
Enhanced Supersaturation via Fusion-Assisted Amorphization during FDM 3D Printing of Crystalline Poorly Soluble Drug Loaded Filaments


Abstract
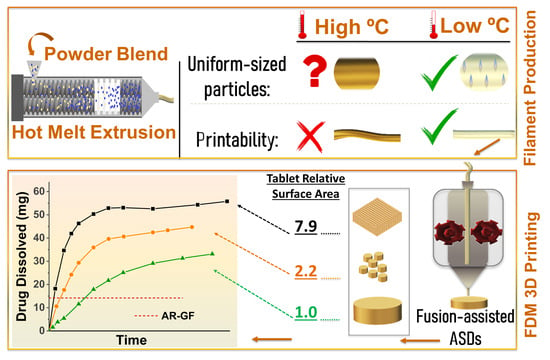
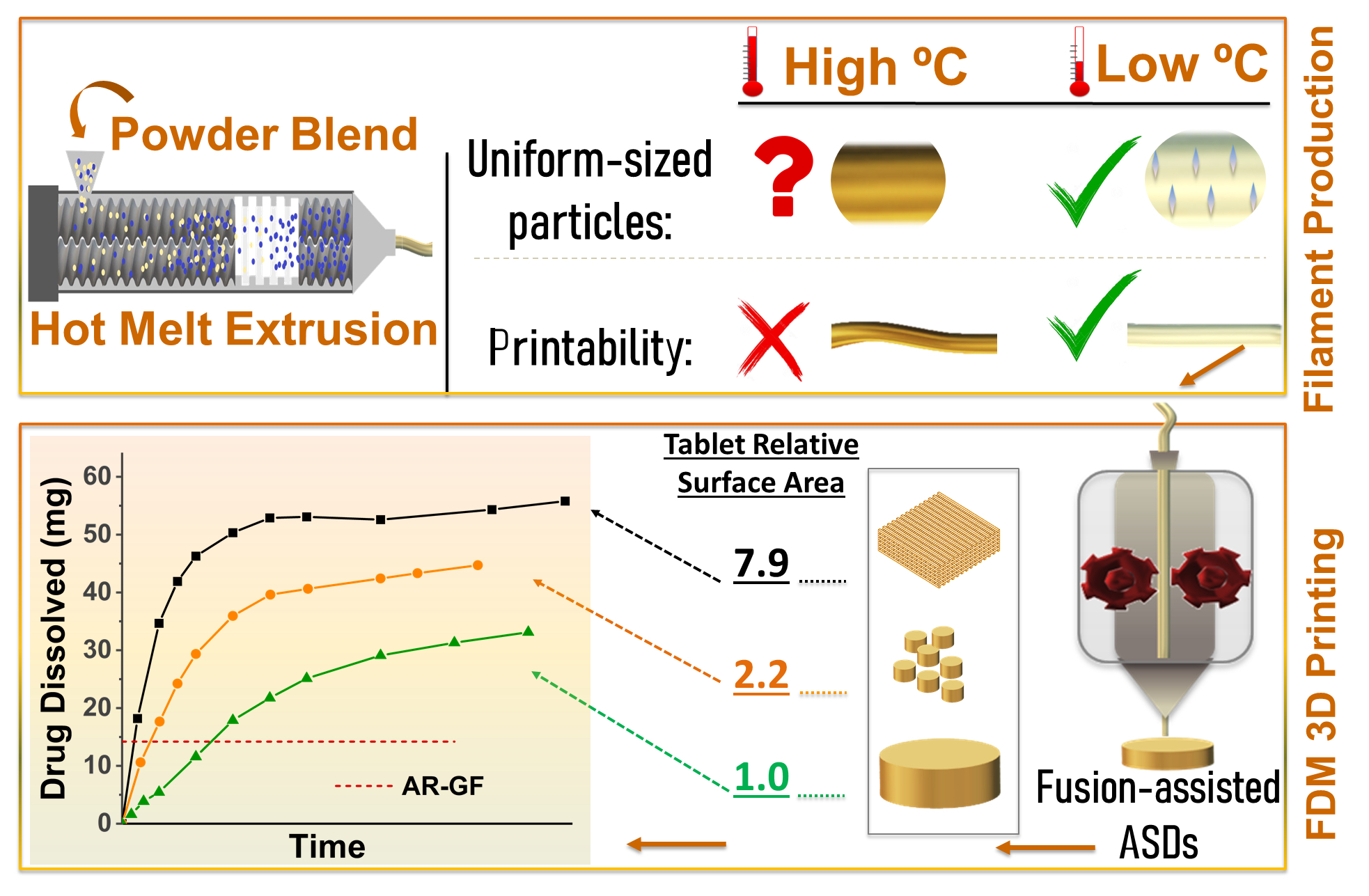
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Feed Materials (Filaments)
2.3. Mechanical Properties of Filaments
2.4. Printing Dosage Forms
2.5. Fourier Transform Infrared (FT-IR) Spectroscopy
2.6. Solid State Characterization
2.7. Morphology
2.8. Thermal Analysis
2.9. Determination of Drug Concentration
2.10. In-Vitro Dissolution, Supersaturation and Release Kinetics of GF Tablets
2.11. Stability Testing
3. Results
3.1. Fabrication of Filaments
3.2. Filaments Quality
3.3. FT-IR Spectroscopy
3.4. Crystallinity
3.5. SEM Images
3.6. Thermal Stability
3.7. Content Uniformity of Printed Tablets
3.8. Drug Supersaturation and Dissolution Kinetics
3.8.1. Effect of Printing Temperature
3.8.2. Effects of Tablet Designs and Agitator Speed
3.8.3. Release Kinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gala, U.H.; Miller, D.A.; Williams III, R.O. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2020, 1873, 188319. [Google Scholar] [CrossRef]
- Lipinski, C. Poor aqueous solubility—an industry wide problem in drug discovery. Am. Pharm. Rev. 2002, 5, 82–85. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Rumondor, A.C.; Ivanisevic, I.; Bates, S.; Alonzo, D.E.; Taylor, L.S. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm. Res. 2009, 26, 2523–2534. [Google Scholar] [CrossRef]
- Moseson, D.E.; Parker, A.S.; Beaudoin, S.P.; Taylor, L.S. Amorphous solid dispersions containing residual crystallinity: Influence of seed properties and polymer adsorption on dissolution performance. Eur. J. Pharm. Sci. 2020, 105276. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Lakshman, J.P.; Cao, Y.; Kowalski, J.; Serajuddin, A.T. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Mol. Pharm. 2008, 5, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Ju, T.-c.R.; Lee, P.I. Enhanced kinetic solubility profiles of indomethacin amorphous solid dispersions in poly (2-hydroxyethyl methacrylate) hydrogels. Eur. J. Pharm. Biopharm. 2012, 81, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Knipp, G.; Zografi, G. Assessing the performance of amorphous solid dispersions. J. Pharm. Sci. 2012, 101, 1355–1377. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Paradkar, A.; Ambike, A.A.; Jadhav, B.K.; Mahadik, K. Characterization of curcumin–PVP solid dispersion obtained by spray drying. Int. J. Pharm. 2004, 271, 281–286. [Google Scholar] [CrossRef]
- Mendonsa, N.; Almutairy, B.; Kallakunta, V.R.; Sarabu, S.; Thipsay, P.; Bandari, S.; Repka, M.A. Manufacturing strategies to develop amorphous solid dispersions: An overview. J. Drug Deliv. Sci. Technol. 2020, 55, 101459. [Google Scholar] [CrossRef]
- Sarode, A.L.; Sandhu, H.; Shah, N.; Malick, W.; Zia, H. Hot melt extrusion for amorphous solid dispersions: Temperature and moisture activated drug–polymer interactions for enhanced stability. Mol. Pharm. 2013, 10, 3665–3675. [Google Scholar] [CrossRef]
- Solanki, N.; Gupta, S.S.; Serajuddin, A.T. Rheological analysis of itraconazole-polymer mixtures to determine optimal melt extrusion temperature for development of amorphous solid dispersion. Eur. J. Pharm. Sci. 2018, 111, 482–491. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniruzzaman, M. Pharmaceutical applications of hot-melt extrusion: Continuous manufacturing, twin-screw granulations, and 3D printing. Pharmaceutics 2019, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Solanki, N.G.; Vasoya, J.M.; Shah, A.V.; Serajuddin, A.T. Development of 3D Printed Tablets by Fused Deposition Modeling Using Polyvinyl Alcohol as Polymeric Matrix for Rapid Drug Release. J. Pharm. Sci. 2020, 109, 1558–1572. [Google Scholar] [CrossRef] [Green Version]
- Arafat, B.; Qinna, N.; Cieszynska, M.; Forbes, R.T.; Alhnan, M.A. Tailored on demand anti-coagulant dosing: An in vitro and in vivo evaluation of 3D printed purpose-designed oral dosage forms. Eur. J. Pharm. Biopharm. 2018, 128, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.G.; Tahsin, M.; Shah, A.V.; Serajuddin, A.T. Formulation of 3D printed tablet for rapid drug release by fused deposition modeling: Screening polymers for drug release, drug-polymer miscibility and printability. J. Pharm. Sci. 2018, 107, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.V.; Repka, M.A. Coupling 3D printing with hot-melt extrusion to produce controlled-release tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef]
- Chai, X.; Chai, H.; Wang, X.; Yang, J.; Li, J.; Zhao, Y.; Cai, W.; Tao, T.; Xiang, X. Fused deposition modeling (FDM) 3D printed tablets for intragastric floating delivery of domperidone. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilyés, K.; Balogh, A.; Casian, T.; Igricz, T.; Borbás, E.; Démuth, B.; Vass, P.; Menyhárt, L.; Kovács, N.K.; Marosi, G. 3D floating tablets: Appropriate 3D design from the perspective of different in vitro dissolution testing methodologies. Int. J. Pharm. 2019, 567, 118433. [Google Scholar] [CrossRef] [PubMed]
- Gioumouxouzis, C.I.; Baklavaridis, A.; Katsamenis, O.L.; Markopoulou, C.K.; Bouropoulos, N.; Tzetzis, D.; Fatouros, D.G. A 3D printed bilayer oral solid dosage form combining metformin for prolonged and glimepiride for immediate drug delivery. Eur. J. Pharm. Sci. 2018, 120, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamróz, W.; Kurek, M.; Łyszczarz, E.; Szafraniec, J.; Knapik-Kowalczuk, J.; Syrek, K.; Paluch, M.; Jachowicz, R. 3D printed orodispersible films with Aripiprazole. Int. J. Pharm. 2017, 533, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Palekar, S.; Nukala, P.K.; Mishra, S.M.; Kipping, T.; Patel, K. Application of 3D printing technology and quality by design approach for development of age-appropriate pediatric formulation of baclofen. Int. J. Pharm. 2019, 556, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Scoutaris, N.; Ross, S.A.; Douroumis, D. 3D printed “Starmix” drug loaded dosage forms for paediatric applications. Pharm. Res. 2018, 35, 34. [Google Scholar] [CrossRef] [PubMed]
- Kempin, W.; Domsta, V.; Brecht, I.; Semmling, B.; Tillmann, S.; Weitschies, W.; Seidlitz, A. Development of a dual extrusion printing technique for an acid-and thermo-labile drug. Eur. J. Pharm. Sci. 2018, 123, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, W.; Vo, A.Q.; Feng, X.; Ye, X.; Kim, D.W.; Repka, M.A. Hydroxypropyl methylcellulose-based controlled release dosage by melt extrusion and 3D printing: Structure and drug release correlation. Carbohydr. Polym. 2017, 177, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Govender, R.; Abrahmsén-Alami, S.; Folestad, S.; Larsson, A. High Content Solid Dispersions for Dose Window Extension: A Basis for Design Flexibility in Fused Deposition Modelling. Pharm. Res. 2020, 37, 9. [Google Scholar] [CrossRef] [Green Version]
- Jamróz, W.; Kurek, M.; Czech, A.; Szafraniec, J.; Gawlak, K.; Jachowicz, R. 3D printing of tablets containing amorphous aripiprazole by filaments co-extrusion. Eur. J. Pharm. Biopharm. 2018, 131, 44–47. [Google Scholar] [CrossRef]
- Nasereddin, J.M.; Wellner, N.; Alhijjaj, M.; Belton, P.; Qi, S. Development of a simple mechanical screening method for predicting the feedability of a pharmaceutical FDM 3D printing filament. Pharm. Res. 2018, 35, 151. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chen, Y.; Wei, Y.; Li, Y. 3D printing of shape memory polymer for functional part fabrication. Int. J. Adv. Manuf. Technol. 2016, 84, 2079–2095. [Google Scholar] [CrossRef]
- Isreb, A.; Baj, K.; Wojsz, M.; Isreb, M.; Peak, M.; Alhnan, M.A. 3D printed oral theophylline doses with innovative ‘radiator-like’design: Impact of polyethylene oxide (PEO) molecular weight. Int. J. Pharm. 2019, 564, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kempin, W.; Franz, C.; Koster, L.-C.; Schneider, F.; Bogdahn, M.; Weitschies, W.; Seidlitz, A. Assessment of different polymers and drug loads for fused deposition modeling of drug loaded implants. Eur. J. Pharm. Biopharm. 2017, 115, 84–93. [Google Scholar] [CrossRef]
- Solanki, N.G.; Lam, K.; Tahsin, M.; Gumaste, S.G.; Shah, A.V.; Serajuddin, A.T. Effects of surfactants on itraconazole-HPMCAS solid dispersion prepared by hot-melt extrusion I: Miscibility and drug release. J. Pharm. Sci. 2019, 108, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Sarode, A.; Wang, P.; Cote, C.; Worthen, D.R. Low-viscosity hydroxypropylcellulose (HPC) grades SL and SSL: Versatile pharmaceutical polymers for dissolution enhancement, controlled release, and pharmaceutical processing. Aaps Pharmscitech 2013, 14, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aho, J.; Van Renterghem, J.; Arnfast, L.; De Beer, T.; Rantanen, J. The flow properties and presence of crystals in drug-polymer mixtures: Rheological investigation combined with light microscopy. Int. J. Pharm. 2017, 528, 383–394. [Google Scholar] [CrossRef]
- Censi, R.; Gigliobianco, M.R.; Casadidio, C.; Di Martino, P. Hot melt extrusion: Highlighting physicochemical factors to be investigated while designing and optimizing a hot melt extrusion process. Pharmaceutics 2018, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Uekama, K.; Ikegami, K.; Wang, Z.; Horiuchi, Y.; Hirayama, F. Inhibitory effect of 2-hydroxypropyl-β-cyclodextrin on crystal-growth of nifedipine during storage: Superior dissolution and oral bioavailability compared with polyvinylpyrrolidone K-30. J. Pharm. Pharmacol. 1992, 44, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef]
- Buyukgoz, G.G.; Soffer, D.; Defendre, J.; Pizzano, G.M.; Davé, R.N. Exploring Tablet Design Options for Tailoring Drug Release and Dose via Fused Deposition Modeling (FDM) 3D Printing. Int. J. Pharm. 2020, 591, 119987. [Google Scholar] [CrossRef] [PubMed]
- Cetindag, E.; Pentangelo, J.; Cespedes, T.A.; Davé, R.N. Effect of solvents and cellulosic polymers on quality attributes of films loaded with a poorly water-soluble drug. Carbohydr. Polym. 2020, 250, 117012. [Google Scholar] [CrossRef]
- Griffin, S.R.; Takanti, N.; Sarkar, S.; Song, Z.; Vogt, A.D.; Danzer, G.D.; Simpson, G.J. Disparities of Single-Particle Growth Rates in Buried Versus Exposed Ritonavir Crystals within Amorphous Solid Dispersions. Mol. Pharm. 2020, 17, 4564–4571. [Google Scholar] [CrossRef] [PubMed]
- Brenken, B.; Favaloro, A.; Barocio, E.; DeNardo, N.M.; Pipes, R.B. Development of a model to predict temperature history and crystallization behavior of 3D printed parts made from fiber-reinforced thermoplastic polymers. In Proceedings of the SAMPE Conference Proceeding, Long Beach, CA, USA, 23–26 May 2016. [Google Scholar]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef] [Green Version]
- Goyanes, A.; Martinez, P.R.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Sadia, M.; Arafat, B.; Ahmed, W.; Forbes, R.T.; Alhnan, M.A. Channelled tablets: An innovative approach to accelerating drug release from 3D printed tablets. J. Control. Release 2018, 269, 355–363. [Google Scholar] [CrossRef]
- Rahman, M.; Coelho, A.; Tarabokija, J.; Ahmad, S.; Radgman, K.; Bilgili, E. Synergistic and Antagonistic Effects of Various Amphiphilic Polymer Combinations in Enhancing Griseofulvin Release from Ternary Amorphous Solid Dispersions. Eur. J. Pharm. Sci. 2020, 150, 105354. [Google Scholar] [CrossRef] [PubMed]
- Öblom, H.; Zhang, J.; Pimparade, M.; Speer, I.; Preis, M.; Repka, M.; Sandler, N. 3D-printed isoniazid tablets for the treatment and prevention of tuberculosis—Personalized dosing and drug release. AAPS PharmSciTech 2019, 20, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzak, K.; Isreb, A.; Alhnan, M.A. A flexible-dose dispenser for immediate and extended release 3D printed tablets. Eur. J. Pharm. Biopharm. 2015, 96, 380–387. [Google Scholar] [CrossRef]
- Li, M.; Ioannidis, N.; Gogos, C.; Bilgili, E. A comparative assessment of nanocomposites vs. amorphous solid dispersions prepared via nanoextrusion for drug dissolution enhancement. Eur. J. Pharm. Biopharm. 2017, 119, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Bühler, V. Kollicoat Grades: Functional Polymers for the Pharmaceutical Industry; BASF: Ludwigshafen, Germany, 2007. [Google Scholar]
- Callister, W.D. Materials Science and Engineering: An Introduction, 7th ed.; John Willey & Sons, Inc: New York, NY, USA, 2007. [Google Scholar]
- Rumondor, A.C.; Marsac, P.J.; Stanford, L.A.; Taylor, L.S. Phase behavior of poly (vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol. Pharm. 2009, 6, 1492–1505. [Google Scholar] [CrossRef]
- Goyanes, A.; Allahham, N.; Trenfield, S.J.; Stoyanov, E.; Gaisford, S.; Basit, A.W. Direct powder extrusion 3D printing: Fabrication of drug products using a novel single-step process. Int. J. Pharm. 2019, 567, 118471. [Google Scholar] [CrossRef]
- Rahman, M.; Arevalo, F.; Coelho, A.; Bilgili, E. Hybrid nanocrystal–amorphous solid dispersions (HyNASDs) as alternative to ASDs for enhanced release of BCS Class II drugs. Eur. J. Pharm. Biopharm. 2019, 145, 12–26. [Google Scholar] [CrossRef]
- Purohit, H.S.; Taylor, L.S. Phase behavior of ritonavir amorphous solid dispersions during hydration and dissolution. Pharm. Res. 2017, 34, 2842–2861. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Ahmad, S.; Tarabokija, J.; Bilgili, E. Roles of surfactant and polymer in drug release from spray-dried hybrid nanocrystal-amorphous solid dispersions (HyNASDs). Powder Technol. 2020, 361, 663–678. [Google Scholar] [CrossRef]
- Bhakay, A.; Azad, M.; Bilgili, E.; Dave, R. Redispersible fast dissolving nanocomposite microparticles of poorly water-soluble drugs. Int. J. Pharm. 2014, 461, 367–379. [Google Scholar] [CrossRef]
- Thommes, M.; Ely, D.R.; Carvajal, M.T.; Pinal, R. Improvement of the dissolution rate of poorly soluble drugs by solid crystal suspensions. Mol. Pharm 2011, 8, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Nagata, N.; Hayashi, N.; Ogawa, E.; Fukushige, K.; Sakai, N.; Ozeki, T. Defined drug release from 3D-printed composite tablets consisting of drug-loaded polyvinylalcohol and a water-soluble or water-insoluble polymer filler. Int. J. Pharm. 2018, 543, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wen, H.; Jia, D.; Guan, X.; Pan, H.; Yang, Y.; Yu, S.; Zhu, Z.; Xiang, R.; Pan, W. Preparation and investigation of controlled-release glipizide novel oral device with three-dimensional printing. Int. J. Pharm. 2017, 525, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Alhijjaj, M.; Nasereddin, J.; Belton, P.; Qi, S. Impact of Processing Parameters on the Quality of Pharmaceutical Solid Dosage Forms Produced by Fused Deposition Modeling (FDM). Pharmaceutics 2019, 11, 633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, C.; Quodbach, J. Formulation development and process analysis of drug-loaded filaments manufactured via hot-melt extrusion for 3D-printing of medicines. Pharm. Dev. Technol. 2018, 23, 1117–1127. [Google Scholar] [CrossRef]
- Verstraete, G.; Samaro, A.; Grymonpré, W.; Vanhoorne, V.; Van Snick, B.; Boone, M.; Hellemans, T.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. 3D printing of high drug loaded dosage forms using thermoplastic polyurethanes. Int. J. Pharm. 2018, 536, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.C.; Keen, J.M.; Bi, Y.; Porter, S.; Dürig, T.; McGinity, J.W. Investigation of the interactions of enteric and hydrophilic polymers to enhance dissolution of griseofulvin following hot melt extrusion processing. J. Pharm. Pharmacol. 2015, 67, 918–938. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, A. Effect of different stabilizers and polymers on spherical agglomerates of gresiofulvine by emulsion solvent diffusion (ESD) system. Int. J. Pharm. Tech. Res 2009, 1, 149–150. [Google Scholar]
- Sarode, A.L.; Malekar, S.A.; Cote, C.; Worthen, D.R. Hydroxypropyl cellulose stabilizes amorphous solid dispersions of the poorly water soluble drug felodipine. Carbohydr. Polym. 2014, 112, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Sathigari, S.K.; Radhakrishnan, V.K.; Davis, V.A.; Parsons, D.L.; Babu, R.J. Amorphous-state characterization of efavirenz—polymer hot-melt extrusion systems for dissolution enhancement. J. Pharm. Sci. 2012, 101, 3456–3464. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, H.; Buckton, G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech 2009, 10, 1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żarów, A.; Zhou, B.; Wang, X.; Pinal, R.; Iqbal, Z. Spectroscopic and X-ray diffraction study of structural disorder in cryomilled and amorphous griseofulvin. Appl. Spectrosc. 2011, 65, 135–143. [Google Scholar] [CrossRef]
- Gogos, C.G.; Liu, H.; Wang, P. Laminar Dispersive and Distributive Mixing with Dissolution and Applications to Hot-Melt Extrusion. Hot Melt Extrus. Pharm. Appl. 2012, 261–284. [Google Scholar]
- Tidau, M.; Kwade, A.; Finke, J.H. Influence of High, Disperse API Load on Properties along the Fused-Layer Modeling Process Chain of Solid Dosage Forms. Pharmaceutics 2019, 11, 194. [Google Scholar] [CrossRef] [Green Version]
- Loftsson, T.; Fri, H.; Gu, T.K. The effect of water-soluble polymers on aqueous solubility of drugs. Int. J. Pharm. 1996, 127, 293–296. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.; Hatton, G.B.; Gaisford, S.; Basit, A.W. 3D printing of modified-release aminosalicylate (4-ASA and 5-ASA) tablets. Eur. J. Pharm. Biopharm. 2015, 89, 157–162. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Probing the mechanisms of drug release from amorphous solid dispersions in medium-soluble and medium-insoluble carriers. J. Control. Release 2015, 211, 85–93. [Google Scholar] [CrossRef]
- Augustijns, P.; Brewster, M.E. Supersaturating drug delivery systems: Fast is not necessarily good enough. J. Pharm. Sci. 2012, 101, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Mendyk, A.; Pacławski, A.; Szlek, J.; Jachowicz, R. PhEq_bootstrap: Open-source software for the simulation of f2 distribution in cases of large variability in dissolution profiles. Dissolution Technol. 2013, 20, 13–17. [Google Scholar] [CrossRef]
- Paixão, P.; Gouveia, L.F.; Silva, N.; Morais, J.A. Evaluation of dissolution profile similarity–Comparison between the f2, the multivariate statistical distance and the f2 bootstrapping methods. Eur. J. Pharm. Biopharm. 2017, 112, 67–74. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blend Composition | Processing Parameters in HME | Final Product | ||||
|---|---|---|---|---|---|---|
| Run | Drug (Wt%) | Polymer (s) (Wt%) | Temperature (°C) | Screw Speed (Rpm) | Feed Rate (g/min) | |
| H160 | - | Placebo | 160 | 30 | 0.7 | Filament |
| H165 | 15% GF | 75% HPC + 10% KP | 165 | 35 | 1.1 | |
| H180 | 180 | 50 | 1.8 | |||
| H190 | 190 | 60 | 2.4 | |||
| H210 | 210 | 250 | 3.0 | |||
| Run | Filament Used for Printing | FDM Printing Temperature (°C) | Final Product |
|---|---|---|---|
| F165 | H-165 | 165 | FDM 3D Printed Tablet |
| F210 | 210 | ||
| F230 | 230 | ||
| F240 | 240 | ||
| P165 | H-160 | 165 | |
| P240 | 240 |
| Run | 3D Printed Tablets | Agitator Speed | |||
|---|---|---|---|---|---|
| Design | Size 1 | Relative SA 2 | Quantity | (Rpm) | |
| F240 | Cylinder | 16 × 2 | 1.0 | 1 | 50 |
| F240a | Cylinder | 16 × 2 | 1.0 | 1 | 250 |
| F240b | Multi-mini w sinker | 3 × 2 | 2.2 | 33 | 50 |
| F240c | Structured w sinker | 16 × 16 × 4.2 | 7.9 | 1 | 50 |
| F240d | Structured w/o sinker | 16 × 16 × 4.2 | 7.9 | 1 | 50 |
| Run | Diameter (mm) | RSD | Height (mm) | RSD | Tablet Weight (mg) | RSD | DC 1 (%) | RSD |
|---|---|---|---|---|---|---|---|---|
| F165 | 5.57 | 1.11 | 1.29 | 3.31 | 26.46 | 1.33 | 15.79 | 0.33 |
| F210 | 5.68 | 1.07 | 1.21 | 2.66 | 30.33 | 3.43 | 15.98 | 0.32 |
| F230 | 5.47 | 0.95 | 1.60 | 1.44 | 34.73 | 1.09 | 15.66 | 0.64 |
| F240 | 5.47 | 2.25 | 1.56 | 5.42 | 33.86 | 5.36 | 15.81 | 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buyukgoz, G.G.; Kossor, C.G.; Davé, R.N. Enhanced Supersaturation via Fusion-Assisted Amorphization during FDM 3D Printing of Crystalline Poorly Soluble Drug Loaded Filaments. Pharmaceutics 2021, 13, 1857. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111857
Buyukgoz GG, Kossor CG, Davé RN. Enhanced Supersaturation via Fusion-Assisted Amorphization during FDM 3D Printing of Crystalline Poorly Soluble Drug Loaded Filaments. Pharmaceutics. 2021; 13(11):1857. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111857
Chicago/Turabian StyleBuyukgoz, Guluzar Gorkem, Christopher Gordon Kossor, and Rajesh N. Davé. 2021. "Enhanced Supersaturation via Fusion-Assisted Amorphization during FDM 3D Printing of Crystalline Poorly Soluble Drug Loaded Filaments" Pharmaceutics 13, no. 11: 1857. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111857