Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Basic Concepts of Enzyme, Substrate, and Inhibitor
2.1. Active Site or Orthosteric Site of the Enzyme
2.2. Allosteric Site
2.3. Substrates
2.4. Binding Affinity
2.5. Inhibitors
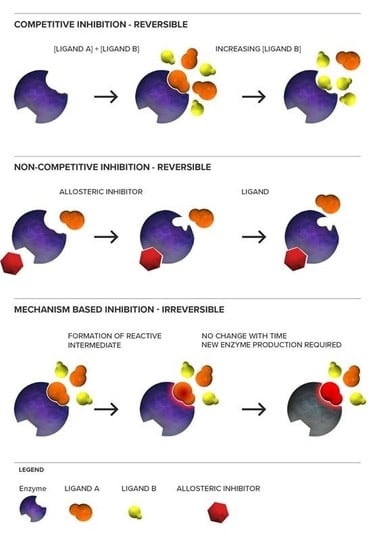
3. Mechanism of CYP450 Inhibition
3.1. Reversible CYP450 Inhibition
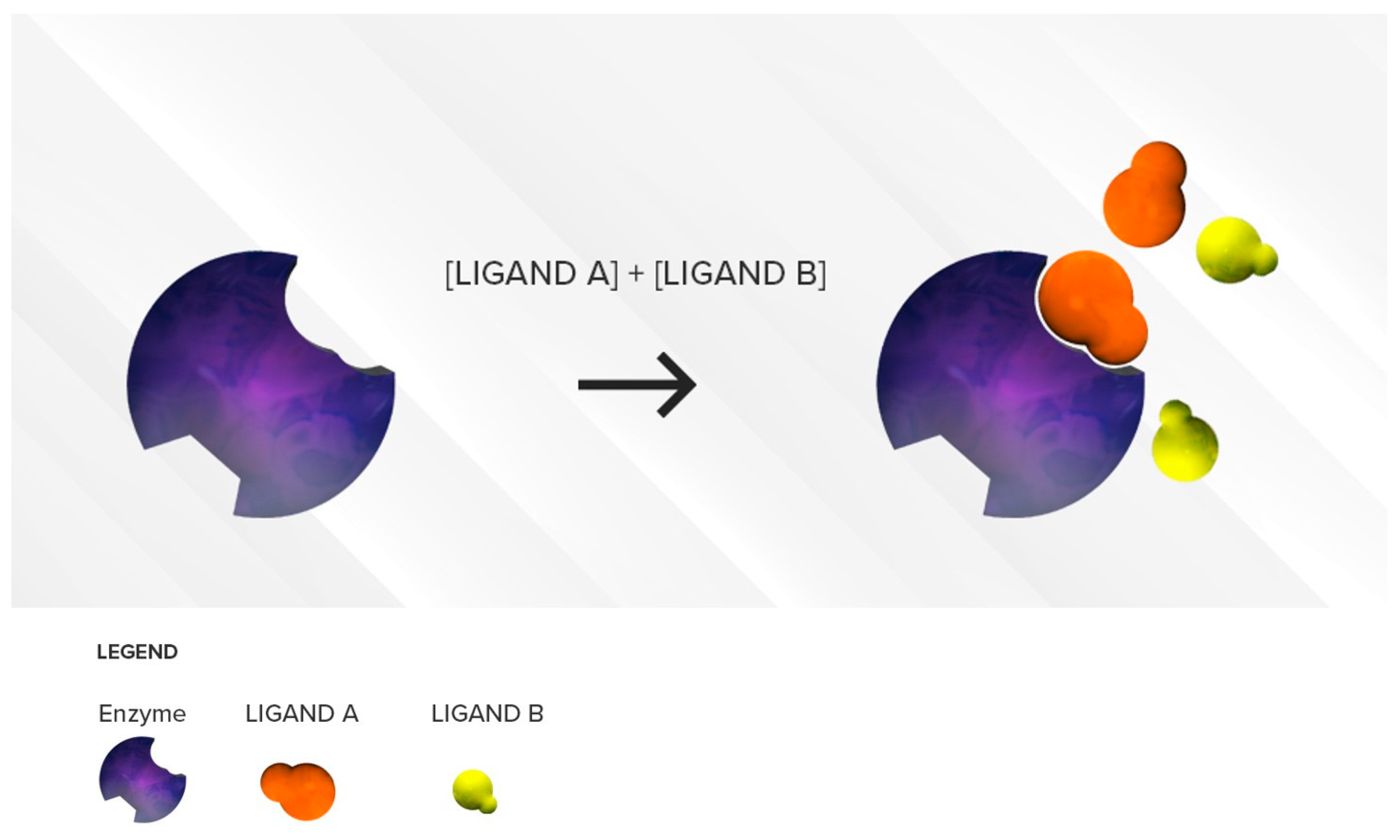
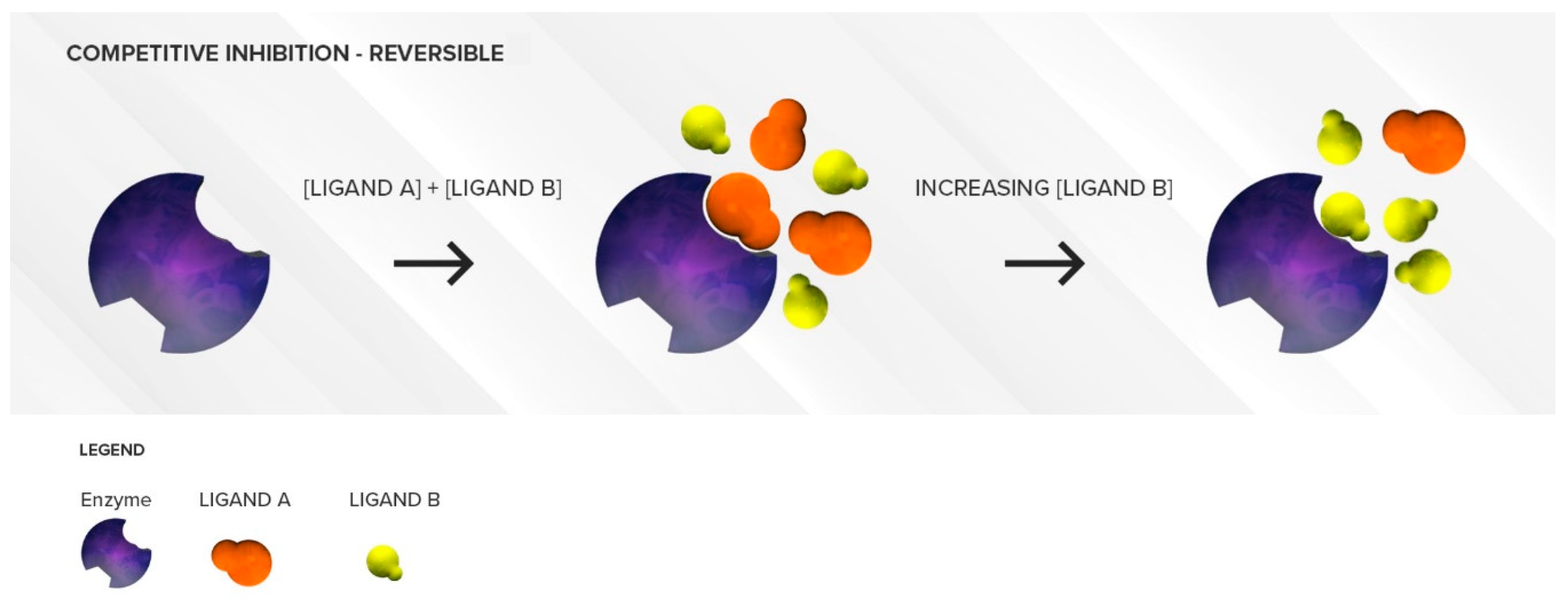
3.1.1. Competitive Inhibition
Two Substrates with Different Affinities Administered Concomitantly
Two Substrates with Largely Different Concentrations
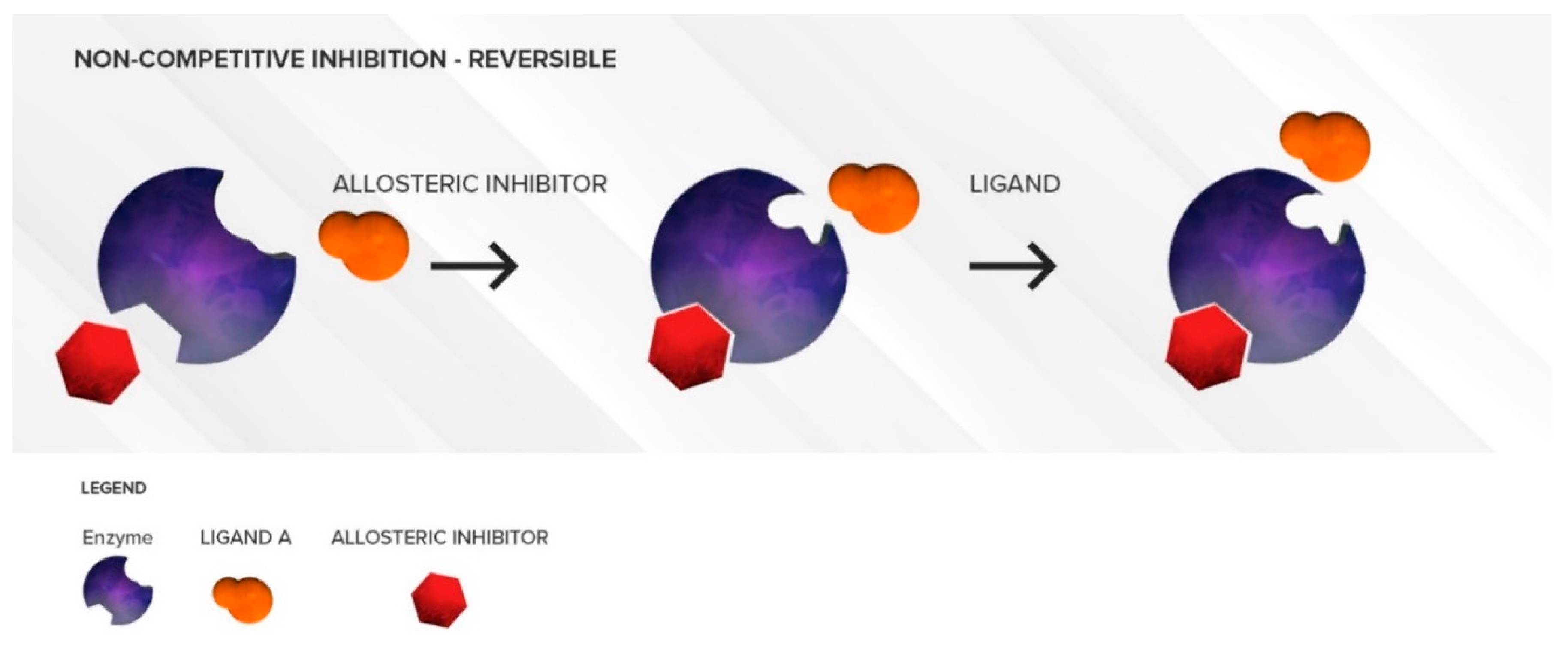
3.1.2. Non-Competitive Inhibition
3.1.3. Mixed inhibition
3.2. Irreversible CYP450 Inhibition
3.2.1. Mechanism-Based Inhibition
Metabolic–Intermediate Complex Formation (or Alternate Substrate Inhibition)
Protein and/or Heme Alkylation (or Suicide Inhibition)
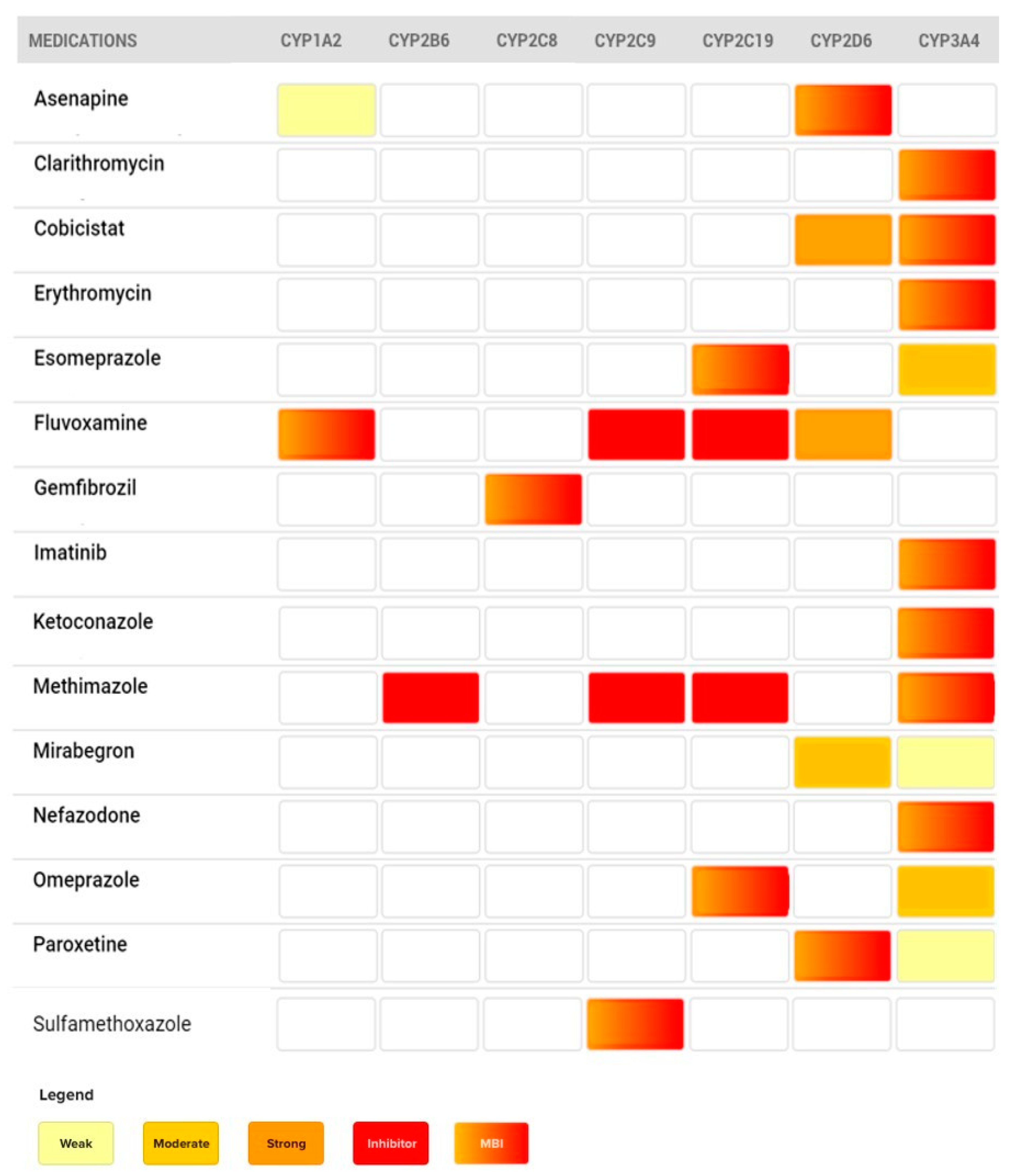
4. Clinical Cases
4.1. The Case of Omeprazole and Clopidogrel
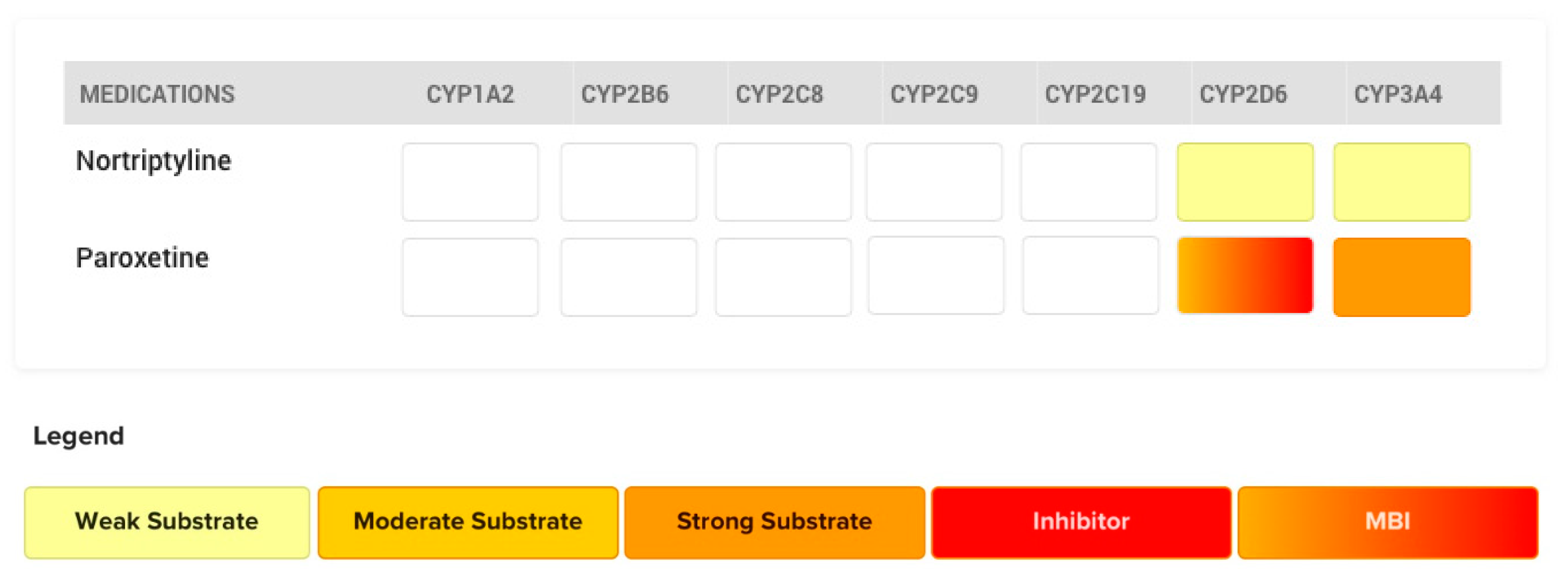
4.2. The Case of Paroxetine
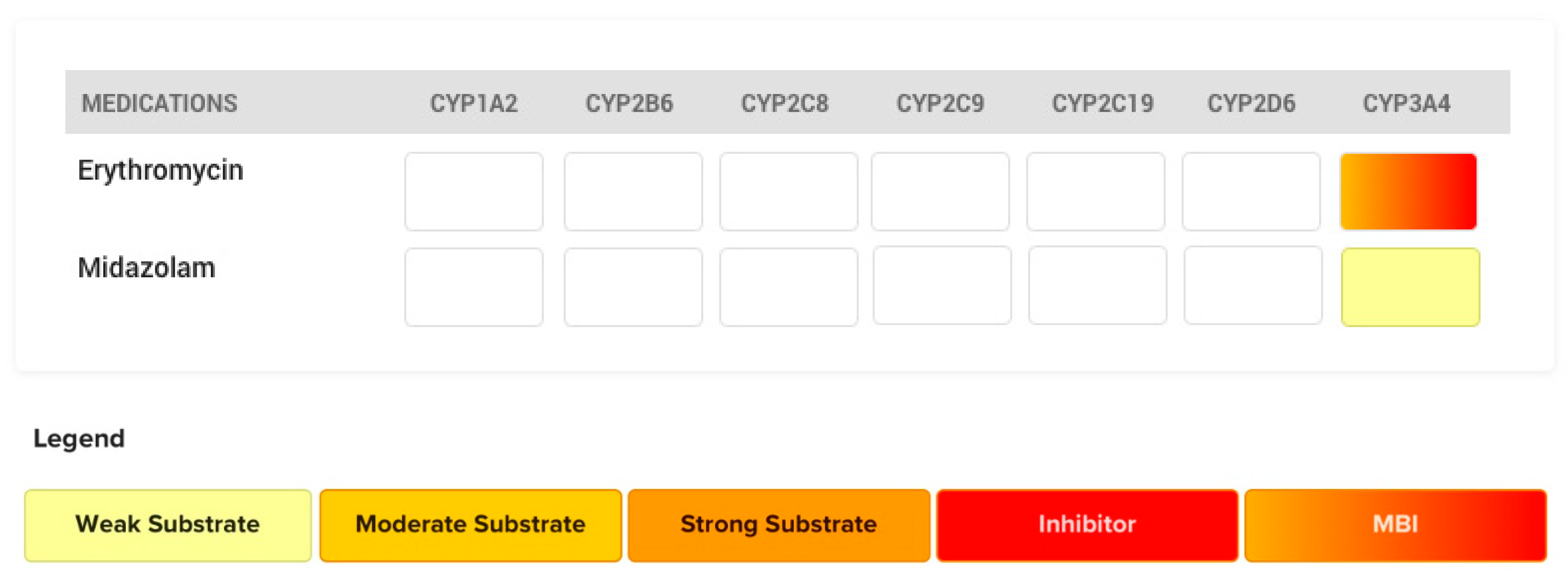
4.3. The Case of Erythromycin
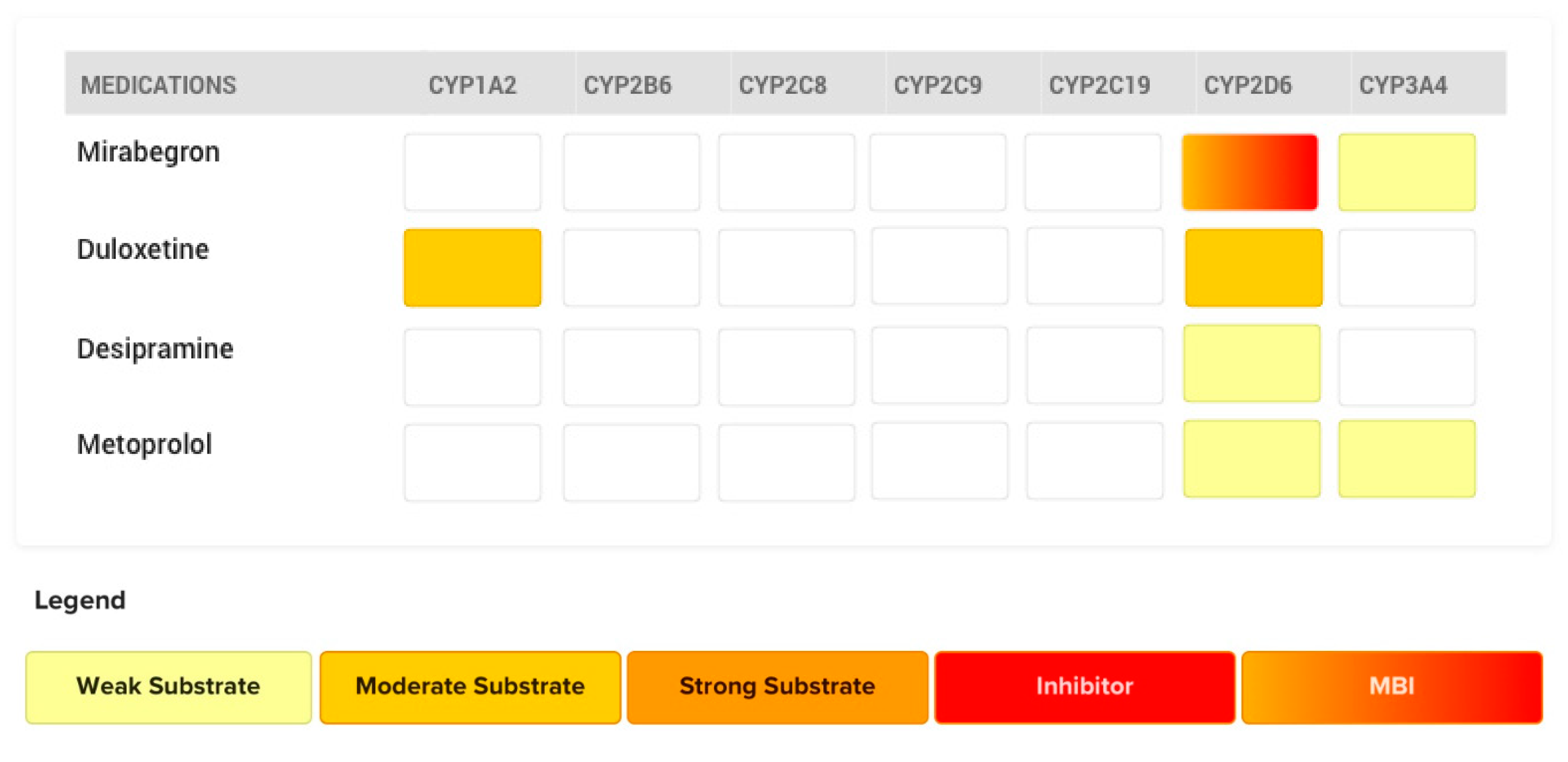
4.4. The Case of Mirabegron
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Doan, J.; Zakrzewski-Jakubiak, H.; Roy, J.; Turgeon, J.; Tannenbaum, C. Prevalence and risk of potential cytochrome P450-mediated drug-drug interactions in older hospitalized patients with polypharmacy. Ann. Pharmacother. 2013, 47, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Mallet, L.; Spinewine, A.; Huang, A. The challenge of managing drug interactions in elderly people. Lancet 2007, 370, 185–191. [Google Scholar] [CrossRef]
- Bankes, D.L.; Jin, H.; Finnel, S.; Michaud, V.; Knowlton, C.H.; Turgeon, J.; Stein, A. Association of a Novel Medication Risk Score with Adverse Drug Events and Other Pertinent Outcomes Among Participants of the Programs of All-Inclusive Care for the Elderly. Pharmacy 2020, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Wen, X.; Wang, J.S.; Backman, J.T.; Kivistö, K.T.; Neuvonen, P.J. Gemfibrozil is a potent inhibitor of human cytochrome P450 2C9. Drug Metab. Dispos. 2001, 29, 1359–1361. [Google Scholar]
- Shitara, Y.; Hirano, M.; Sato, H.; Sugiyama, Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: Analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J. Pharmacol. Exp. Ther. 2004, 311, 228–236. [Google Scholar]
- Lilja, J.J.; Backman, J.T.; Neuvonen, P.J. Effect of gemfibrozil on the pharmacokinetics and pharmacodynamics of racemic warfarin in healthy subjects. Br. J. Clin. Pharmacol. 2005, 59, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Backman, J.T.; Kyrklund, C.; Neuvonen, M.; Neuvonen, P.J. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin. Pharmacol. Ther. 2002, 72, 685–691. [Google Scholar] [CrossRef]
- Varma, M.V.S.; Lin, J.; Bi, Y.-A.; Kimoto, E.; Rodrigues, A.D. Quantitative Rationalization of Gemfibrozil Drug Interactions: Consideration of Transporters-Enzyme Interplay and the Role of Circulating Metabolite Gemfibrozil 1-O-β-Glucuronide. Drug Metab. Dispos. 2015, 43, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Niemi, M.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: Potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia 2003, 46, 347–351. [Google Scholar] [CrossRef]
- Jaakkola, T.; Backman, J.T.; Neuvonen, M.; Neuvonen, P.J. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin. Pharmacol. Ther. 2005, 77, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, B.W.; Zhang, D.; Li, W.; Rodrigues, A.D.; Gipson, A.E.; Holsapple, J.; Toren, P.; Parkinson, A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: Implications for drug-drug interactions. Drug Metab. Dispos. 2006, 34, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, N.; Liu, T.; Fang, Y.; Qi, B.; Wen, Q.; Zhou, J.; Jia, L.; Qiao, H. Effect of Cytochrome b5 Content on the Activity of Polymorphic CYP1A2, 2B6, and 2E1 in Human Liver Microsomes. PLoS ONE 2015, 10, e0128547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bart, A.G.; Scott, E.E. Structural and functional effects of cytochrome b(5) interactions with human cytochrome P450 enzymes. J. Biol. Chem. 2017, 292, 20818–20833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Rahman, S.M.; Gotschall, R.R.; Kauffman, R.E.; Leeder, J.S.; Kearns, G.L. Investigation of terbinafine as a CYP2D6 inhibitor in vivo. Clin. Pharmacol. Ther. 1999, 65, 465–472. [Google Scholar] [CrossRef]
- Vickers, A.E.M.; Sinclair, J.R.; Zollinger, M.; Heitz, F.; Glänzel, U.; Johanson, L.; Fischer, V. Multiple Cytochrome P-450s Involved in the Metabolism of Terbinafine Suggest a Limited Potential for Drug-Drug Interactions. Drug Metab. Dispos. 1999, 27, 1029–1038. [Google Scholar] [PubMed]
- Rasmussen, B.B.; Nielsen, T.L.; Brøsen, K. Fluvoxamine inhibits the CYP2C19-catalysed metabolism of proguanil in vitro. Eur. J. Clin. Pharmacol. 1998, 54, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.W.; Alfaro, C.L.; Ereshefsky, L.; Miller, M. Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J. Clin. Pharmacol. 2003, 43, 1274–1282. [Google Scholar] [CrossRef]
- Drolet, B.; Khalifa, M.; Daleau, P.; Hamelin, B.A.; Turgeon, J. Block of the rapid component of the delayed rectifier potassium current by the prokinetic agent cisapride underlies drug-related lengthening of the QT interval. Circulation 1998, 97, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Naritomi, Y.; Teramura, Y.; Terashita, S.; Kagayama, A. Utility of microtiter plate assays for human cytochrome P450 inhibition studies in drug discovery: Application of simple method for detecting quasi-irreversible and irreversible inhibitors. Drug Metab. Pharmacokinet. 2004, 19, 55–61. [Google Scholar] [CrossRef]
- Uttamsingh, V.; Gallegos, R.; Liu, J.F.; Harbeson, S.L.; Bridson, G.W.; Cheng, C.; Wells, D.S.; Graham, P.B.; Zelle, R.; Tung, R. Altering Metabolic Profiles of Drugs by Precision Deuteration: Reducing Mechanism-Based Inhibition of CYP2D6 by Paroxetine. J. Pharmacol. Exp. Ther. 2015, 354, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, K.M.; Venkatakrishnan, K.; Von Moltke, L.L.; Obach, R.S.; Greenblatt, D.J. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: Comparison with fluoxetine and quinidine. Drug Metab. Dispos. 2003, 31, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouladjian, L.; Chen, T.F.; Gnjidic, D.; Hilmer, S.N. Education and Assessment of Pharmacists on the Use of the Drug Burden Index in Older Adults Using a Continuing Professional Development Education Method. Am. J. Pharm. Educ. 2016, 80, 63. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, B.W. An In Vitro Investigation into the Mechanism of the Clinically Relevant Drug-Drug Interaction between Omeprazole or Esomeprazole and Clopidogrel. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 23 April 2015. [Google Scholar]
- Ray, W.A.; Murray, K.T.; Griffin, M.R.; Chung, C.P.; Smalley, W.E.; Hall, K.; Daugherty, J.R.; Kaltenbach, L.A.; Stein, C.M. Outcomes with concurrent use of clopidogrel and proton-pump inhibitors: A cohort study. Ann. Int. Med. 2010, 152, 337–345. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Cryer, B.L.; Contant, C.F.; Cohen, M.; Lanas, A.; Schnitzer, T.J.; Shook, T.L.; Lapuerta, P.; Goldsmith, M.A.; Laine, L.; et al. Clopidogrel with or without omeprazole in coronary artery disease. N. Engl. J. Med. 2010, 363, 1909–1917. [Google Scholar] [CrossRef] [Green Version]
- Sidney, M.; Wolfe, M.D. Proton Pump Inhibitors: Dangerous and Habit-Forming Heartburn Drugs; Citizen, P., Ed.; The Citizen: Washington, DC, USA, 2011; Volume 27, p. 9. [Google Scholar]
- Karaźniewicz-Łada, M.; Danielak, D.; Burchardt, P.; Kruszyna, L.; Komosa, A.; Lesiak, M.; Główka, F. Clinical pharmacokinetics of clopidogrel and its metabolites in patients with cardiovascular diseases. Clin. Pharmacokinet. 2014, 53, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Zvyaga, T.; Chang, S.-Y.; Chen, C.; Yang, Z.; Vuppugalla, R.; Hurley, J.; Thorndike, D.; Wagner, A.; Chimalakonda, A.; Rodrigues, A.D. Evaluation of Six Proton Pump Inhibitors As Inhibitors of Various Human Cytochromes P450: Focus on Cytochrome P450 2C19. Drug Metab. Dispos. 2012, 40, 1698–1711. [Google Scholar] [CrossRef] [Green Version]
- Angiolillo, D.J.; Gibson, C.M.; Cheng, S.; Ollier, C.; Nicolas, O.; Bergougnan, L.; Perrin, L.; LaCreta, F.P.; Hurbin, F.; Dubar, M. Differential Effects of Omeprazole and Pantoprazole on the Pharmacodynamics and Pharmacokinetics of Clopidogrel in Healthy Subjects: Randomized, Placebo-Controlled, Crossover Comparison Studies. Clin. Pharmacol. Ther. 2011, 89, 65–74. [Google Scholar] [CrossRef]
- Furtado, R.H.M.; Giugliano, R.P.; Strunz, C.M.C.; Filho, C.C.; Ramires, J.A.F.; Filho, R.K.; Neto, P.A.L.; Pereira, A.C.; Rocha, T.R.; Freire, B.T.; et al. Drug Interaction Between Clopidogrel and Ranitidine or Omeprazole in Stable Coronary Artery Disease: A Double-Blind, Double Dummy, Randomized Study. Am. J. Cardiovasc. Drugs 2016, 16, 275–284. [Google Scholar] [CrossRef]
- Gilard, M.; Arnaud, B.; Cornily, J.-C.; Le Gal, G.; Lacut, K.; Le Calvez, G.; Mansourati, J.; Mottier, D.; Abgrall, J.-F.; Boschat, J. Influence of Omeprazole on the Antiplatelet Action of Clopidogrel Associated With Aspirin: The Randomized, Double-Blind OCLA (Omeprazole CLopidogrel Aspirin) Study. J. Am. Coll. Cardiol. 2008, 51, 256–260. [Google Scholar] [CrossRef]
- Frelinger, A.L., III; Lee, R.D.; Mulford, D.J.; Wu, J.; Nudurupati, S.; Nigam, A.; Brooks, J.K.; Bhatt, D.L.; Michelson, A.D. A randomized, 2-period, crossover design study to assess the effects of dexlansoprazole, lansoprazole, esomeprazole, and omeprazole on the steady-state pharmacokinetics and pharmacodynamics of clopidogrel in healthy volunteers. J. Am. Coll. Cardiol. 2012, 59, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. Information for Healthcare Professionals: Update to the Labeling of Clopidogrel Bisulfate (Marketed as Plavix) to Alert Healthcare Professionals about a Drug Interaction with Omeprazole (Marketed as Prilosec and Prilosec OTC). Available online: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm190787 (accessed on 25 July 2020).
- Bundhun, P.K.; Teeluck, A.R.; Bhurtu, A.; Huang, W.-Q. Is the concomitant use of clopidogrel and Proton Pump Inhibitors still associated with increased adverse cardiovascular outcomes following coronary angioplasty? A systematic review and meta-analysis of recently published studies (2012–2016). BMC Cardiovasc. Disord. 2017, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhurke, S.M.; Martin, B.C.; Li, C.; Franks, A.M.; Bursac, Z.; Said, Q. Effect of the clopidogrel-proton pump inhibitor drug interaction on adverse cardiovascular events in patients with acute coronary syndrome. Pharmacotherapy 2012, 32, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahabaleshwarkar, R.K.; Yang, Y.; Datar, M.V.; Bentley, J.P.; Strum, M.W.; Banahan, B.F.; Null, K.D. Risk of adverse cardiovascular outcomes and all-cause mortality associated with concomitant use of clopidogrel and proton pump inhibitors in elderly patients. Curr. Med. Res. Opin. 2013, 29, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.M.; Maddox, T.M.; Wang, L.; Fihn, S.D.; Jesse, R.L.; Peterson, E.D.; Rumsfeld, J.S. Risk of Adverse Outcomes Associated With Concomitant Use of Clopidogrel and Proton Pump Inhibitors Following Acute Coronary Syndrome. JAMA 2009, 301, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.F.; Shen, L.J.; Wu, F.L.; Bai, C.H.; Gau, C.S. Cardiovascular outcomes associated with concomitant use of clopidogrel and proton pump inhibitors in patients with acute coronary syndrome in Taiwan. Br. J. Clin. Pharmacol. 2012, 74, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaduganathan, M.; Cannon, C.P.; Cryer, B.L.; Liu, Y.; Hsieh, W.H.; Doros, G.; Cohen, M.; Lanas, A.; Schnitzer, T.J.; Shook, T.L.; et al. Efficacy and Safety of Proton-Pump Inhibitors in High-Risk Cardiovascular Subsets of the COGENT Trial. Am. J. Med. 2016, 129, 1002–1005. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, M.W.; Melloni, C.; Jones, W.S.; Washam, J.B.; Hasselblad, V.; Dolor, R.J. Individual Proton Pump Inhibitors and Outcomes in Patients with Coronary Artery Disease on Dual Antiplatelet Therapy: A Systematic Review. J. Am. Heart Assoc. 2015, 4, e002245. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Tsukahara, K.; Morita, S.; Endo, T.; Sugano, T.; Hibi, K.; Himeno, H.; Fukui, K.; Umemura, S.; Kimura, K. Influence of omeprazole and famotidine on the antiplatelet effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes: A prospective, randomized, multicenter study. Circ. J. 2012, 76, 2673–2680. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.C.; Gurbel, P.A. The drug–drug interaction between proton pump inhibitors and clopidogrel. Can. Med. Assoc. J. 2009, 180, 699–700. [Google Scholar] [CrossRef] [Green Version]
- Juurlink, D.N.; Gomes, T.; Ko, D.T.; Szmitko, P.E.; Austin, P.C.; Tu, J.V.; Henry, D.A.; Kopp, A.; Mamdani, M.M. A population-based study of the drug interaction between proton pump inhibitors and clopidogrel. Can. Med. Assoc. J. 2009, 180, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.R.; Wang, D.Q.; Du, J.; Qu, G.S.; Du, J.L.; Deng, S.B.; Liu, Y.J.; Cai, J.X.; She, Q. Efficacy of Clopidogrel and Clinical Outcome When Clopidogrel Is Coadministered With Atorvastatin and Lansoprazole: A Prospective, Randomized, Controlled Trial. Medicine 2015, 94, e2262. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Shen, K.; Hu, P. Single- and multiple-dose pharmacokinetics and tolerability of a paroxetine controlled-release tablet in healthy Chinese subjects. Int. J. Clin. Pharmacol. Ther. 2017, 55, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Laine, K.; Tybring, G.; Härtter, S.; Andersson, R.N.K.; Svensson, J.-O.; Widén, J.; Bertilsson, L. Inhibition of cytochrome P4502D6 activity with paroxetine normalizes the ultrarapid metabolizer phenotype as measured by nortriptyline pharmacokinetics and the debrisoquin test. Clin. Pharmacol. Ther. 2001, 70, 327–335. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Paxil [Drug Label]. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020031s060,020936s037,020710s024lbl.pdf (accessed on 31 July 2020).
- Bartkowski, R.R.; Goldberg, M.E.; Larijani, G.E.; Boerner, T. Inhibition of alfentanil metabolism by erythromycin. Clin. Pharmacol. Ther. 1989, 46, 99–102. [Google Scholar] [CrossRef]
- Bartkowski, R.R.; McDonnell, T.E. Prolonged Alfentanil Effect Following Erythromycin Administration. Anesthesiology 1990, 73, 566–567. [Google Scholar] [CrossRef]
- Yate, P.M.; Short, S.M.; Sebel, P.S.; Thomas, D.; Morton, J. Comparison of infusions of alfentanil or pethidine for sedation of ventilated patients on THE ITU. Br. J. Anaesth. 1986, 58, 1091–1099. [Google Scholar] [CrossRef]
- Okudaira, T.; Kotegawa, T.; Imai, H.; Tsutsumi, K.; Nakano, S.; Ohashi, K. Effect of the Treatment Period With Erythromycin on Cytochrome P450 3A Activity in Humans. J. Clin. Pharmacol. 2007, 47, 871–876. [Google Scholar] [CrossRef]
- Patroneva, A.; Connolly, S.M.; Fatato, P.; Pedersen, R.; Jiang, Q.; Paul, J.; Guico-Pabia, C.; Isler, J.A.; Burczynski, M.E.; Nichols, A.I. An assessment of drug-drug interactions: The effect of desvenlafaxine and duloxetine on the pharmacokinetics of the CYP2D6 probe desipramine in healthy subjects. Drug Metab. Dispos. 2008, 36, 2484–2491. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Consultation for NDA 202611. In Clinical Pharmacology and Biopharmaceutics Review(s), Center for Drug Evaluation and Research Division of Cardiovascular and Renal Products; U.S. Food and Drug Administration: Pharr, TX, USA, 2012; p. 218. [Google Scholar]
- Krauwinkel, W.; Dickinson, J.; Schaddelee, M.; Meijer, J.; Tretter, R.; van de Wetering, J.; Strabach, G.; van Gelderen, M. The effect of mirabegron, a potent and selective β3-adrenoceptor agonist, on the pharmacokinetics of CYP2D6 substrates desipramine and metoprolol. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 43–52. [Google Scholar] [CrossRef]
- Bramer, S.L.; Suri, A. Inhibition of CYP2D6 by Quinidine and its Effects on the Metabolism of Cilostazol. Clin. Pharmacokinet. 1999, 37, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Storelli, F.; Matthey, A.; Lenglet, S.; Thomas, A.; Desmeules, J.; Daali, Y. Impact of CYP2D6 Functional Allelic Variations on Phenoconversion and Drug-Drug Interactions. Clin. Pharmacol. Ther. 2018, 104, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Storelli, F.; Desmeules, J.; Daali, Y. Genotype-sensitive reversible and time-dependent CYP2D6 inhibition in human liver microsomes. Basic Clin. Pharmacol. Toxicol. 2019, 124, 170–180. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Inhibitor Type | ||
|---|---|---|---|
| Competitive | Non-Competitive Non-Mechanism Based | Mechanism-Based | |
| Metabolism required | No | No | Yes |
| Active site mediated | Yes | No | Yes |
| Time dependent | No | No | Yes |
| Substrate concentration dependent | Yes | No | Yes |
| Km (victim drug) | ↑ | ↔ | ↔ |
| Vmax (victim drug) | ↔ | ↓ | ↓ |
| CLint (victim drug) | ↓ | ↓ | ↓ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics 2020, 12, 846. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090846
Deodhar M, Al Rihani SB, Arwood MJ, Darakjian L, Dow P, Turgeon J, Michaud V. Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice. Pharmaceutics. 2020; 12(9):846. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090846
Chicago/Turabian StyleDeodhar, Malavika, Sweilem B Al Rihani, Meghan J. Arwood, Lucy Darakjian, Pamela Dow, Jacques Turgeon, and Veronique Michaud. 2020. "Mechanisms of CYP450 Inhibition: Understanding Drug-Drug Interactions Due to Mechanism-Based Inhibition in Clinical Practice" Pharmaceutics 12, no. 9: 846. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090846