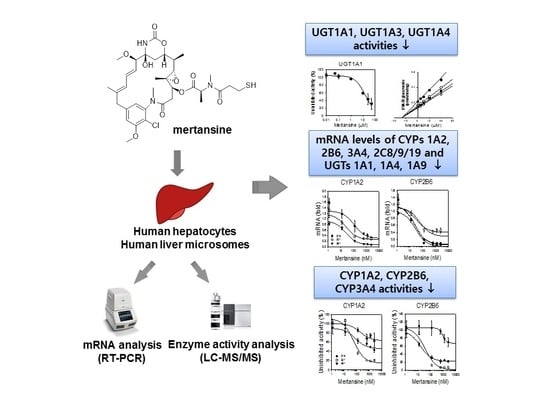
Mertansine Inhibits mRNA Expression and Enzyme Activities of Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyltransferases in Human Hepatocytes and Liver Microsomes




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Inhibitory Potential of Mertansine on Human Major UGTs in Human Liver Microsomes
2.3. Kinetic Analysis for the Inhibition of UGT1A1, UGT1A3, and UGT1A4 by Mertansine
2.4. Induction of Mertansine on Human Major CYPs and UGTs in Human Hepatocytes
2.4.1. Cytotoxicity of Mertansine in Human Hepatocytes
2.4.2. Treatment of Mertansine in Human Hepatocytes
2.4.3. CYP1A2, CYP2B6, and CYP3A4 Activity Measurement
2.4.4. RNA Purification and RT-PCR Analysis
2.5. Data Analysis
3. Results
3.1. Inhibition of UGT Enzyme Activities by Mertansine in Human Liver Microsomes
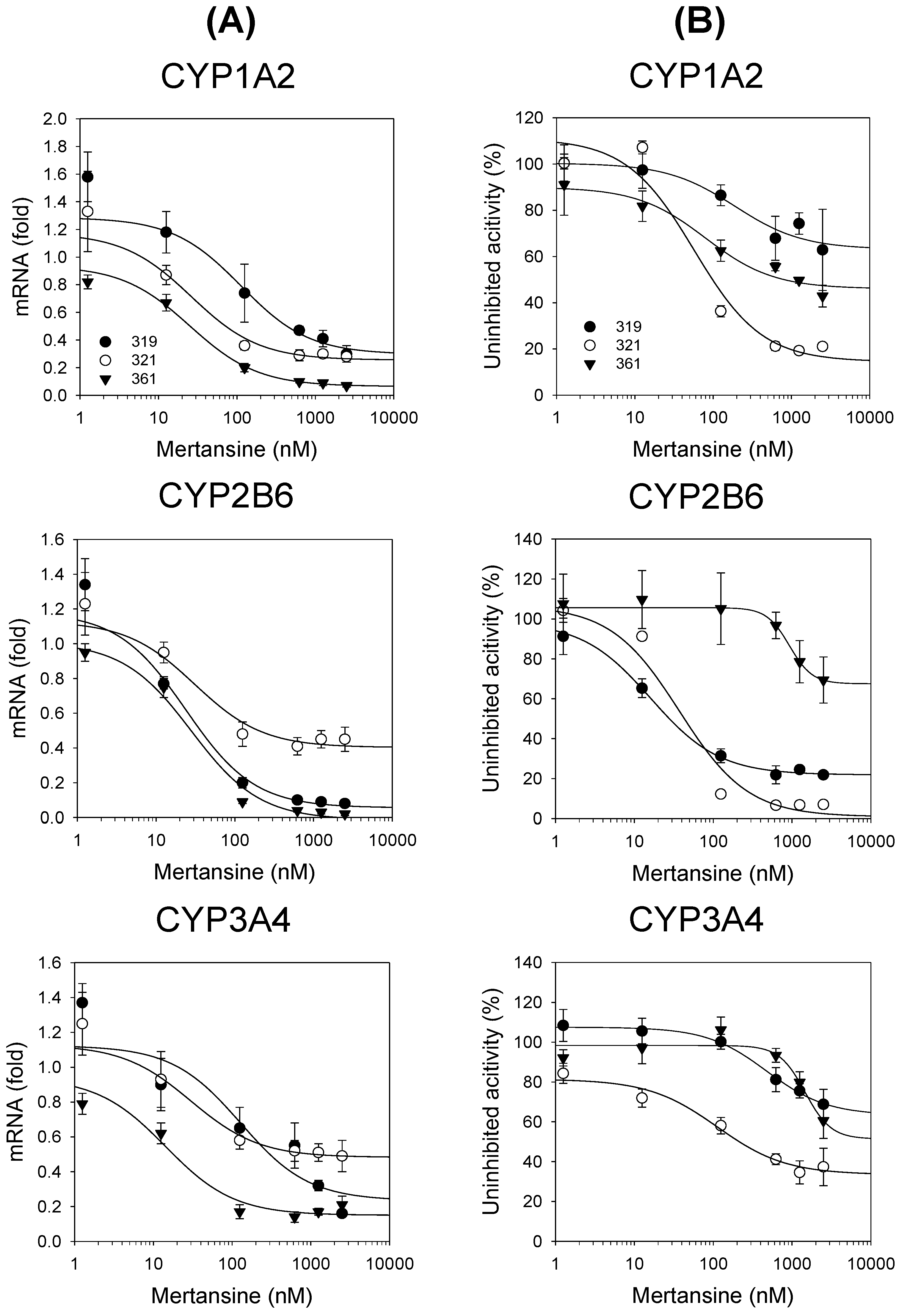
3.2. Effects of Mertansine on CYP and UGT mRNA Levels in Human Hepatocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Tumor inhibitors. LXXIII. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef]
- Huang, A.B.; Lin, C.M.; Hamel, E. Maytansine inhibits nucleotide binding at the exchangeable site of tubulin. Biochem. Biophys. Res. Commun. 1985, 128, 1239–1246. [Google Scholar] [CrossRef]
- Hamel, E. Natural products which interact with tubulin in the vinca domain: Maytansine, rhizoxin, phomopsin A, dolastatins 10 and 15 and halichondrin B. Pharm. Ther. 1992, 55, 31–51. [Google Scholar] [CrossRef]
- Sawada, T.; Kato, Y.; Kobayashi, H.; Hashimoto, Y.; Watanabe, T.; Sugiyama, Y.; Iwasaki, S. A fluorescent probe and a photoaffinity labeling reagent to study the binding site of maytansine and rhizoxin on tubulin. Bioconjugate Chem. 1993, 4, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Taplin, S.; Vashisht, K.; Walles, M.; Calise, D.; Kluwe, W.; Bouchard, P.; Johnson, R. Hepatotoxicity with antibody maytansinoid conjugates: A review of preclinical and clinical findings. J. Appl. Toxicol. 2018, 38, 600–615. [Google Scholar] [CrossRef] [PubMed]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent developments in the maytansinoid antitumor agents. Chem. Pharm. Bull. 2004, 52, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Widdison, W.; Mayo, M.; Wilhelm, S.; Leece, B.; Chari, R.; Singh, R.; Erickson, H. Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconj. Chem. 2011, 22, 728–735. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin inhibitor-based antibody-drug conjugates for cancer therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [Green Version]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-drug conjugates for cancer therapy: Chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Morris, C.Q. Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: A review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucci, F.; Casterta, C.A.; Romeo, E.; Rumio, C. Antibody-drug conjugates (ADC) against cancer stem-like cells (CSC): Is there still room for optimism? Front. Oncol. 2019, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Wolska-Washer, A.; Robak, T. Safety and tolerability of antibody-drug conjugates in cancer. Drug Saf. 2019, 42, 295–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired resistance to antibody-drug conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S.; et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J. Clin. Oncol. 2011, 29, 398–405. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ando, M.; Aogi, K.; Iwata, H.; Tamura, K.; Yonemori, K.; Shimizu, C.; Hara, F.; Takabatake, D.; Hattori, M.; et al. Phase I and pharmacokinetic study of trastuzumab emtansine in Japanese patients with HER2-positive metastatic breast cancer. Jpn. J. Clin. Oncol. 2015, 45, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Girish, S.; Gupta, M.; Wang, B.; Lu, D.; Krop, I.E.; Vogel, C.L.; Burris, H.A., 3rd; LoRusso, P.M.; Yi, J.H.; Saad, O.; et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): An antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother. Pharmacol. 2012, 69, 1229–1240. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Yu, K.; Zhang, K.; Liu, L.; Li, Y. Efficacy and safety of trastuzumab emtansine (T-DM1) in the treatment of HER2-positive metastatic breast cancer (MBC): A meta-analysis of randomized controlled trial. Oncotarget 2017, 8, 102458–102467. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Sychev, D.A.; Ashraf, G.M.; Svistunov, A.A.; Maksimov, M.L.; Tarasov, V.V.; Chubarev, V.N.; Otdelenov, V.A.; Denisenko, N.P.; Barreto, G.E.; Aliev, G. The cytochrome P450 isoenzyme and some new opportunities for the prediction of negative drug interaction in vivo. Drug Des. Dev. 2018, 12, 1147–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manikandan, P.; Nagini, S. Cytochrome P450 structure, function and clinical significance: A review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.; Morcos, P.N.; Cleary, Y.; Martin-Facklam, M.; Parrott, N.; Gertz, M.; Yu, L. Progress in prediction and interpretation of clinically relevant metabolic drug–drug interactions: A minireview illustrating recent developments and current opportunities. Curr. Pharmacol. Rep. 2017, 3, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.G.; Hulin, J.U.; Nair, P.C.; Haines, A.Z.; McKinnon, R.A.; Mackenzie, P.I.; Meech, R. The UGTome: The expanding diversity of UDP glycosyltransferases and its impact on small molecule metabolism. Pharmacol. Ther. 2019, 204, 107414. [Google Scholar] [CrossRef]
- Hariparsad, N.; Ramsden, D.; Palamanda, J.; Dekeyser, J.G.; Fahmi, O.A.; Kenny, J.R.; Einolf, H.; Mohutsky, M.; Pardon, M.; Amy Siu, Y.; et al. Considerations from the IQ induction working group in response to drug-drug interaction guidance from regulatory agencies: Focus on downregulation, CYP2C induction, and CYP2B6 positive control. Drug Metab. Dispos. 2017, 45, 1049–1059. [Google Scholar] [CrossRef]
- Cerny, M.A. Prevalence of non-cytochrome P450-mediated metabolism in food and drug administration-approved oral and intravenous drugs: 2006–2015. Drug Metab. Dispos. 2016, 44, 1246–1252. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.Q.; Bumbaca, D.; Yue, Q.; Saad, O.; Tibbitts, J.; Khojasteh, S.C.; Girish, S. Non-clinical disposition and metabolism of DM1, a component of trastuzumab emtansine (T-DM1), in Sprague Dawley rats. Drug Metab. Lett. 2015, 9, 119–131. [Google Scholar] [CrossRef]
- Shen, B.Q.; Bumbaca, D.; Saad, O.; Yue, Q.; Pastuskovas, C.V.; Khojasteh, S.C.; Tibbitts, J.; Kaur, S.; Wang, B.; Chu, Y.W.; et al. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T-DM1): An emphasis on preclinical and clinical catabolism. Curr. Drug Metab. 2012, 13, 901–910. [Google Scholar] [CrossRef]
- Erickson, H.K.; Lambert, J.M. ADME of antibody-maytansinoid conjugates. AAPS J. 2012, 14, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Han, T.H.; Zhao, B. Absorption, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab. Dispos. 2014, 42, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.A.; Rock, D.A.; Wienkers, L.C.; Pearson, J.T. In vitro characterization of the drug-drug interaction potential of catabolites of antibody-maytansinoid conjugates. Drug Metab. Dispos. 2012, 40, 1927–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolenski, F.S.; Xia, C.Q.; Ma, B.; Han, T.H.; Shyu, W.C.; Balani, S.K. CYP suppression in human hepatocytes by monomethyl auristatin E, the payload in brentuximab vedotin (Adcetris®), is associated with microtubule disruption. Eur. J. Drug Metab. Pharmacok. 2018, 43, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, Z.; Modriansky, M.; Pichard-Garcia, L.; Balaguer, P.; Vilarem, M.J.; Ulrichová, J.; Maurel, P.; Pascussi, J.M. Colchicine down-regulates Cytochrome P450 2B6, 2C8, 2C9, and 3A4 in human hepatocytes by affecting their glucocorticoid receptor-mediated regulation. Mol. Pharm. 2003, 64, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, Z.; Ulrichova, J.; Modriansky, M. Role of microtubules network in CYP gene expression. Curr. Drug Metab. 2005, 6, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.S.; Kim, J.H.; Jeong, H.U.; Cho, Y.Y.; Oh, S.R.; Lee, H.S. Inhibitory effects of aschantin on cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2016, 21, 554. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.Y.; Jeong, H.U.; Kim, J.H.; Lee, H.S. Effect of honokiol on the induction of drug metabolizing enzymes in human hepatocytes. Drug Des. Devel. Ther. 2014, 8, 2137–2145. [Google Scholar]
- Yoshinari, K.; Ueda, R.; Kusano, K.; Yoshimura, T.; Nagata, K.; Yamazoe, Y. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem. Pharm. 2008, 76, 139–145. [Google Scholar] [CrossRef]
- Luo, G.; Cunningham, M.; Kim, S.; Burn, T.; Lin, J.; Sinz, M.; Hamilton, G.; Rizzo, C.; Jolley, S.; Gilbert, D.; et al. CYP3A4 induction by drugs: Correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab. Dispos. 2002, 30, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Faucette, S.; Moore, R.; Sueyoshi, T.; Negishi, M.; LeCluyse, E. Human constitutive androstane receptor mediates induction of CYP2B6 gene expression by phenytoin. J. Biol. Chem. 2004, 279, 29295–29301. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walles, M.; Rudolph, B.; Wolf, T.; Bourgailh, J.; Suetterlin, M.; Moenius, T.; Peraus, G.; Heudi, O.; Elbast, W.; Lanshoeft, C.; et al. New insights in tissue distribution, metabolism, and excretion of [3H]-labeled antibody maytansinoid conjugates in female tumor-bearing nude rats. Drug Metab. Dispos. 2016, 44, 897–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]


 ); 0.5 μM (
); 0.5 μM (  ); 1 μM (▼); 2 μM (▽); (B) chenodeoxycholic acid; 0.5 μM ( ); 1 μM ( ); 2 μM (▼); 5 μM (▽); and (C) trifluoperazine; 0.2 μM ( ); 0.5 μM ( ); 1 μM (▼); 2 μM (▽). Data are expressed as means ± SD (n = 3).
); 0.5 μM ( ); 1 μM (▼); 2 μM (▽); (B) chenodeoxycholic acid; 0.5 μM ( ); 1 μM ( ); 2 μM (▼); 5 μM (▽); and (C) trifluoperazine; 0.2 μM ( ); 0.5 μM ( ); 1 μM (▼); 2 μM (▽). Data are expressed as means ± SD (n = 3).
); 1 μM (▼); 2 μM (▽); (B) chenodeoxycholic acid; 0.5 μM ( ); 1 μM ( ); 2 μM (▼); 5 μM (▽); and (C) trifluoperazine; 0.2 μM ( ); 0.5 μM ( ); 1 μM (▼); 2 μM (▽). Data are expressed as means ± SD (n = 3).
); 0.5 μM ( ); 1 μM (▼); 2 μM (▽); (B) chenodeoxycholic acid; 0.5 μM ( ); 1 μM ( ); 2 μM (▼); 5 μM (▽); and (C) trifluoperazine; 0.2 μM ( ); 0.5 μM ( ); 1 μM (▼); 2 μM (▽). Data are expressed as means ± SD (n = 3). ), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3). ), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).
), 321 ( ), and 361 (▼). Data are expressed as means ± SD (n = 3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UGTs | Enzyme Activities | IC50 (µM) | Ki (µM) | Inhibition Mode |
|---|---|---|---|---|
| 1A1 | SN-38 glucuronidation | 16.2 | 13.5 | Noncompetitive |
| 1A3 | Chenodeoxycholic acid 24-acyl-β-glucuronidation | 6.4 | 4.3 | Competitive |
| 1A4 | Trifluoperazine N-β-d-glucuronidation | 23.3 | 21.2 | Competitive |
| 1A6 | N-acetylserotonin β-d-glucuronidation | No inhibition | - | - |
| 1A9 | Mycophenolic acid β-d-glucuronidation | No inhibition | - | - |
| 2B7 | Naloxone 3-β-d-glucuronidation | No inhibition | - | - |
| Enzymes | mRNA (Fold Change) | Enzyme Activities (pmol/106 Cells/min) | ||||
|---|---|---|---|---|---|---|
| Lot 319 | Lot 321 | Lot 361 | Lot 319 | Lot 321 | Lot 361 | |
| Omeprazole 50 μM | ||||||
| Vehicle control | 1.0 | 1.0 | 1.0 | 1.6 ± 0.40 a | 3.10 ± 0.26 a | 1.3 ± 0.16 a |
| CYP1A2 | 132.4 ± 0.22 | 58.7 ± 4.7 | 299.3 ± 49.0 | 18.5 ± 3.3 a | 89.45 ± 4.52 a | 81.0 ± 0.53 a |
| UGT1A1 | 6.97 ± 0.94 | 4.05 ± 0.53 | 3.9 ± 0.24 | - | - | - |
| UGT1A4 | 3.31 ± 0.35 | 4.13 ± 0.66 | 3.7 ± 0.62 | - | - | - |
| CITCO 10 nM | ||||||
| Vehicle control | 1.0 | 1.0 | 1.0 | 0.68 ± 0.23 b | 0.72 ± 0.10 b | 3.43 ± 0.48 b |
| CYP2B6 | 7.4 ± 1.9 | 5.6 ± 1.6 | 8.7 ± 0.32 | 9.2 ± 0.28 b | 11.3 ± 0.49 b | 12.9 ± 0.51 b |
| Rifampin 10 μM | ||||||
| Vehicle control | 1.00 | 1.00 | 1.00 | 16.6 ± 3.78 c | 4.3 ± 0.09 c | 7.1 ± 0.67 c |
| CYP3A4 | 74.0 ± 10.3 | 146.7 ± 24.9 | 129.8 ± 0.5 | 74.9 ± 3.36 c | 42.0 ± 9.8 c | 25.6 ± 1.0 c |
| CYP2C8 | 3.7 ± 0.57 | 4.8 ± 0.84 | 4.0 ± 0.16 | - | - | - |
| CYP2C9 | 5.3 ± 0.02 | 2.9 ± 0.11 | 3.7 ± 0.36 | - | - | - |
| CYP2C19 | 2.2 ± 0.09 | 2.0 ± 0.18 | 2.1 ± 0.25 | - | - | - |
| UGT1A1 | 2.9 ± 0.13 | 3.0 ± 0.11 | 2.5 ± 0.37 | - | - | - |
| UGT1A4 | 4.0 ± 0.22 | 4.5 ± 0.59 | 4.5 ± 0.87 | - | - | - |
| UGT1A9 | 2.0 ± 0.23 | 2.2 ± 0.31 | 2.1 ± 0.23 | - | - | - |
| Enzymes | IC50 (nM) | |||
|---|---|---|---|---|
| Lot 319 | Lot 321 | Lot 361 | Mean ± SD | |
| CYP1A2 | 219.7 | 34.3 | 27.3 | 93.7 ± 109.1 |
| CYP2B6 | 25.0 | 57.8 | 27.6 | 36.8 ± 18.3 |
| CYP3A4 | 344.5 | 120.5 | 16.8 | 160.6 ± 167.4 |
| CYP2C8 | 48.7 | 20.0 | 27.5 | 32.1 ± 14.9 |
| CYP2C9 | 788.2 | 887.5 | 59.6 | 578.4 ± 452.0 |
| CYP2C19 | 573.5 | 754.1 | 291.0 | 539.5 ± 233.4 |
| UGT1A1 | 784.5 | 113.4 | 1672.1 | 856.7 ± 781.9 |
| UGT1A4 | >2500 | >2500 | >2500 | >2500 |
| UGT1A9 | 86.9 | 31.4 | 44.1 | 54.1 ± 29.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, W.-G.; Park, R.; Kim, D.K.; Shin, Y.; Cho, Y.-Y.; Lee, H.S. Mertansine Inhibits mRNA Expression and Enzyme Activities of Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyltransferases in Human Hepatocytes and Liver Microsomes. Pharmaceutics 2020, 12, 220. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030220
Choi W-G, Park R, Kim DK, Shin Y, Cho Y-Y, Lee HS. Mertansine Inhibits mRNA Expression and Enzyme Activities of Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyltransferases in Human Hepatocytes and Liver Microsomes. Pharmaceutics. 2020; 12(3):220. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030220
Chicago/Turabian StyleChoi, Won-Gu, Ria Park, Dong Kyun Kim, Yongho Shin, Yong-Yeon Cho, and Hye Suk Lee. 2020. "Mertansine Inhibits mRNA Expression and Enzyme Activities of Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyltransferases in Human Hepatocytes and Liver Microsomes" Pharmaceutics 12, no. 3: 220. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030220