; Kim, J.O.; Lee, Y.I.; Kim, D.D.; Jee, J.P.; Lee, Y.B.; Woo, J.S.; Yong, C.S.; et al. Enhanced Oral Bioavailability of Coenzyme Q10 by Self-Emulsifying Drug Delivery Systems. Int. J. Pharm. 2009, 374, 66–72. [Google Scholar] [CrossRef]
- Milović, M.; Simović, S.; Lošić, D.; Dashevskiy, A.; Ibrić, S. Solid Self-Emulsifying Phospholipid Suspension (SSEPS) with Diatom as a Drug Carrier. Eur. J. Pharm. Sci. 2014, 63, 226–232. [Google Scholar] [CrossRef]
- Chavan, R.B.; Modi, S.R.; Bansal, A.K. Role of Solid Carriers in Pharmaceutical Performance of Solid Supersaturable SEDDS of Celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Soliman, K.A.B.; Ibrahim, H.K.; Ghorab, M.M. Formulation of Avanafil in a Solid Self-Nanoemulsifying Drug Delivery System for Enhanced Oral Delivery. Eur. J. Pharm. Sci. 2016, 93, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Aleksovski, A.; van Bockstal, P.J.; Roškar, R.; Sovány, T.; Regdon, G.; de Beer, T.; Vervaet, C.; Dreu, R. Comparison of Metoprolol Tartrate Multiple-Unit Lipid Matrix Systems Produced by Different Technologies. Eur. J. Pharm. Sci. 2016, 88, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, S.; Katare, O.P.; Saini, S.; Garg, B.; Khurana, R.K.; Singh, B. Solid Self-Nanoemulsifying Systems of Olmesartan Medoxomil: Formulation Development, Micromeritic Characterization, in Vitro and in Vivo Evaluation. Powder Technol. 2016, 294, 93–104. [Google Scholar] [CrossRef]
- Vohra, A.M.; Patel, C.V.; Kumar, P.; Thakkar, H.P. Development of Dual Drug Loaded Solid Self Microemulsifying Drug Delivery System: Exploring Interfacial Interactions Using QbD Coupled Risk Based Approach. J. Mol. Liq. 2017, 242, 1156–1168. [Google Scholar] [CrossRef]
- Yan, Y.D.; Kim, J.A.; Kwak, M.K.; Yoo, B.K.; Yong, C.S.; Choi, H.G. Enhanced Oral Bioavailability of Curcumin via a Solid Lipid-Based Self-Emulsifying Drug Delivery System Using a Spray-Drying Technique. Biol. Pharm. Bull. 2011, 34, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Oh, D.H.; Oh, Y.K.; Yong, C.S.; Choi, H.G. Effects of Solid Carriers on the Crystalline Properties, Dissolution and Bioavailability of Flurbiprofen in Solid Self-Nanoemulsifying Drug Delivery System (solid SNEDDS). Eur. J. Pharm. Biopharm. 2012, 80, 289–297. [Google Scholar] [CrossRef]
- Tang, B.; Cheng, G.; Gu, J.C.; Xu, C.H. Development of Solid Self-Emulsifying Drug Delivery Systems: Preparation Techniques and Dosage Forms. Drug Discov. Today 2008, 13, 606–612. [Google Scholar] [CrossRef]
- Nanda Kishore, R.; Yalavarthi, P.R.; Vadlamudi, H.C.; Vandana, K.R.; Rasheed, A.; Sushma, M. Solid Self Microemulsification of Atorvastatin Using Hydrophilic Carriers: A Design. Drug Dev. Ind. Pharm. 2015, 41, 1213–1222. [Google Scholar] [CrossRef]
- Tarate, B.; Chavan, R.; Bansal, A. Oral Solid Self-Emulsifying Formulations: A Patent Review. Recent Pat. Drug Deliv. Formul. 2014, 8, 126–143. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming Lipid-Based Oral Drug Delivery Systems into Solid Dosage Forms: An Overview of Solid Carriers, Physicochemical Properties, and Biopharmaceutical Performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Szlęk, J.; Jany, B.R.; Jachowicz, R. Preformulation Studies on Solid Self-Emulsifying Systems in Powder Form Containing Magnesium Aluminometasilicate as Porous Carrier. AAPS Pharmscitech 2015, 16, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Kusawake, T.; Ishida, M.; Tawa, R.; Shibata, N.; Takada, K. Oral Solid Gentamicin Preparation Using Emulsifier and Adsorbent. J. Control. Release 2005, 105, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mandić, J.; Zvonar Pobirk, A.; Vrečer, F.; Gašperlin, M. Overview of Solidification Techniques for Self-Emulsifying Drug Delivery Systems from Industrial Perspective. Int. J. Pharm. 2017, 533, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yi, T.; Lam, C.W.K. Effects of Spray-Drying and Choice of Solid Carriers on Concentrations of Labrasol®® and Transcutol®® in Solid Self-Microemulsifying Drug Delivery Systems (SMEDDS). Molecules 2013, 18, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Baek, I.H.; Ha, E.S.; Yoo, J.W.; Jung, Y.; Kim, M.S. Design of a Gelatin Microparticle-Containing Self-Microemulsifying Formulation for Enhanced Oral Bioavailability of Dutasteride. Drug Des. Dev. Ther. 2015, 9, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Čerpnjak, K.; Pobirk, A.Z.; Vrečer, F.; Gašperlin, M. Tablets and Minitablets Prepared from Spray-Dried SMEDDS Containing Naproxen. Int. J. Pharm. 2015, 495, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral Lipid-Based Drug Delivery Systems—An Overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.; Schæfer, T. Melt Agglomeration with Polyethylene Glycol Beads at a Low Impeller Speed in a High Shear Mixer. Eur. J. Pharm. Biopharm. 2001, 52, 315–325. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-Emulsifying Oral Lipid Drug Delivery Systems: Advances and Challenges. AAPS Pharmscitech 2019, 20, 129. [Google Scholar] [CrossRef]
- Breitenbach, J. Melt Extrusion: From Process to Drug Delivery Technology. Eur. J. Pharm. Biopharm. 2002, 54, 107–117. [Google Scholar] [CrossRef]
- GlattGmbH Home Page. Available online: https://www.glatt.com/en/processes/%20pelletizing/extrusion-spheronization/ (accessed on 5 February 2020).
- Abdalla, A.; Mäder, K. Preparation and Characterization of a Self-Emulsifying Pellet Formulation. Eur. J. Pharm. Biopharm. 2007, 66, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, J.; Wang, Y.; Liu, X.; Liu, Y.; Fu, Q.; Meng, P.; He, Z. Solid Self-Emulsifying Nitrendipine Pellets: Preparation and in Vitro/in Vivo Evaluation. Int. J. Pharm. 2010, 383, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Chapter 8—Lipid-Based Nanoparticles for Drug-Delivery Systems. Nanocarriers Drug Deliv. 2019, 249–284. [Google Scholar] [CrossRef]
- Dorset, D.L. X-ray Diffraction: A Practical Approach. Microsc. Microanal. 1998, 4, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gonzalez, M.F.S. Solid Lipid Nanoparticles and Applications. Nanotechnol. Funct. Foods Eff. Deliv. Bioact. Ingred. 2015, 47, 214–223. [Google Scholar] [CrossRef]
- Silva, C.O.; Reis, C.P. Drug Nanocarriers Based on Biomacromolecules: How Far We’ve Come? Nanotechnology 2014, 11, 484. [Google Scholar]
- Westbrook, J.D.; Burley, S.K. How Structural Biologists and the Protein Data Bank Contributed to Recent FDA New Drug Approvals. Structure 2019, 27, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2020 Update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Bernkop-Schnürch, A. Self-Emulsifying Drug Delivery Systems in Oral (poly)peptide Drug Delivery. Expert Opin. Drug Deliv. 2015, 12, 1703–1716. [Google Scholar] [CrossRef]
- Hauptstein, S.; Prüfert, F.; Bernkop-Schnürch, A. Self-Nanoemulsifying Drug Delivery Systems as Novel Approach for pDNA Drug Delivery. Int. J. Pharm. 2015, 487, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Prüfert, F.; Efiana, N.A.; Ashraf, M.I.; Hermann, M.; Hussain, S.; Bernkop-Schnürch, A. Cell-Penetrating Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Oral Gene Delivery. Expert Opin. Drug Deliv. 2016, 13, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nielsen, H.M.; Müllertz, A. Impact of Lipid-Based Drug Delivery Systems on the Transport and Uptake of Insulin across Caco-2 Cell Monolayers. J. Pharm. Sci. 2016, 105, 2743–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Alfaro, D.A.; Muñoz-Correa, M.O.F.; Santos-Luna, D.; Toro-Vazquez, J.F.; Cano-Sarmiento, C.; García-Varela, R.; García, H.S. Encapsulation of an Insulin-Modified Phosphatidylcholine Complex in a Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Oral Insulin Delivery. J. Drug Deliv. Sci. Technol. 2020, 57, 101622. [Google Scholar] [CrossRef]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. Role of Self-Emulsifying Drug Delivery Systems in Optimizing the Oral Delivery of Hydrophilic Macromolecules and Reducing Interindividual Variability. Colloids Surfaces B Biointerfaces 2018, 167, 82–92. [Google Scholar] [CrossRef]
- O’Driscoll, C.M.; Bernkop-Schnürch, A.; Friedl, J.D.; Préat, V.; Jannin, V. Oral Delivery of Non-Viral Nucleic Acid-Based Therapeutics - Do We Have the Guts for This? Eur. J. Pharm. Sci. 2019, 133, 190–204. [Google Scholar] [CrossRef]
- Rao, S.V.R.; Shao, J. Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Oral Delivery of Protein Drugs. I. Formulation Development. Int. J. Pharm. 2008, 362, 2–9. [Google Scholar] [CrossRef]
- Meyer, J.D.; Manning, M.C. Altering Properties of Biomolecules.Pdf. Pharm. Res. 1998, 15, 188–192. [Google Scholar] [CrossRef]
- Zhang, Q.; He, N.; Zhang, L.; Zhu, F.; Chen, Q.; Qin, Y.; Zhang, Z.; Zhang, Q.; Wang, S.; He, Q. The in Vitro and in Vivo Study on Self-Nanoemulsifying Drug Delivery System (SNEDDS) Based on Insulin-Phospholipid Complex. J. Biomed. Nanotechnol. 2012, 8, 90–97. [Google Scholar] [CrossRef]
- Mahjub, R.; Dorkoosh, F.A.; Rafiee-Tehrani, M.; Bernkop Schnürch, A. Oral Self-Nanoemulsifying Peptide Drug Delivery Systems: Impact of Lipase on Drug Release. J. Microencapsul. 2015, 32, 401–407. [Google Scholar] [CrossRef]
- Ijaz, M.; Bonengel, S.; Zupančič, O.; Yaqoob, M.; Hartl, M.; Hussain, S.; Huck, C.W.; Bernkop-Schnürch, A. Development of Oral Self Nano-Emulsifying Delivery System(s) of Lanreotide with Improved Stability against Presystemic Thiol-Disulfide Exchange Reactions. Expert Opin. Drug Deliv. 2016, 13, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Soltani, Y.; Goodarzi, N.; Mahjub, R. Preparation and Characterization of Self Nano-Emulsifying Drug Delivery System (SNEDDS) for Oral Delivery of Heparin Using Hydrophobic Complexation by Cationic Polymer of β-Cyclodextrin. Drug Dev. Ind. Pharm. 2017, 43, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Wang, L.; Zhu, J.; Hu, Z.; Zhang, J. Self-Double-Emulsifying Drug Delivery System (SDEDDS): A New Way for Oral Delivery of Drugs with High Solubility and Low Permeability. Int. J. Pharm. 2011, 409, 245–251. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, S.; Wang, X.; Liao, J.; Yin, Z. Preparation and Evaluation of Nattokinase-Loaded Self-Double-Emulsifying Drug Delivery System. Asian J. Pharm. Sci. 2015, 10, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Shima, M.; Tanaka, M.; Fujii, T.; Egawa, K.; Kimura, Y.; Adachi, S.; Matsuno, R. Oral Administration of Insulin Included in Fine W/O/W Emulsions to Rats. Food Hydrocoll. 2006, 20, 523–531. [Google Scholar] [CrossRef]
- Siddhartha, T.; Senthil, V.; Kishan, I.; Khatwal, R.; Madhunapantula, S. Design and Development of Oral Nanoparticulated Insulin in Multiple Emulsion. Curr. Drug Deliv. 2014, 11, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Winarti, L.; Suwaldi; Martien, R.; Hakim, L. Formulation of Self-Nanoemulsifying Drug Delivery System of Bovine Serum Albumin Using HLB (Hydrophilic-Lypophilic Balance) Approach. Indones. J. Pharm. 2016, 27, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Cole, E.T.; Cadé, D.; Benameur, H. Challenges and Opportunities in the Encapsulation of Liquid and Semi-Solid Formulations into Capsules for Oral Administration. Adv. Drug Deliv. Rev. 2008, 60, 747–756. [Google Scholar] [CrossRef]
- Wang, J.; Wu, D.; Shen, W.C. Structure-Activity Relationship of Reversibly Lipidized Peptides: Studies of Fatty Acid-Desmopressin Conjugates. Pharm. Res. 2002, 19, 609–614. [Google Scholar] [CrossRef]
- Wang, J.; Chow, D.; Heiati, H.; Shen, W.C. Reversible Lipidization for the Oral Delivery of Salmon Calcitonin. J. Control. Release 2003, 88, 369–380. [Google Scholar] [CrossRef]
- Wang, J.; Shen, W.C. Gastric Retention and Stability of Lipidized Bowman-Birk Protease Inhibitor in Mice. Int. J. Pharm. 2000, 204, 111–116. [Google Scholar] [CrossRef]
- Ekrami, H.M.; Kennedy, A.R.; Shen, W.C. Water-Soluble Fatty Acid Derivatives as Acylating Agents for Reversible Lipidization of Polypeptides. FEBS Lett. 1995, 371, 283–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupančič, O.; Rohrer, J.; Thanh Lam, H.; Grießinger, J.A.; Bernkop-Schnürch, A. Development and in Vitro Characterization of Self-Emulsifying Drug Delivery System (SEDDS) for Oral Opioid Peptide Delivery. Drug Dev. Ind. Pharm. 2017, 43, 1694–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Saravanakumar, G.; Kim, K.; Kwon, I.C. Targeted Delivery of Low Molecular Drugs Using Chitosan and Its Derivatives. Adv. Drug Deliv. Rev. 2010, 62, 28–41. [Google Scholar] [CrossRef]
- Stout, D.B.; Suckow, C.E. MicroCT Liver Contrast Agent Enhancement over Time, Dose, and Mouse Strain. Mol. Imaging Biol. 2008, 10, 114–120. [Google Scholar] [CrossRef]
- Hallouard, F.; Anton, N.; Choquet, P.; Constantinesco, A.; Vandamme, T. Iodinated Blood Pool Contrast Media for Preclinical X-ray Imaging Applications—A Review. Biomaterials 2010, 31, 6249–6268. [Google Scholar] [CrossRef]
- Dierling, A.M.; Cui, Z. Targeting Primaquine into Liver Using Chylomicron Emulsions for Potential Vivax Malaria Therapy. Int. J. Pharm. 2005, 303, 143–152. [Google Scholar] [CrossRef]
- Jang, J.H.; Kim, C.K.; Choi, H.G.; Sung, J.H. Preparation and Evaluation of 2-(allylthio)pyrazine-Loaded Lipid Emulsion with Enhanced Stability and Liver Targeting. Drug Dev. Ind. Pharm. 2009, 35, 363–368. [Google Scholar] [CrossRef]
- Nikonenko, B.; Reddy, V.M.; Bogatcheva, E.; Protopopova, M.; Einck, L.; Nacy, C.A. Therapeutic Efficacy of SQ641-NE against Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Shegokar, R.; Singh, K.K. Stavudine Entrapped Lipid Nanoparticles for Targeting Lymphatic HIV Reservoirs. Pharmazie 2011, 66, 264–271. [Google Scholar] [CrossRef]
- Shahnaz, G.; Hartl, M.; Barthelmes, J.; Leithner, K.; Sarti, F.; Hintzen, F.; Rahmat, D.; Salvenmoser, W.; Bernkop-Schnürch, A. Uptake of Phenothiazines by the Harvested Chylomicrons Ex Vivo Model: Influence of Self-Nanoemulsifying Formulation Design. Eur. J. Pharm. Biopharm. 2011, 79, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Padera, T.P.; Meijer, E.F.J.; Munn, L.L. The Lymphatic System in Disease Processes and Cancer Progression. Annu. Rev. Biomed. Eng. 2016, 18, 125–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohya, Y.; Shiratani, M.; Kobayashi, H.; Ouchi, T. Release Behavior of 5-Fluorouracil from Chitosan-Gel Nanospheres Immobilizing 5-Fluorouracil Coated with Polysaccharides and Their Cell Specific Cytotoxicity. J. Macromol. Sci. Part A 1994, 31, 629–642. [Google Scholar] [CrossRef]
- Anton, N.; Vandamme, T.F. The Universality of Low-Energy Nano-Emulsification. Int. J. Pharm. 2009, 377, 142–147. [Google Scholar] [CrossRef]
- Rehman, J.; Dillow, J.M.; Carter, S.M.; Chou, J.; Le, B.; Maisel, A.S. Increased Production of Antigen-Specific Immunoglobulins G and M Following in Vivo Treatment with the Medicinal Plants Echinacea Angustifolia and Hydrastis Canadensis. Immunol. Lett. 1999, 68, 391–395. [Google Scholar] [CrossRef]
- Wiseman, N. Traditional Chinese Medicine: A Brief Outline. J. Chem. Inf. Comput. Sci. 2002, 42, 445–455. [Google Scholar] [CrossRef]
- Rivera, E.; Hu, S.; Concha, C. Ginseng and Aluminium Hydroxide Act Synergistically as Vaccine Adjuvants. Vaccine 2003, 21, 1149–1157. [Google Scholar] [CrossRef]
- Borchers, A.T.; Hackman, R.M.; Keen, C.L.; Stern, J.S.; Gershwin, M.E. Complementary Medicine: A Review of Immunomodulatory Effects of Chinese Herbal Medicines. Am. J. Clin. Nutr. 1997, 66, 1303–1312. [Google Scholar] [CrossRef] [Green Version]
- Cragg, G.M. Natural Product Drug Discovery and Development: The United States National Cancer Institute Role. P. R. Health Sci. J. 2002, 21, 97–111. [Google Scholar] [CrossRef]
- Jang, M.H.; Piao, X.L.; Kim, J.M.; Kwon, S.W.; Park, J.H. Inhibition of Cholinesterase and Amyloid-beta Aggregation by Resveratrol Oligomers from Vitis Amurensis. Phyther. Res. 2008, 22, 544–549. [Google Scholar] [CrossRef]
- Licciardi, P.V.; Underwood, J.R. Plant-Derived Medicines: A Novel Class of Immunological Adjuvants. Int. Immunopharmacol. 2011, 11, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Atul Bhattaram, V.; Graefe, U.; Kohlert, C.; Veit, M.; Derendorf, H. Pharmacokinetics and Bioavailability of Herbal Medicinal Products. Phytomedicine 2002, 9, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Byeon, J.C.; Ahn, J.B.; Jang, W.S.; Lee, S.E.; Choi, J.S.; Park, J.S. Recent Formulation Approaches to Oral Delivery of Herbal Medicines. J. Pharm. Investig. 2019, 49, 17–26. [Google Scholar] [CrossRef]
- Lagoa, R.; Silva, J.; Rodrigues, J.R.; Bishayee, A. Advances in Phytochemical Delivery Systems for Improved Anticancer Activity. Biotechnol. Adv. 2020, 38, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.X.; Zhou, J.T.; Wang, T.T.; Huang, Y.F.; Chen, V.P.; Xie, Y.L.; Lin, Z.X.; Gao, J.S.; Su, Z.R.; Zeng, H.F. Self-Nanoemulsifying Drug Delivery System of Bruceine D: A New Approach for Anti-Ulcerative Colitis. Int. J. Nanomed. 2018, 13, 5887–5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.T.; Tran, C.S.; Nguyen, H.A.; Nguyen, T.D.; Chi, S.C.; Pham, D.V.; Bui, Q.D.; Ho, X.H. Formulation and Biopharmaceutical Evaluation of Supersaturatable Self-Nanoemulsifying Drug Delivery Systems Containing Silymarin. Int. J. Pharm. 2019, 555, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Baskaran, R.; Balakrishnan, P.; Thapa, P.; Yong, C.S.; Yoo, B.K. Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) Containing Phosphatidylcholine for Enhanced Bioavailability of Highly Lipophilic Bioactive Carotenoid Lutein. Eur. J. Pharm. Biopharm. 2011, 79, 250–257. [Google Scholar] [CrossRef]
- Kazi, M.; Shahba, A.A.; Alrashoud, S.; Alwadei, M.; Sherif, A.Y.; Alanazi, F.K. Bioactive Self-Nanoemulsifying Drug Delivery Systems (Bio-SNEDDS) for Combined Oral Delivery of Curcumin and Piperine. Molecules 2020, 25, 1703. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Tang, J.; Ji, H.; Liu, H.; Liu, Y.; Zhu, D.; Wu, L. Enhanced Oral Bioavailability of Wurenchun (Fructus Schisandrae Chinensis Extracts) by Self-Emulsifying Drug Delivery Systems. Drug Dev. Ind. Pharm. 2010, 36, 1356–1363. [Google Scholar] [CrossRef]
- Setthacheewakul, S.; Kedjinda, W.; Maneenuan, D.; Wiwattanapatapee, R. Controlled Release of Oral Tetrahydrocurcumin from a Novel Self-Emulsifying Floating Drug Delivery System (SEFDDS). AAPS Pharmscitech 2011, 12, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zhao, Q.; Wang, Y.; Guo, T.; An, Y.; Shi, G. Design and Evaluation of Self-Emulsifying Drug Delivery Systems of Rhizoma Corydalis Decumbentis Extracts. Drug Dev. Ind. Pharm. 2012, 38, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Bi, J.; Tian, H.; Jin, Y.; Wang, Y.; Yang, X.; Yang, Z.; Kou, J.; Li, F. Preparation and Evaluation of a Self-Nanoemulsifying Drug Delivery System Loaded with Akebia Saponin D-Phospholipid Complex. Int. J. Nanomed. 2016, 11, 4919–4929. [Google Scholar] [CrossRef] [Green Version]
- Kalantari, A.; Kósa, D.; Nemes, D.; Ujhelyi, Z.; Fehér, P.; Vecsernyés, M.; Váradi, J.; Fenyvesi, F.; Kuki, Á.; Gonda, S.; et al. Self-Nanoemulsifying Drug Delivery Systems Containing Plantago Lanceolata—An Assessment of Their Antioxidant and Antiinflammatory Effects. Molecules 2017, 22, 1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, J.; Yang, X.; Jin, Y.; Yang, Z.; Wang, R.; Zhang, F.; Linhardt, R.J. Mechanism of Enhanced Oral Absorption of Akebia Saponin D by a Self-Nanoemulsifying Drug Delivery System Loaded with Phospholipid Complex. Drug Dev. Ind. Pharm. 2019, 45, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Chairuk, P.; Tubtimsri, S.; Jansakul, C.; Sriamornsak, P.; Weerapol, Y. Enhancing Oral Absorption of Poorly Water-Soluble Herb (Kaempferia Parviflora) Extract Using Self-Nanoemulsifying Formulation. Pharm. Dev. Technol. 2020, 25, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, K.; Alanazi, F. The Fate of Paclitaxel during in Vitro Dispersion Testing of Different Lipid-Based Formulations. J. Drug Deliv. Sci. Technol. 2012, 22, 197–204. [Google Scholar] [CrossRef]
- Shahba, A.A.W.; Mohsin, K.; Alanazi, F.K. Novel Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for Oral Delivery of Cinnarizine: Design, Optimization, and in-Vitro Assessment. AAPS Pharmscitech 2012, 13, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Kuentz, M. Drug Supersaturation during Formulation Digestion, Including Real-Time Analytical Approaches. Adv. Drug Deliv. Rev. 2019, 142, 50–61. [Google Scholar] [CrossRef]
- Cserháti, T.; Forgács, E.; Oros, G. Biological Activity and Environmental Impact of Anionic Surfactants. Environ. Int. 2002, 28, 337–348. [Google Scholar] [CrossRef]
- Bandopadhyay, S.; Manchanda, S.; Chandra, A.; Ali, J.; Deb, P.K. Overview of Different Carrier Systems for Advanced Drug Delivery; Drug Delivery Systems, Academic Press: Cambridge, MA, USA, 2020; pp. 179–233. ISBN 9780128144879. [Google Scholar] [CrossRef]
- Halbaut, L.; Barbé, C.; Aróztegui, M.; De La Torre, C. Oxidative Stability of Semi-Solid Excipient Mixtures with Corn Oil and Its Implication in the Degradation of Vitamin A. Int. J. Pharm. 1997, 147, 31–40. [Google Scholar] [CrossRef]
- Wasylaschuk, W.R.; Harmon, P.A.; Wagner, G.; Amy, B.H.; Allen, C.T.; Huix, X.; Robert, A.A. Evaluation of hydroperoxides in common pharmaceutical excipients. J. Pharm. Sci. 2007, 96, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Chambin, O.; Jannin, V.; Champion, D.; Chevalier, C.; Rochat-Gonthier, M.H.; Pourcelot, Y. Influence of Cryogenic Grinding on Properties of a Self-Emulsifying Formulation. Int. J. Pharm. 2004, 278, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Feng, N.; Xu, J. Preparation and Evaluation of Self-Microemulsifying Drug Delivery System of Oridonin. Int. J. Pharm. 2008, 355, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Sunesen, V.H.; Pedersen, B.L.; Kristensen, H.G.; Müllertz, A. In Vivo in Vitro Correlations for a Poorly Soluble Drug, Danazol, Using the Flow-through Dissolution Method with Biorelevant Dissolution Media. Eur. J. Pharm. Sci. 2005, 24, 305–313. [Google Scholar] [CrossRef]
- Stillhart, C.; Kuentz, M. Trends in the Assessment of Drug Supersaturation and Precipitation In Vitro Using Lipid-Based Delivery Systems. J. Pharm. Sci. 2016, 105, 2468–2476. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mcclements, D.J. New Mathematical Model for Interpreting Ph-Stat Digestion Profiles: Impact of Lipid Droplet Characteristics on in Vitro Digestibility. J. Agric. Food Chem. 2010, 58, 8085–8092. [Google Scholar] [CrossRef]
- Buyukozturk, F.; Di Maio, S.; Budil, D.E.; Carrier, R.L. Effect of Ingested Lipids on Drug Dissolution and Release with Concurrent Digestion: A Modeling Approach. Pharm. Res. 2013, 30, 3131–3144. [Google Scholar] [CrossRef] [Green Version]
- Berthelsen, R.; Klitgaard, M.; Rades, T.; Müllertz, A. In Vitro Digestion Models to Evaluate Lipid Based Drug Delivery Systems; Present Status and Current Trends. Adv. Drug Deliv. Rev. 2019, 142, 35–49. [Google Scholar] [CrossRef]
- Crommelin, J.A.D.; Metselaar, M.J.; Storm, G. Non-Biological Complex Drugs: The Science and the Regulatory Landscape; Springer: Cham, Switzerland, 2015; pp. 77–185. ISBN 978-3-319-16241-6. [Google Scholar] [CrossRef]
- Ragelle, H.; Danhier, F.; Préat, V.; Langer, R.; Anderson, D.G. Nanoparticle-Based Drug Delivery Systems: A Commercial and Regulatory Outlook as the Field Matures. Expert Opin. Drug Deliv. 2017, 14, 851–864. [Google Scholar] [CrossRef]
- Nardin, I.; Köllner, S. Successful Development of Oral SEDDS: Screening of Excipients from the Industrial Point of View. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration Home Page. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (accessed on 12 May 2020).
- US Food and Drug Administration Home Page. Available online: https://www.fda.gov/media/70837/download (accessed on 12 May 2020).
- US Food and Drug Administration Home Page. Available online: http://home.att.ne.jp/red/akihiro/fda/2635fnl.pdf (accessed on 20 May 2020).
- Eifler, A.C.; Thaxton, C.S. Nanoparticle therapeutics: FDA approval, clinical trials, regulatory pathways, and case study. Methods Mol. Biol. 2011, 726, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration Home Page. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070570.pdf (accessed on 17 May 2020).
- Committee for Medicinal Products for Human Use; European Medicine Agency. Joint MHLW/EMA Reflection Paper on the Development of Block Copolymer Micelle Medicinal Products. Ema/Chmp/13099/2013. 2013, Volume 44, pp. 1–18. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-joint-ministry-health-labour-welfare/european-medicines-agency-reflection-paper-development-block-copolymer-micelle-medicinal-products_en.pdf (accessed on 1 October 2020).
- European Medicine Agency Home Page. Available online: http://www.euronanoforum2015.eu/wpcontent/uploads/2015/06/2_NanomedicinesEMA-experienceperspective_DoloresHernan_10042015.pdf (accessed on 9 May 2020).










{kind=link}
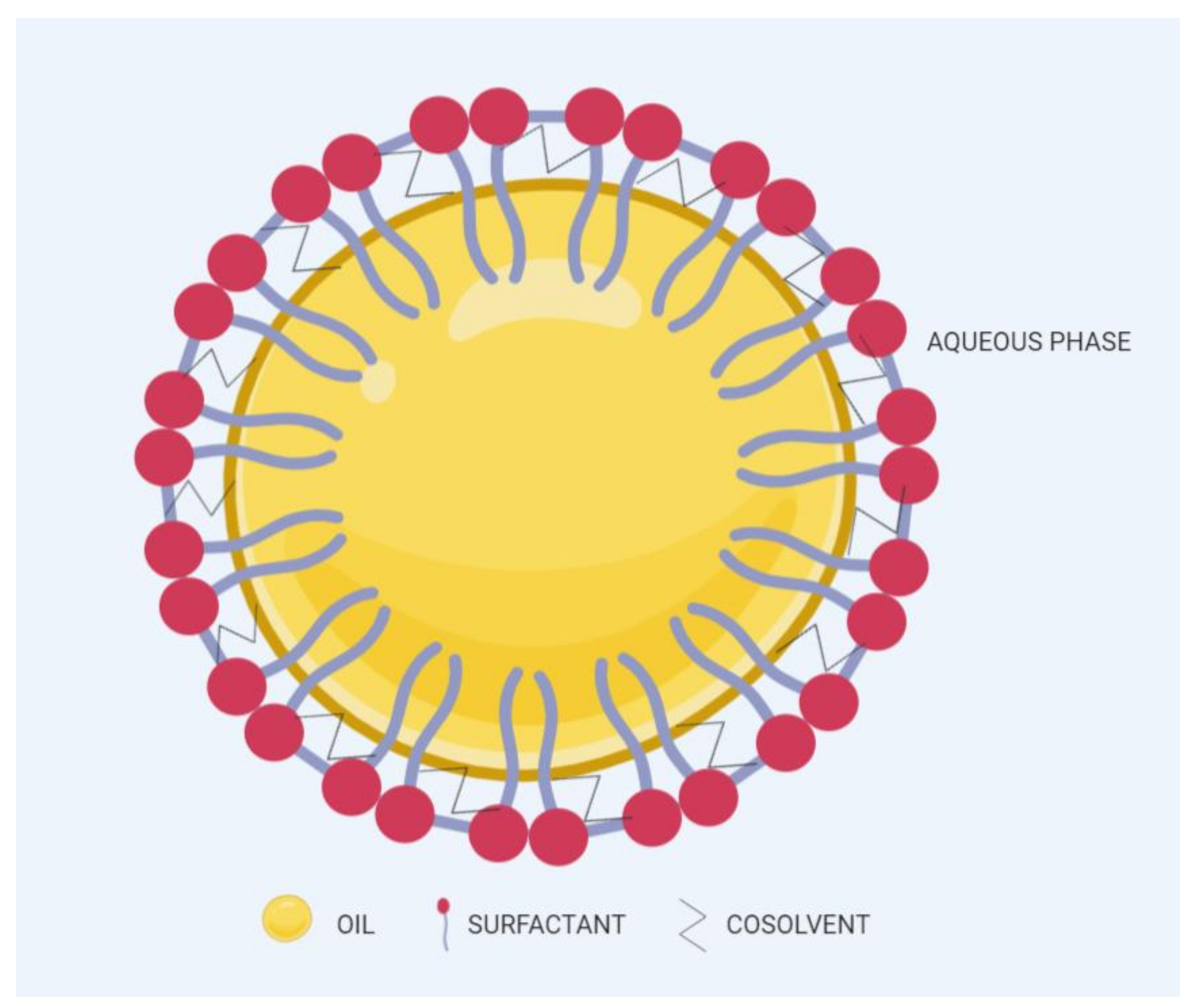{kind=link}
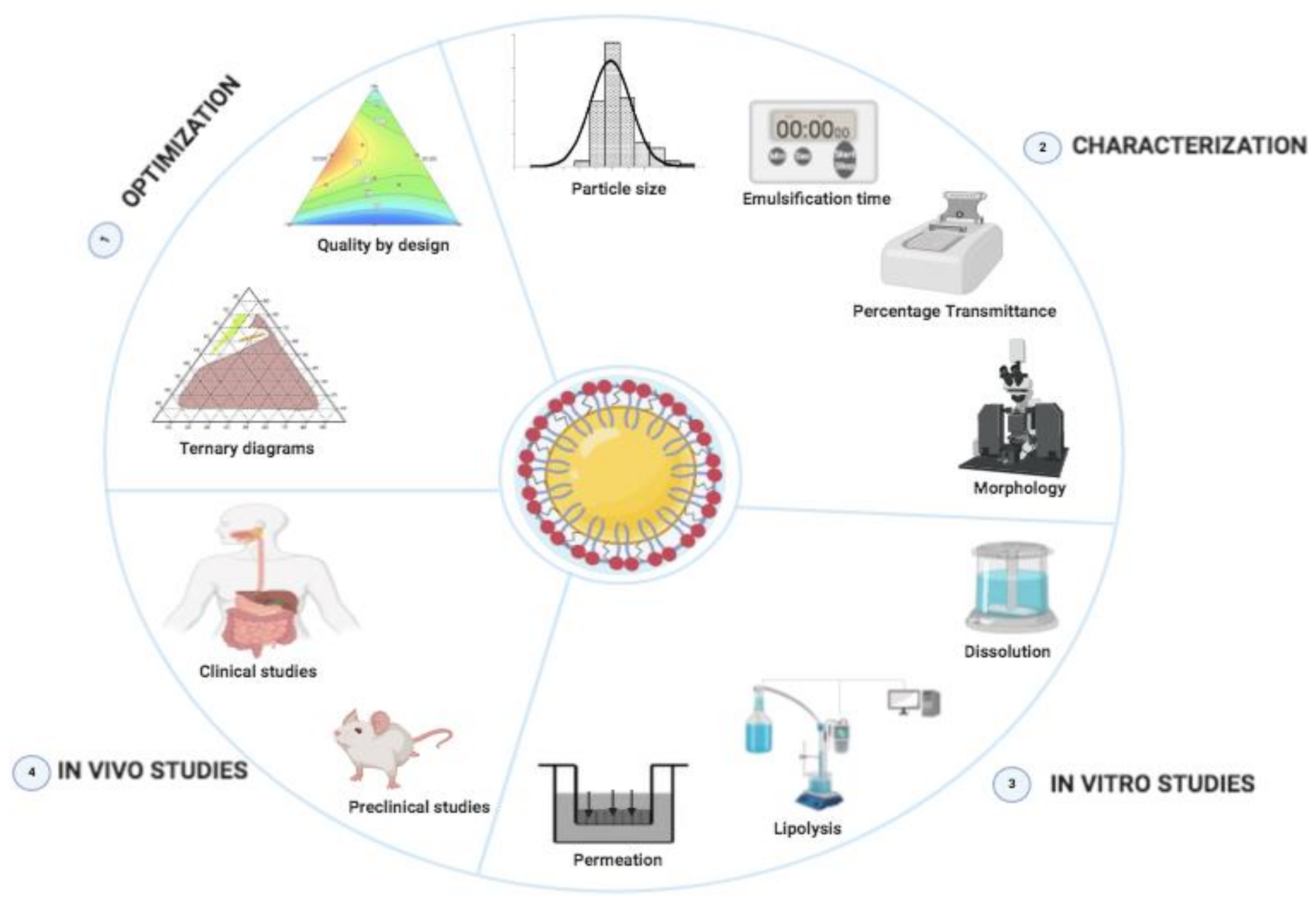{kind=link}
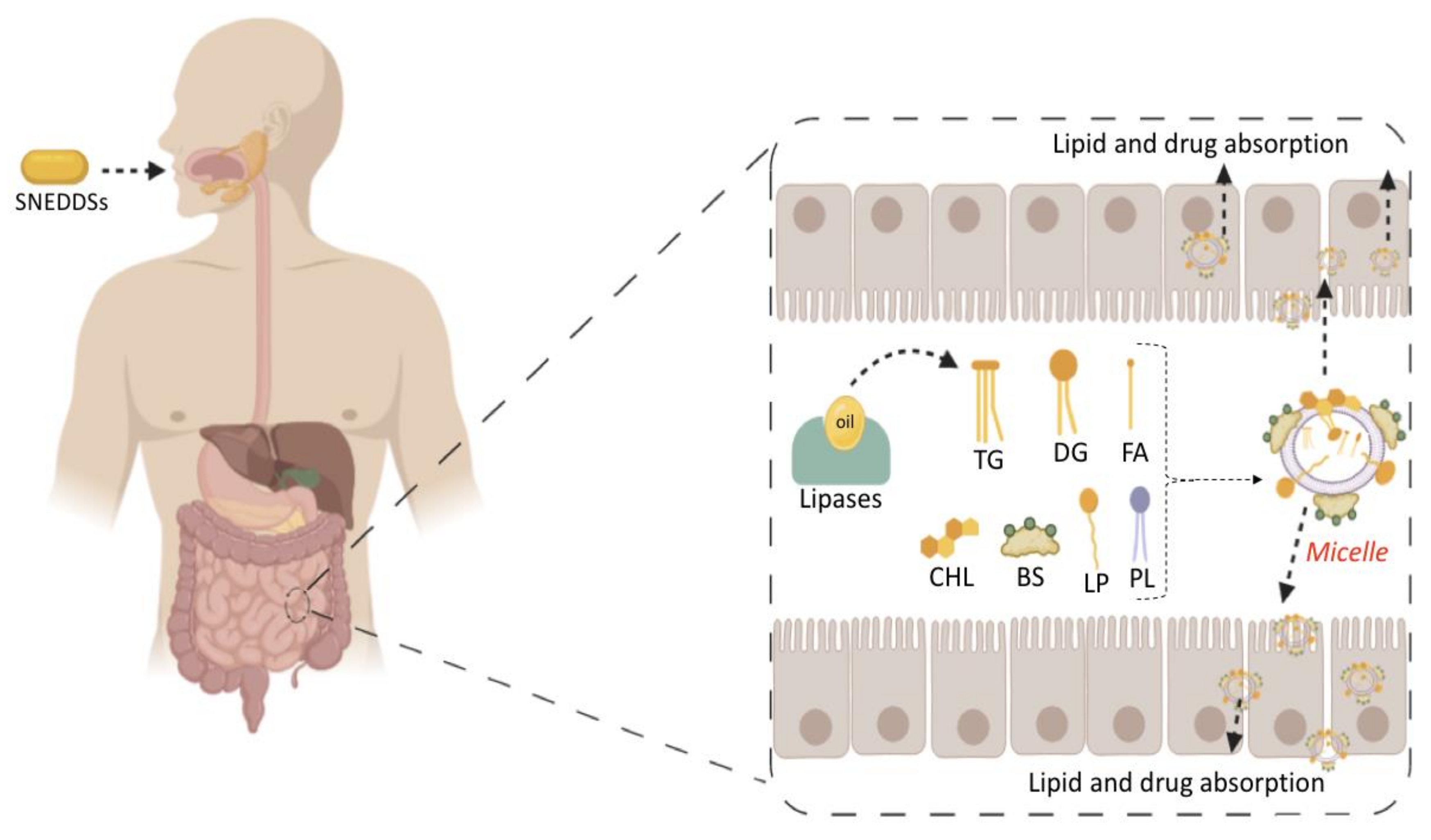{kind=link}
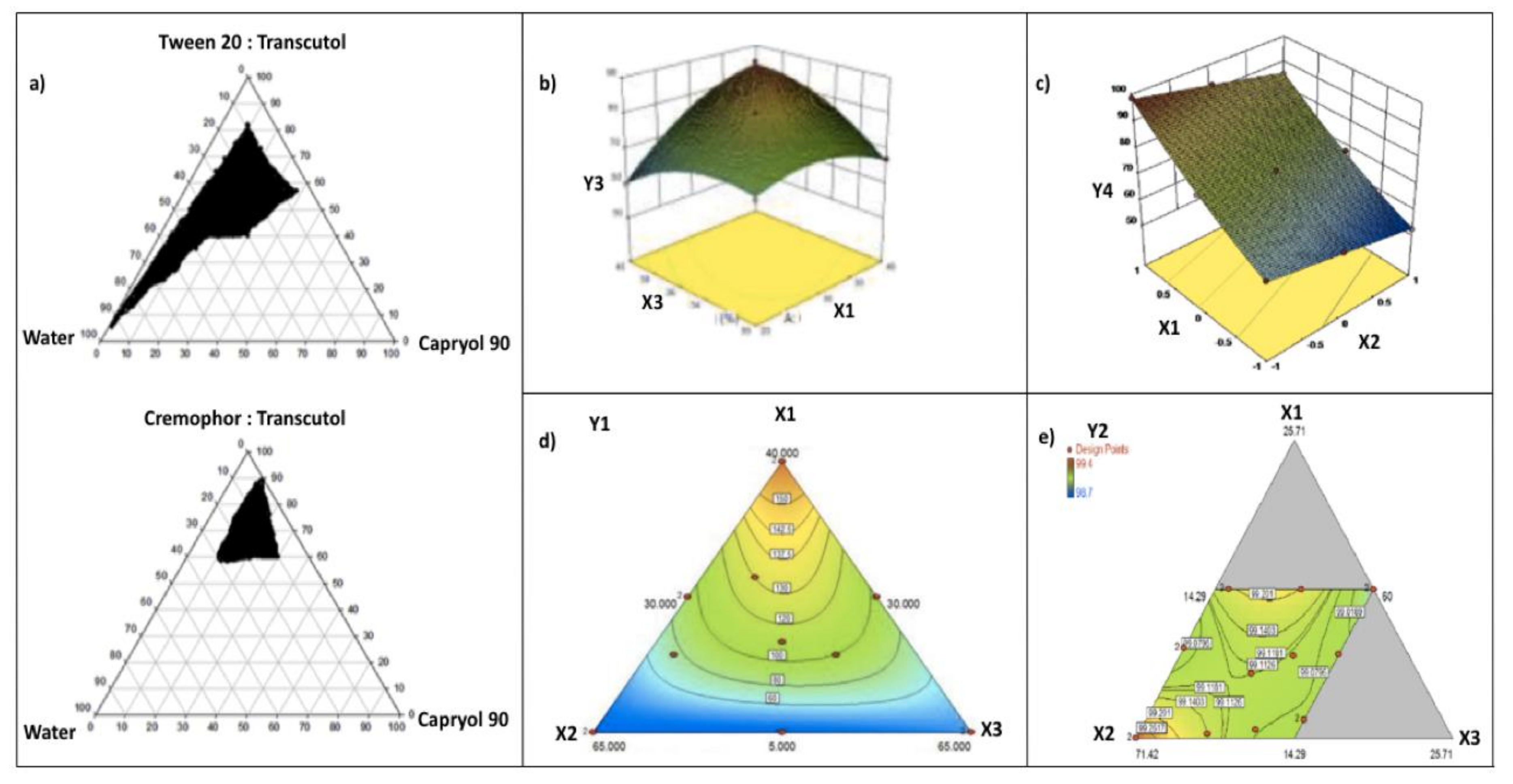{kind=link}
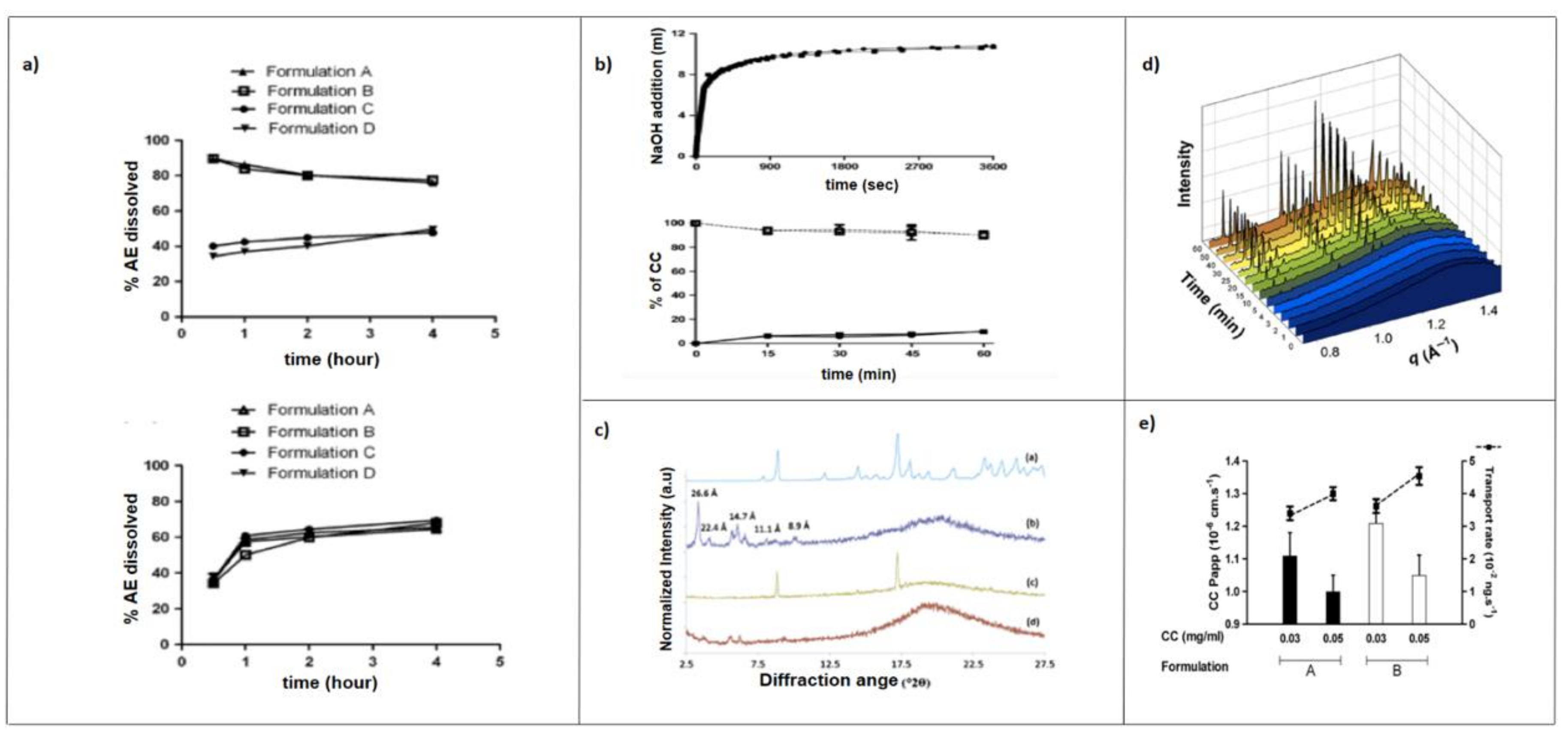{kind=link}
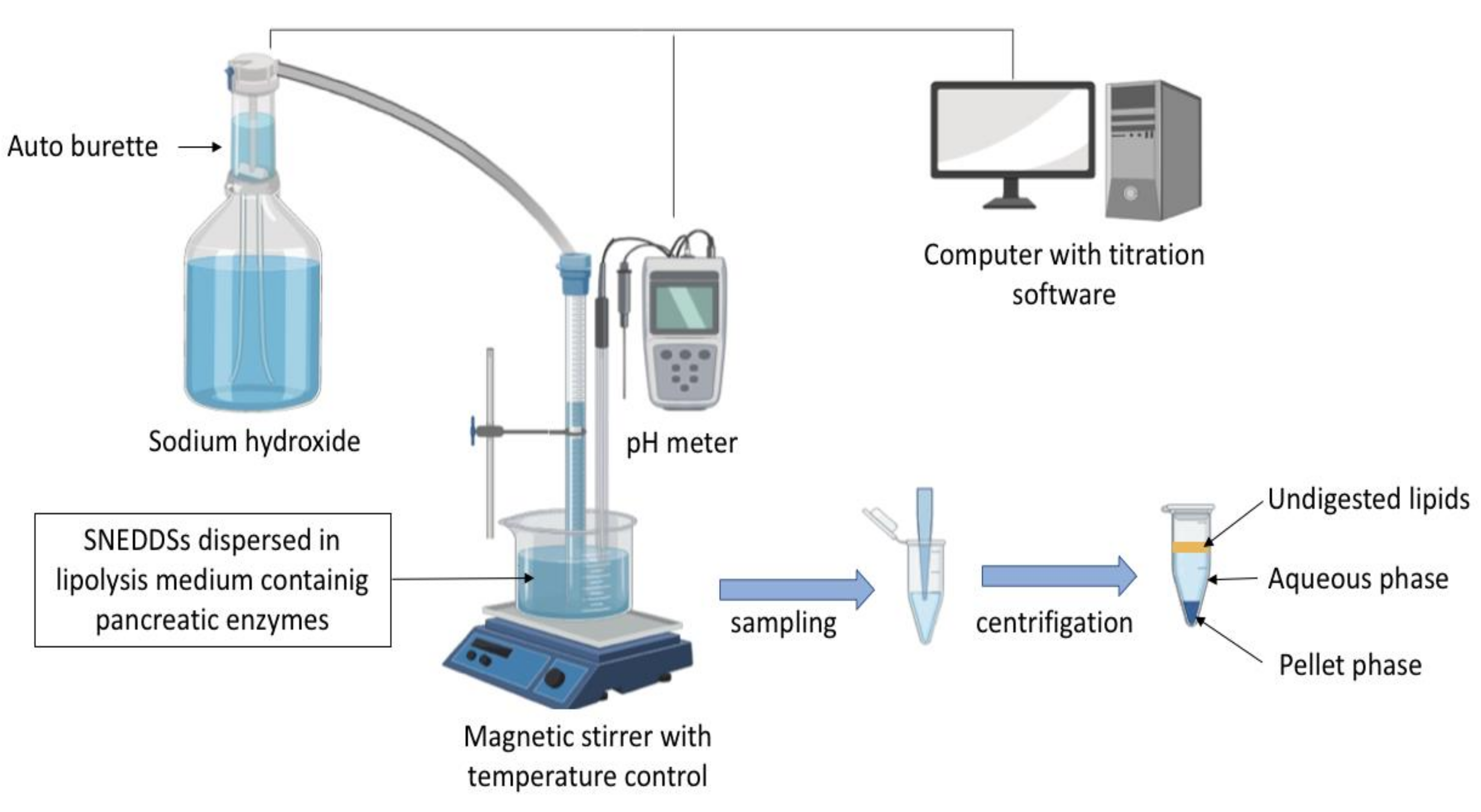{kind=link}
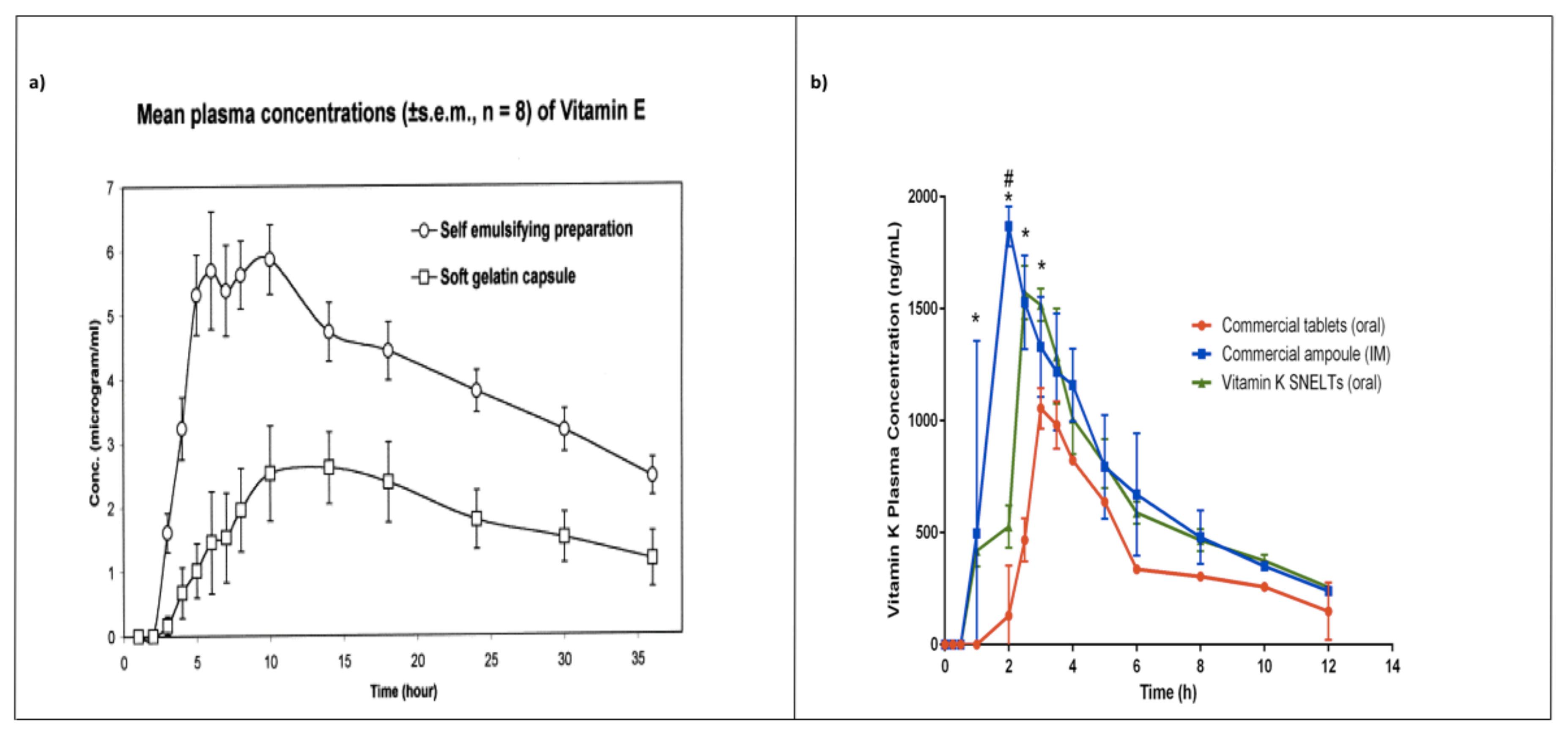{kind=link}
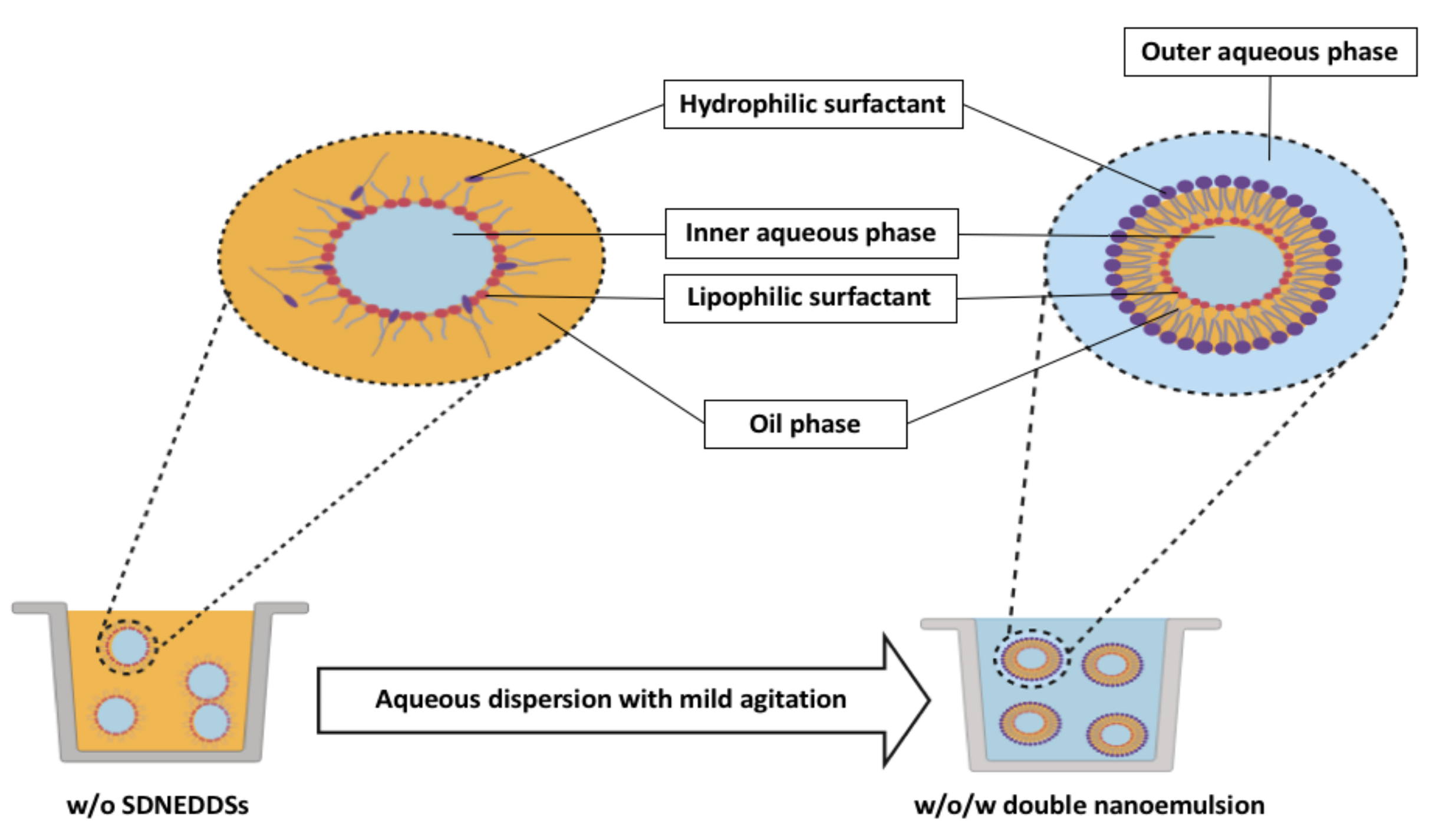{kind=link}
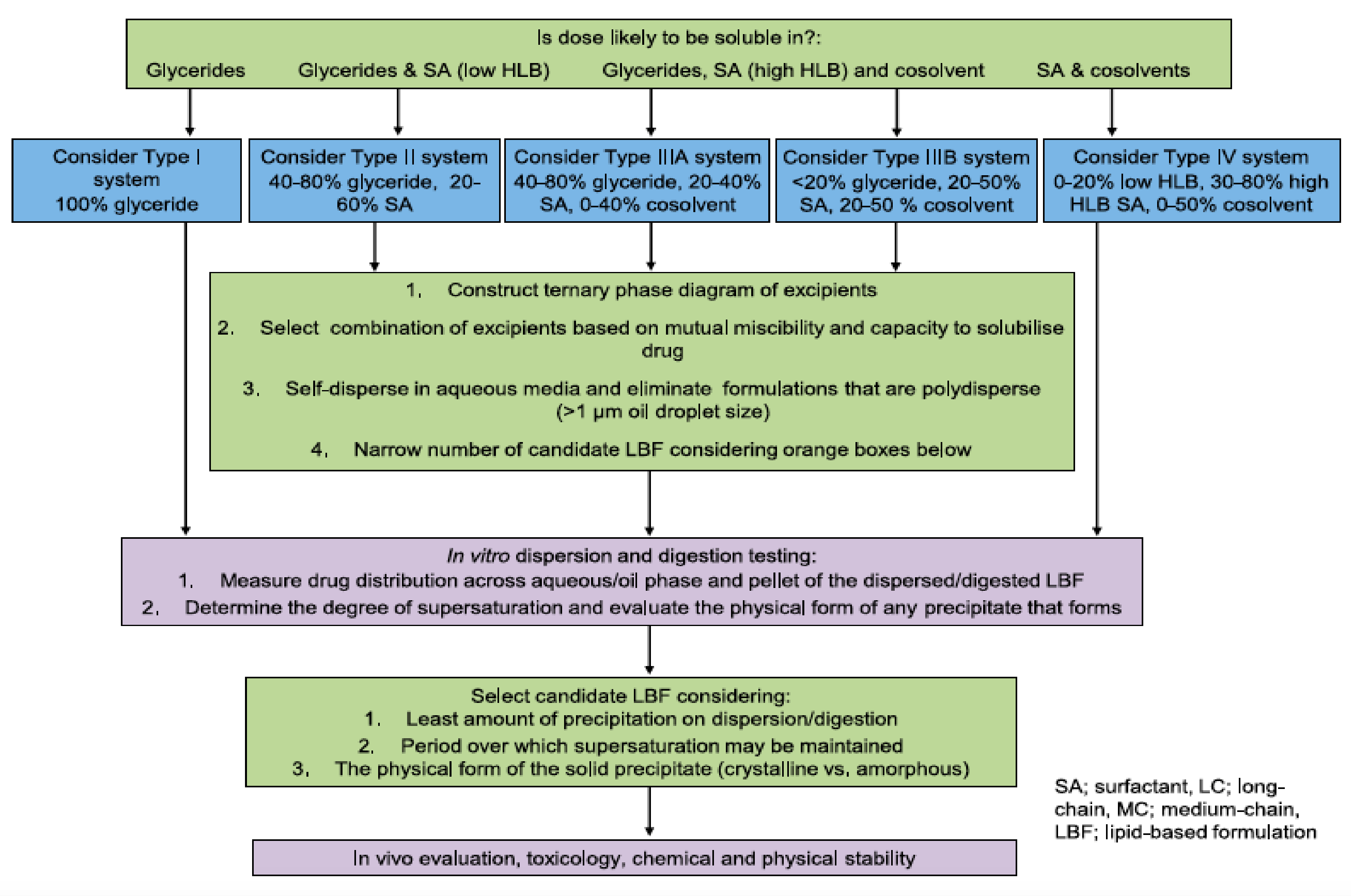{kind=link}
| General Class | Example | Molecular Structure | Commercial Name | Acceptability |
|---|---|---|---|---|
| OILS | ||||
| Medium-chain | Triglycerides of capric/caprylic acids |  | Captex® 300, 350, Labrafac® CC, Crodamol GTCC | P/O/T/Oc/M |
| Di-glycerides of capric/caprylic acids |  | Capmul® MCM, Akoline® MCM | O/T | |
| Monoglycerides of capric/caprylic acids |  | Capryol® 90, Capryol® PGMC, Imwitor® 742 | O/T | |
| Long-chain | Glyceryl monooleate |  | Peceol®, Capmul®-GMO | O/T |
| Glyceryl monolinoleate |  | Maisine®-35 | O/T | |
| Propylene glycol fatty acid esters | Propylene glycol monocaprylate |  | Capmul® PG-8, Sefsol 218 | O/T |
| Propylene glycol dicaprylate/caprate |  | Miglyol® 840, Captex® 200 | O/T | |
| Propylene glycol Monolaurate |  | Lauroglycol® 90, Capmul® PG-12, Lauroglycol® FCC | O/T | |
| SURFACTANTS | ||||
| Polysorbates | Polysorbate esters |  | Tween® 20, Tween® 80 | P/O/T/Oc M |
| Sorban esters | Sorban esters |  | Span® 20,80, Crill® 4 | P/O/T/Oc M |
| Castor oil esters | Ethoxylated castor oil |  | Cremophor®-EL, Etocas® 35 HV | O/T |
| Hydrogenated castor oil |  | Cremophor® RH40, 60, Croduret® 40 | O/T | |
| Polyglycolyzed glycerides | Linoleoyl/Oleoyl Macrogol glycerides |  | Labrafil® 1944, 2121 CS | O/T |
| Caprylocaproyl macrogol glycerides |  | Labrasol® | O/T | |
| COSOLVENTS | ||||
| Alcohols | Short chain Alcohols | R-OH | Ethanol, benzyl alcohol | P/T/Oc/M |
| Alkane diols |  | Propylene glycol | P/T/Oc/M | |
| Polyethylene glycols | Polyethylene glycols |  | PEG 400, 600 | P/T/Oc/M |
| Esters | Glycerol esters |  | Transcutol® | O/T |
| Method/Model | Information Provided | |
|---|---|---|
| DLS | Droplet size, PDI, thermodynamic stability | |
| Physico-chemical characterization | Electrophoretic velocimetry | Zeta potential |
| Spectrophotometry | Transmittance percentage, cloud point, thermodynamic stability | |
| TEM, SEM | Morphology, droplet size | |
| Viscosimeter | Viscosity, thermodynamic stability | |
| Dissolution apparatus | Drug dissolution, emulsification time | |
| Preclinical in vitro and ex vivo evaluation | pH-stat unit | Formulation digestion, drug distribution across aqueous/oil phase |
| PAMPA | Permeation across intestinal barrier | |
| SPIP | Permeation across intestinal barrier | |
| IRP | Permeation across intestinal barrier | |
| CaCO-2 | Permeation across intestinal barrier, cytotoxicity | |
| Preclinical In vivo evaluation | Animals | Pharmacokinetic, toxicity, pharmacodynamic |
| Clinical trials | Humans | Pharmacokinetic, bioequivalence toxicity, pharmacodynamic |
| Class | Drug | Components | In Vitro/Vivo Observation | References |
|---|---|---|---|---|
| ANTI-CANCER | Docetaxel | Capryol® 90, Labrasol®, Transcutol® HP | AUC0-t and Cmax increased 6.4 and 6.5-fold, respectively compared to docetaxel aqueous solution. | [196] |
| Erlotinib | Labrafil® M2125 CS, Labrasol®, Transcutol® HP, Aerosil® 200, Dextran 40 | AUC0–t and Cmax increased 2.1 and 2.4-fold, respectively in case of dextran-based solid SEDDS compared to erlotinib powder. | [197] | |
| Paclitaxel | Sesame oil, Labrasol®, Sodium deoxycholate | AUC0–t and Cmax increased to 2.7 and 3.99-fold, respectively compared to drug suspension. | [194] | |
| Lycopene | LCT, Tween® 85, Cremophor® RH, Gelucire® | AUC0–t and Cmax increased 2.3 and 2.85-fold, respectively compared to Lycovit®. | [198] | |
| Methotrexate | Ethyl oleate, Tween® 80, Propylene glycol | AUC0–24 and Cmax increased 1.57 and 1.68-fold, respectively compared to native drug. | [199] | |
| Irinotecan | Capmul® CM-C8, Cremophor® EL, Pluronic L-121 | AUC0–t and Cmax increased 4.2 and 1.7-fold, respectively compared to drug suspension. | [200] | |
| CARDIOVASCULAR AND ANTI-HYPERTENSIVE | Carvedilol | Labrafil® M1944CS, Tween® 80, Transcutol® | Relative bio-availability enhanced by 4.1 times compared with tablet. | [201] |
| Felodipine | Miglyol® 812, Cremophor® RH 40, Tween® 80, Transcutol® HP, Silicon dioxide | AUC0–t increased 2-fold compared to conventional tablets. | [202] | |
| Clinidipine | Capryol® 90, Tween® 80, Transcutol® | The absorption of the drug was enhanced from liquid-SEDDS as 99 % of the drug was transported from mucosal to serosal side of the rat intestine within 90 min from SEDDS in comparison to only 42.2% from that of the pure drug suspension. | [203] | |
| Valsartan | Triacetin or Castor oil, Tween® 80, PEG 600 | For triacetin-SNEDDS 5 and 2.4-fold increase in Cmax and AUC, respectively; for castor oil SNEDDS 8 and 3.6-fold increase in Cmax and AUC, respectively. | [204] | |
| Rosuvastatin | Peceol®, Tween® 80, Transcutol® HP | In vivo pharmacokinetic studies revealed 1.8 and 5.7-fold enhancement in AUC0-t and Cmax, respectively, and 0.33-fold reduction in Tmax of drug from the SNEDDS vis-à-vis the pure drug suspension. | [173] | |
| Atenolol | Tartaric acid, Captex®, Span® 80, Oleic acid | Ex vivo intestinal permeability studies revealed that atenolol SDEDDS exhibited better drug permeation compared to atenolol or atenolol-tartaric acid suspension. | [205] | |
| Ramipril | Sefsol, Tween® 80, Carbitol | 2.29-fold improvement in oral bio-availability compared with free drug suspension. | [104] | |
| ANTI-DIABETIC | Insulin | Miglyol®, Cremophor® RH40, MCM C-10, Ethanol | AUC0–t increased 2.7-fold compared to insulin solution. | [206] |
| Glibenclamide | Cotton oil, Tween® 80, Propylene glycol | AUC0–t increased 1.4-fold compared to free drug. | [207] | |
| Trans-cinnamic acid | Isopropyl myristate, Cremophor® EL, PEG 400 | The efficacy of trans-cinnamic acid in both hyperglycemia and glucolipid metabolic disorder was enhanced in SNEDDS compared to the drug suspension. | [208] | |
| Gliclazide | Capryol® 90, Cremophor® EL, Akoline® MCM | Enhancement in oral bio-availability as compared to the free drug. | [209] | |
| Exenatide | Cremophor® EL, Labrafil® 1944, Capmul®-PG 8, propylene glycol | 14.6-fold higher relative bio-availability versus subcutaneous exenatide solution. | [210] | |
| ANTIOXIDANT | Quercetin | Capmul®, Tween® 20, Ethanol | 23.7-fold increase in the cell uptake of quercetin when incorporated in SEDDS compared to free drug. | [211] |
| Resveratrol | Miglyol® 812, Montanox, Labrasol®, Gelucire®, Ethanol | The absorptive fluxes through the intestinal epithelium from the nano-emulsions were significantly increased compared to an ethanolic control solution. | [212] | |
| Genistein | Labrafac® lipophile 1349, Maisine®-35, Cremophor® EL, Labrasol®, Transcutol® | 95% of drug release in 5 min. | [213] | |
| Retinol acetate | Soybean oil, Capmul®, Cremophor® EL | Improved in dissolution rate. | [214] | |
| Coenzyme Q10 | Lauroglycol® FCC, Witepsol® H335, Solutol® HS 15 | 5-fold improvement in oral bio-availability compared to free drug. | [39] | |
| ANTI-VIRAL, ANTI-BACTERIAL, ANTI-FUNGAL, AND ANTIPROTOZOAL | Darunavir | Lauroglycol® 90, Tween® 80, Transcutol® HP | Enhancement in AUC0-t, oral bio-availability and Cmax, 1.45,5.8 and 7.5-fold, respectively compared to free drug. | [215] |
| Nelfinavir mesylate | Maisine® 35-1, Tween® 80, Transcutol® HP | 4.5-fold improvement in permeability and 3.6-fold improvement in bio-availability. | [113] | |
| Lopinavir | Maisine®, Tween®-80, Transcutol® HP | Enhanced oral bio-availability (3.9-fold) compared to the pure drug. | [216] | |
| Acyclovir | Sunflower oil, Tween® 60, Glycerol | 3.5-fold increase in oral bio-availability compared to the pure drug suspension. | [217] | |
| Rifampicin | Capmul® MCM C, Cremophor®-EL, Labrasol® | 3.72 and 5.22-fold improvement in AUC0–t and Cmax, respectively compared to drug suspension. | [218] | |
| Amphotericin B | Peceol®, PEG-200, Distearoylphos-phatidylethanolamine | Amphotericin B-SEDD treatment significantly decreases total fungal colony forming unit concentrations compared to non-treated controls without significant changes in plasma creatinine levels in the A. fumigatus infected rats. | [219] | |
| Satranidazole | Oleic acid, Tween® 20, PEG 400 | SNEDDSs formulations showed a drug release of greater than 70% in 45 minutes whereas marketed preparation showed more than 70% of drug release in 90 min. | [220] |
| Drug | Components | In Vivo Observation | References |
|---|---|---|---|
| Vitamin E | Palm oil, Tween® 80, Span® 80 | 3-fold higher oral bio-availability from SEDDSs. | [223] |
| Cyclosporin | Corn oil glycerides, Cremophor® RH40, PG, DL-α-tocopherol and ethanol | AUC0–t and Cmax increased 1.18 and 1.17-fold, respectively from SEDDSs. | [224] |
| Tocotrienols | Tocomin, Soybean oil Tween® 80 Labrasol® | 2 to 3-fold higher oral bio-availability from SEDDSs. | [82] |
| Saquinavir (Fortovase®) | Medium-chain mono- and di-glycerides | Increased oral bio-availability up to 331% from Fortovase® compared to Invirase®. | [198] |
| Simvastatin | Labrafil®, Tween® 80, Transcutol® HP | 1.55 and 1.5 increased in Cmax and AUC0–t, respectively from SNEDDSs. | [225] |
| Vitamin K | Vitamin K, Labrasol®, Transcutol® HP | Enhancement in vitamin K relative bio-availability from SNEDDSs. | [222] |
| Drug Name | Trade Name (Company) | Composition | Dosage Form |
|---|---|---|---|
| Ritonavir | Norvir® Abbott Laboratories | Ole Oleic acid, Cremophor®-EL, ethanol, butylated hydroxytoluene | Soft capsules |
| Tipranavir | Aptivus® (Boehringer Ingelheim) | Mono/di-glycerides of caprylic acids, Cremophor® EL ethanol, propylene glycol | Soft capsules |
| Cyclosporine | Sandimmune® (Novartis) | Corn oil/olive oil, Labrafil® M 1944 CS, ethanol, α-tocopherol | Soft capsule |
| Neoral® (Novartis) | Mono-, di- and triglycerides of corn oil, Cremophor® RH40, propylene glycol, ethanol, D-α-tocopherol | Oral solution and soft capsules | |
| Isotretinoin | Accutane® (Roche) | Beeswax, hydrogenated soybean oil flakes, hydrogenated vegetable oil, soybean oil Olive, polyoxyethylated oleic glycerides, ethanol | Soft capsules |
| Sirolymus | Rapamune® (Wyeth-Ayerst) | Phosphatidylcholine, mono- and di-glycerides, soy fatty acids, Tween® 80, ethanol, propylene glycol, ascorbyl palmitate | Oral solution |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buya, A.B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery. Pharmaceutics 2020, 12, 1194. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121194
Buya AB, Beloqui A, Memvanga PB, Préat V. Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery. Pharmaceutics. 2020; 12(12):1194. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121194
Chicago/Turabian StyleBuya, Aristote B., Ana Beloqui, Patrick B. Memvanga, and Véronique Préat. 2020. "Self-Nano-Emulsifying Drug-Delivery Systems: From the Development to the Current Applications and Challenges in Oral Drug Delivery" Pharmaceutics 12, no. 12: 1194. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121194