Stem Cells as Drug-like Biologics for Mitochondrial Repair in Stroke


Abstract
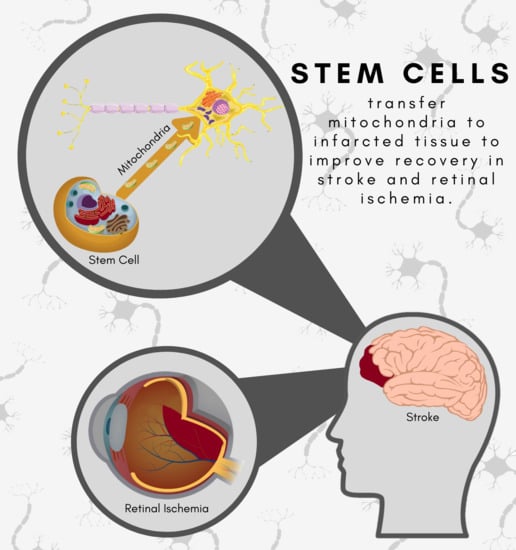
:
1. Stroke: A Trilogy of Cell Death Events
2. May the Force of Mitochondria Be with Stroke
3. The Return of the Force: Enhancing Mitochondrial Function in Stroke
4. Force in the Outer Rim: Stroke Extends to the Retina but may be Repaired by Stem Cell-Mediated Mitochondria Transfer
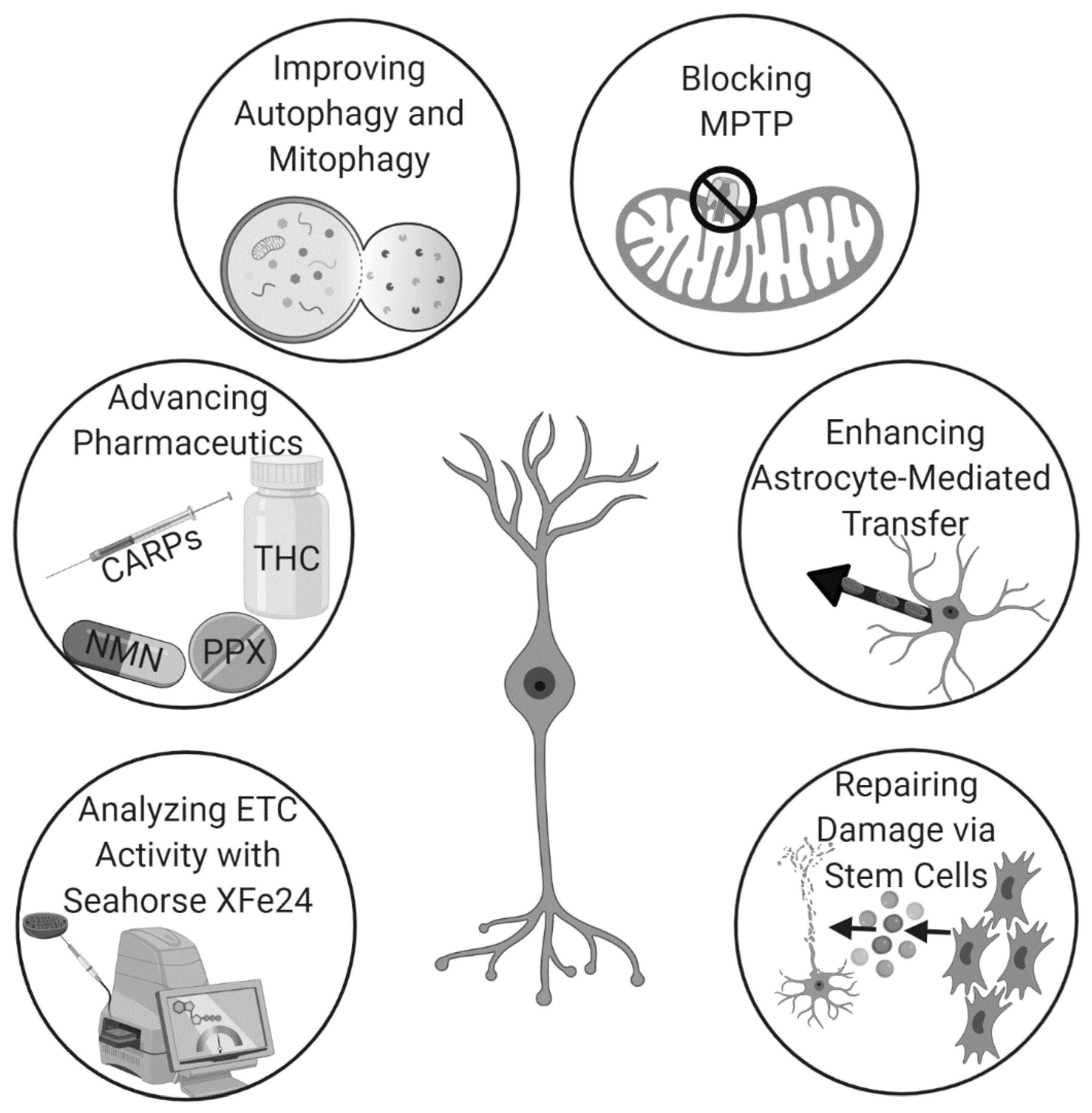
5. The Rise of the Force: New Horizons in Mitochondrial Repair for Stroke
5.1. Pharmacological Treatment
5.2. Autophagy/Mitophagy
5.3. Molecular and Other Mechanisms
5.4. Stem Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ovbiagele, B.; Goldstein, L.B.; Higashida, R.T.; Howard, V.J.; Johnston, S.C.; Khavjou, O.A.; Lackland, D.T.; Lichtman, J.H.; Mohl, S.; Sacco, R.L.; et al. Forecasting the Future of Stroke in the United States. Stroke 2013, 44, 2361–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zou, Z.; Tian, H.; Zhang, Y.; Zhou, H.; Liu, L. Stem cell-based therapies for ischemic stroke. Biomed. Res. Int. 2014, 2014, 468748. [Google Scholar] [CrossRef] [Green Version]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Stonesifer, C.; Corey, S.; Ghanekar, S.; Diamandis, Z.; Acosta, S.A.; Borlongan, C.V. Stem cell therapy for abrogating stroke-induced neuroinflammation and relevant secondary cell death mechanisms. Prog Neurobiol. 2017, 158, 94–131. [Google Scholar] [CrossRef]
- Kingsbury, C.; Heyck, M.; Bonsack, B.; Lee, J.Y.; Borlongan, C.V. Stroke gets in your eyes: Stroke-induced retinal ischemia and the potential of stem cell therapy. Neural Regen. Res. 2020, 15, 1014–1018. [Google Scholar] [CrossRef]
- Osborne, N. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp. Eye Res. 2010, 90, 8. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, K.; Lindsey, J.; Dai, Y.; Heo, H.; Nguyen, D.; Ellisman, M.; Weinreb, R.; Ju, W. A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest. Ophthalmol. Vis. Sci. 2011, 52, 7. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.L.; Mukda, S.; Chen, S.D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., 3rd. CNS energy metabolism as related to function. Brain Res. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyck, M.; Bonsack, B.; Zhang, H.; Sadanandan, N.; Cozene, B.; Kingsbury, C.; Lee, J.Y.; Borlongan, C.V. The brain and eye: Treating cerebral and retinal ischemia through mitochondrial transfer. Exp. Biol. Med. (Maywood) 2019, 244, 1485–1492. [Google Scholar] [CrossRef]
- Sims, N.R.; Muyderman, H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim. Biophys. Acta 2010, 1802, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Saks, V.A.; Rosenshtraukh, L.V.; Smirnov, V.N.; Chazov, E.I. Role of creatine phosphokinase in cellular function and metabolism. Can. J. Physiol. Pharmacol. 1978, 56, 691–706. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Stachowiak, O.; Dolder, M.; Wallimann, T.; Richter, C. Mitochondrial creatine kinase is a prime target of peroxynitrite-induced modification and inactivation. J. Biol. Chem. 1998, 273, 16694–16699. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.; Tse, H.F.; Mak, J.C.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Liou, C.W.; Chen, S.D.; Hsu, T.Y.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; Lin, T.K.; et al. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Hristov, M.; Erl, W.; Weber, P.C. Endothelial progenitor cells: Isolation and characterization. Trends Cardiovasc. Med. 2003, 13, 201–206. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, F.; Frenzel, T.; Zhu, W.; Ye, J.; Liu, J.; Chen, Y.; Su, H.; Young, W.L.; Yang, G.Y. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann. Neurol. 2010, 67, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Aithal, A.P.; Bairy, L.K.; Seetharam, R.N. Safety Assessment of Human Bone Marrow-derived Mesenchymal Stromal Cells Transplantation in Wistar Rats. J. Clin. Diagn. Res. 2017, 11, FF01–FF03. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, H.-Q.; Li, J.; Liu, X.-L. Endothelial progenitor cells promote tumor growth and progression by enhancing new vessel formation. Oncol. Lett. 2016, 12, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slevin, M.; Kumar, P.; Gaffney, J.; Kumar, S.; Krupinski, J. Can angiogenesis be exploited to improve stroke outcome? Mechanisms and therapeutic potential. Clin. Sci. (Lond) 2006, 111, 171–183. [Google Scholar] [CrossRef] [PubMed]
- van der Strate, B.W.; Popa, E.R.; Schipper, M.; Brouwer, L.A.; Hendriks, M.; Harmsen, M.C.; van Luyn, M.J. Circulating human CD34+ progenitor cells modulate neovascularization and inflammation in a nude mouse model. J. Mol. Cell Cardiol. 2007, 42, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Lee, J.Y.; Nguyen, H.; Kaneko, Y.; Borlongan, C.V. Endothelial Progenitor Cells Modulate Inflammation-Associated Stroke Vasculome. Stem Cell Rev. Rep. 2019, 15, 256–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, F. Stroke survivors’ views and experiences on impact of visual impairment. Brain Behav. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Sand, K.M.; Midelfart, A.; Thomassen, L.; Melms, A.; Wilhelm, H.; Hoff, J. Visual impairment in stroke patients-a review. Acta Neurol. Scand. Suppl. 2013, 196, 5. [Google Scholar] [CrossRef]
- Nguyen, H.; Lee, J.; Sanberg, P.; Napoli, E.; Borlongan, C. Eye opener in stroke. Stroke 2019, 50, 10. [Google Scholar] [CrossRef]
- Borlongan, C.; Lind, J.; Dillon-Carter, O.; Yu, G.; Hadman, M.; Cheng, C.; Carroll, J.; Hess, D. Bone marrow grafts restore cerebral blood flow and blood brain barrier in stroke rats. Brain Res. 2004, 1010, 9. [Google Scholar] [CrossRef]
- Taninishi, H.; Jung, J.; Izutsu, M.; Wang, Z.; Sheng, H.; Warner, D. A blinded randomized assessment of laser Doppler flowmetry efficacy in standardizing outcome from intraluminal filament MCAO in the rat. J. Neurosci. Methods 2015, 241, 10. [Google Scholar] [CrossRef]
- Shih, Y.; De La Garza, B.; Huang, S.; Li, G.; Wang, L.; Duong, T. Comparison of retinal and cerebral blood flow between continuous arterial spin labeling MRI and fluorescent microsphere techniques. J. Magn. Reson. Imaging 2014, 40, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, F.; Nguyen, C.; He, Z.; Vingrys, A.; Gurrell, R.; Fish, R.; Bui, B. Retinal and cortical blood flow dynamics following systemic blood-neural barrier disruption. Front. Neurosci. 2017, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritzel, R.; Pan, S.; Verma, R.; Wizeman, J.; Crapser, J.; Patel, A.; Lieberman, R.; Mohan, R.; McCullough, L. Early retinal inflammatory biomarkers in the Middle cerebral artery occlusion model of ischemic stroke. Mol. Vis. 2016, 22, 14. [Google Scholar]
- Allen, R.; Sayeed, I.; Oumarbaeva, Y.; Morrison, K.; Choi, P.; Pardue, M.; Stein, D. Progesterone treatment shows greater protection in brain vs. retina in a rat model of Middle cerebral artery occlusion: Progesterone receptor levels may play an important role. Restor. Neurol. Neurosci. 2016, 34, 17. [Google Scholar] [CrossRef]
- Xiao, J.; Zhou, X.; Jiang, T.; Zhi, Z.; Li, Q.; Qu, J.; Chen, J. Unilateral cerebral ischemia inhibits optomotor responses of the ipsilateral eye in mice. J. Integr. Neurosci. 2012, 11, 8. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, G.; Guo, S.; Ding, J.; Lin, J.; Yang, Q.; Li, Z. Mfn2-mediated preservation of mitochondrial function contributes to the portective effects of BHAPI in response to ischemia. J. Mol. Neurosci. 2017, 63, 8. [Google Scholar] [CrossRef]
- Shi, Y.; Yi, C.; Li, X.; Wang, J.; Zhou, F.; Chen, X. Overexpression of Mitofusin 2 decreased the reactive astrocytes proliferation in vitro induced by oxygen-glucose deprivation/reoxygenation. Neurosci. Lett. 2017, 639, 6. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, S.; Xia, C.; Guo, X.; He, W.; Chen, N. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: The role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 2014, 86, 13. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial approaches for neuroprotection. Ann. NY Acad. Sci. 2008, 1147, 18. [Google Scholar] [CrossRef]
- Prass, K.; Royl, G.; Lindauer, U.; Freyer, D.; Megow, D.; Dirnagl, U.; Stöckler-Ipsiroglu, G.; Wallimann, T.; Priller, J. Improved reperfusion and neuroprotection by creatine in a mouse model of stroke. J. Cereb. Blood Flow Metab 2007, 27, 8. [Google Scholar] [CrossRef]
- Kitzenberg, D.; Colgan, S.; Glover, L. Creatine kinase in ischemic and inflammatory disorders. Clin. Transl. Med. 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamadera, M.; Fujimura, H.; Shimizu, Y.; Matsui, M.; Nakamichi, I.; Yokoe, M.; Sakoda, S. Increased number of mitochondria in capillaries distributed in stroke-like lesions of two patients with MELAS. Neuropathology 2019, 39, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Sperling, J.A.; Sakamuri, S.; Albuck, A.L.; Sure, V.N.; Evans, W.R.; Peterson, N.R.; Rutkai, I.; Mostany, R.; Satou, R.; Katakam, P.V.G. Measuring Respiration in Isolated Murine Brain Mitochondria: Implications for Mechanistic Stroke Studies. Neuromol. Med. 2019, 21, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Ali, M.; Tabassum, H.; Parveen, S.; Parvez, S. Pramipexole prevents ischemic cell death via mitochondrial pathways in ischemic stroke. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Mondal, N.K.; Behera, J.; Kelly, K.E.; George, A.K.; Tyagi, P.K.; Tyagi, N. Tetrahydrocurcumin epigenetically mitigates mitochondrial dysfunction in brain vasculature during ischemic stroke. Neurochem. Int. 2019, 122, 120–138. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Nagana Gowda, G.A.; Tian, R. Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef]
- MacDougall, G.; Anderton, R.S.; Mastaglia, F.L.; Knuckey, N.W.; Meloni, B.P. Mitochondria and neuroprotection in stroke: Cationic arginine-rich peptides (CARPs) as a novel class of mitochondria-targeted neuroprotective therapeutics. Neurobiol. Dis. 2019, 121, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Forte, M.; Bianchi, F.; Cotugno, M.; Marchitti, S.; De Falco, E.; Raffa, S.; Stanzione, R.; Di Nonno, F.; Chimenti, I.; Palmerio, S.; et al. Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy 2019. [Google Scholar] [CrossRef]
- Vargas, J.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 16. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Li, L.; Ying, Z.; Cao, Z.; Ma, Y.; Mao, X.; Li, J.; Qi, X.; Zhang, Z.; Wang, X. A small molecule protects mitochondrial integrity by inhibiting mTOR activity. Proc. Natl. Acad. Sci. USA 2019, 116, 7. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Sideris, D.P.; Giagtzoglou, N.; Ni, L.; Kankel, M.W.; Sen, A.; Bochicchio, L.E.; Huang, C.H.; Nussenzweig, S.C.; Worley, S.H.; et al. PINK1/Parkin Influences Cell Cycle by Sequestering TBK1 at Damaged Mitochondria, Inhibiting Mitosis. Cell Rep. 2019, 29, 225–235.e225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 2019, 73, 1028–1043.e1025. [Google Scholar] [CrossRef] [Green Version]
- Puri, R.; Cheng, X.T.; Lin, M.Y.; Huang, N.; Sheng, Z.H. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 2019, 10, 3645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.S.; Tabassum, H.; Parveen, S.; Parvez, S. Ropinirole induces neuroprotection following reperfusion-promoted mitochondrial dysfunction after focal cerebral ischemia in Wistar rats. Neurotoxicology 2020, 77, 94–104. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 2020, 146, 45–58. [Google Scholar] [CrossRef]
- Cabral-Costa, J.V.; Kowaltowski, A.J. Neurological disorders and mitochondria. Mol. Aspects Med. 2020, 71, 100826. [Google Scholar] [CrossRef]
- Tomar, D.; Jaña, F.; Dong, Z.; Quinn, W.J., 3rd; Jadiya, P.; Breves, S.L.; Daw, C.C.; Srikantan, S.; Shanmughapriya, S.; Nemani, N.; et al. Blockade of MCU-Mediated Ca(2+) Uptake Perturbs Lipid Metabolism via PP4-Dependent AMPK Dephosphorylation. Cell Rep. 2019, 26, 3709–3725.e3707. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e415. [Google Scholar] [CrossRef] [Green Version]
- Mukda, S.; Tsai, C.Y.; Leu, S.; Yang, J.L.; Chan, S.H.H. Pinin protects astrocytes from cell death after acute ischemic stroke via maintenance of mitochondrial anti-apoptotic and bioenergetics functions. J. Biomed. Sci 2019, 26, 43. [Google Scholar] [CrossRef]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 2019, 25, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Nguyen, H.; Lippert, T.; Russo, E.; Tuazon, J.; Xu, K.; Lee, J.Y.; Sanberg, P.R.; Kaneko, Y.; Napoli, E. May the force be with you: Transfer of healthy mitochondria from stem cells to stroke cells. J. Cereb. Blood Flow Metab 2019, 39, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc. Res. 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Surugiu, R.; Olaru, A.; Hermann, D.M.; Glavan, D.; Catalin, B.; Popa-Wagner, A. Recent Advances in Mono- and Combined Stem Cell Therapies of Stroke in Animal Models and Humans. Int. J. Mol. Sci. 2019, 20, 6029. [Google Scholar] [CrossRef] [Green Version]
- Sarmah, D.; Kaur, H.; Saraf, J.; Vats, K.; Pravalika, K.; Wanve, M.; Kalia, K.; Borah, A.; Kumar, A.; Wang, X.; et al. Mitochondrial Dysfunction in Stroke: Implications of Stem Cell Therapy. Transl. Stroke Res. 2019, 10, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, G.; Zhang, Y.-L.; He, H.-W.; Yu, B.-Q.; Hong, Y.-M.; You, W.; Li, X. Transfer of mitochondria from mesenchymal stem cells derived from induced pluripotent stem cells attenuates hypoxia-ischemia-induced mitochondrial dysfunction in PC12 cells. Neural Regen. Res. 2020, 15, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Lee, J.Y.; Nguyen, H.; Corrao, S.; Anzalone, R.; La Rocca, G.; Borlongan, C.V. Energy Metabolism Analysis of Three Different Mesenchymal Stem Cell Populations of Umbilical Cord Under Normal and Pathologic Conditions. Stem Cell Rev. Rep. 2020, 16, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.-Y.; Biswas, S.; Li, J.; Mao, C.-J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.-R.; Liu, C.-F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; He, T.; Zhao, T.; Chen, Y.-Y.; Shen, Y.-L.; Zhang, X.; Wang, L.-L. Mitochondrial Transfer from Bone Marrow Mesenchymal Stem Cells to Motor Neurons in Spinal Cord Injury Rats via Gap Junction. Theranostics 2019, 9, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cheung, M.K.H.; Han, S.; Zhang, Z.; Chen, L.; Chen, J.; Zeng, H.; Qiu, J. Mesenchymal stem cells and their mitochondrial transfer: A double-edged sword. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Milestone Discovery | Reference |
|---|---|
| Measuring mitochondrial respiration | [52,53] |
| Targeting mitochondria via pharmacological treatment | [54,55,56,57] |
| Normalizing autophagy | [58,59,60] |
| Enhancing mitophagy | [61,62,63] |
| Inhibiting MPTP | [64,65,66,67,68] |
| Inducing astrocyte-based transfer | [69,70] |
| Augmenting endogenous tissue repair | [71,72,73,74,75,76,77,78,79,80] |
| Experimentally Demonstrated Therapeutic Effect | Type of Stem Cell |
|---|---|
| Transfer of mitochondria via intracellular mechanisms to injured cells | MSC, iPSC, BM-MSC, EPC, ASC |
| Enhances astrocyte-based transfer | MSC, iPSC, BM-MSC, EPC, ASC |
| Represses apoptosis after IR injury | MSC |
| Reverses mitochondrial swelling | MSC |
| Diminishes mitochondrial cristae dissipation | MSC |
| Restores ischemic mitochondrial function | MSC |
| Ameliorates ganglion cell death within 14 days of stroke | MSC |
| Confers neuroprotection against OGD | MSC |
| Bolsters TNT formation after IR injury | MSC |
| Improves mitochondrial survival rate | hUC-MSC |
| Augments cell bioenergetics and locomotor function | BM-MSC |
| Rescues aerobic respiration in HUVECs | BM-MSC |
| Repairs dopaminergic neuron damage | iPSC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, J.; Park, Y.J.; Cho, J.; Saft, M.; Sadanandan, N.; Cozene, B.; Borlongan, C.V. Stem Cells as Drug-like Biologics for Mitochondrial Repair in Stroke. Pharmaceutics 2020, 12, 615. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12070615
Farooq J, Park YJ, Cho J, Saft M, Sadanandan N, Cozene B, Borlongan CV. Stem Cells as Drug-like Biologics for Mitochondrial Repair in Stroke. Pharmaceutics. 2020; 12(7):615. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12070615
Chicago/Turabian StyleFarooq, Jeffrey, You Jeong Park, Justin Cho, Madeline Saft, Nadia Sadanandan, Blaise Cozene, and Cesar V. Borlongan. 2020. "Stem Cells as Drug-like Biologics for Mitochondrial Repair in Stroke" Pharmaceutics 12, no. 7: 615. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12070615