
Exosome-Mediated Insulin Delivery for the Potential Treatment of Diabetes Mellitus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Culture
2.3. Exosome Isolation
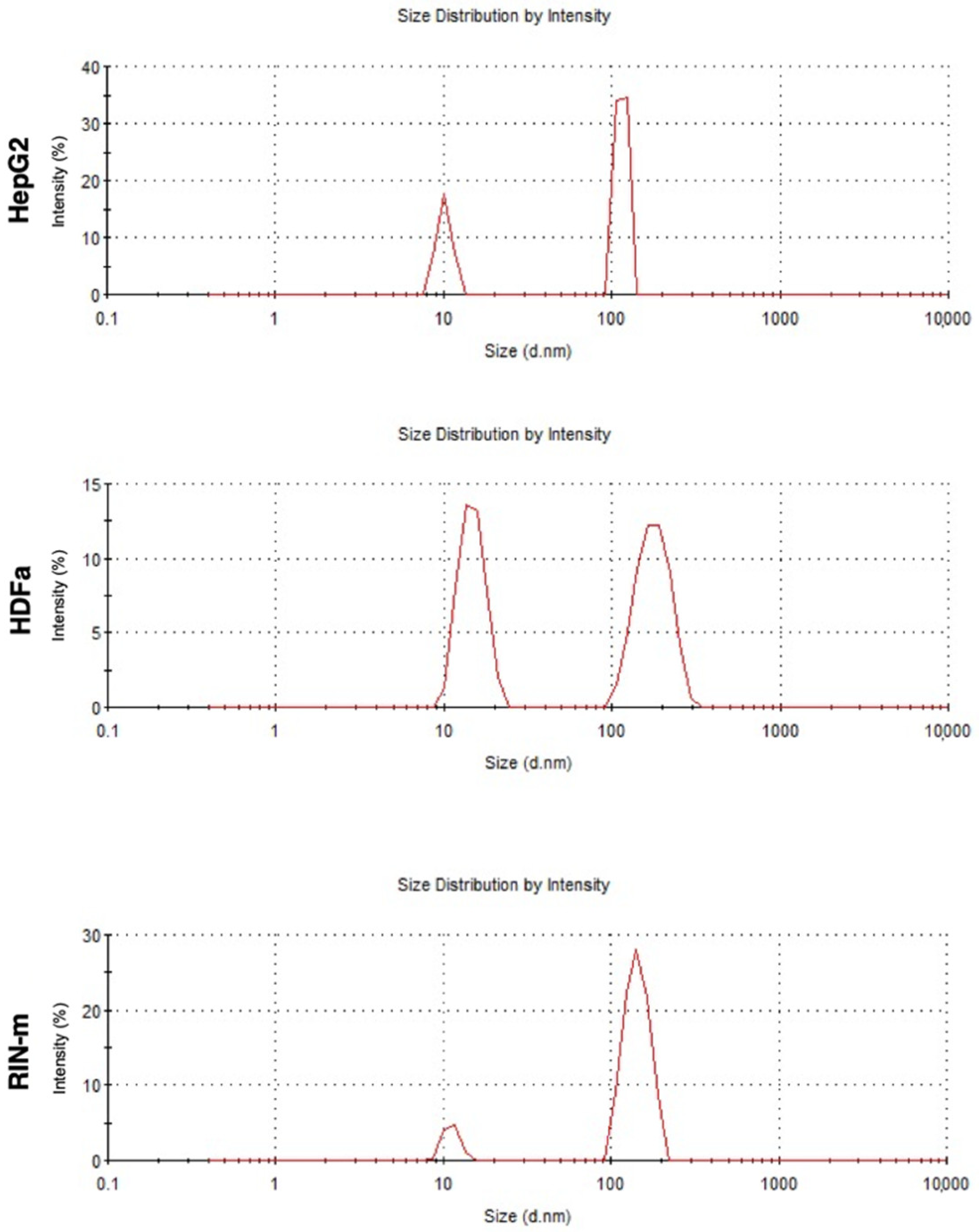
2.4. Exosome Characterization
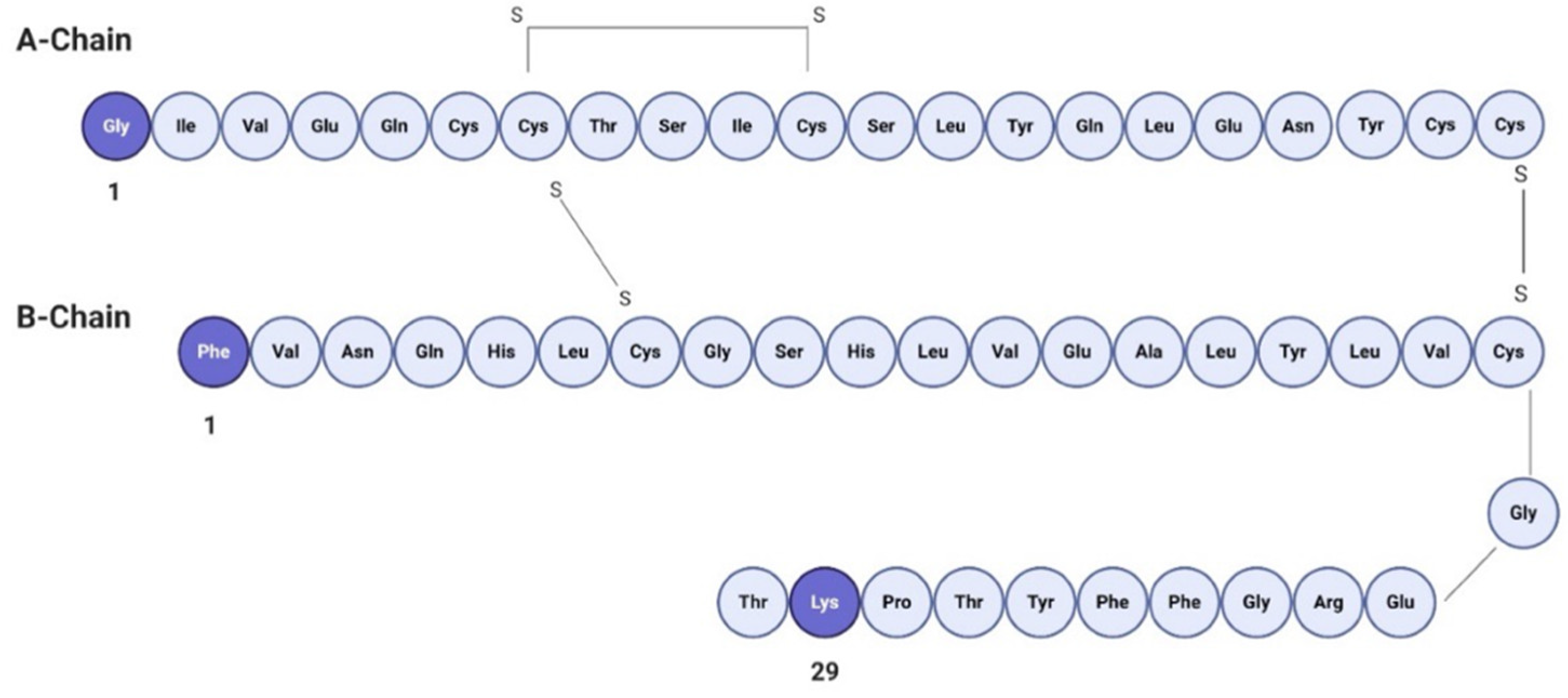
2.5. Human Insulin Labeling and Detection
2.6. FITC–Insulin Exosome Loading
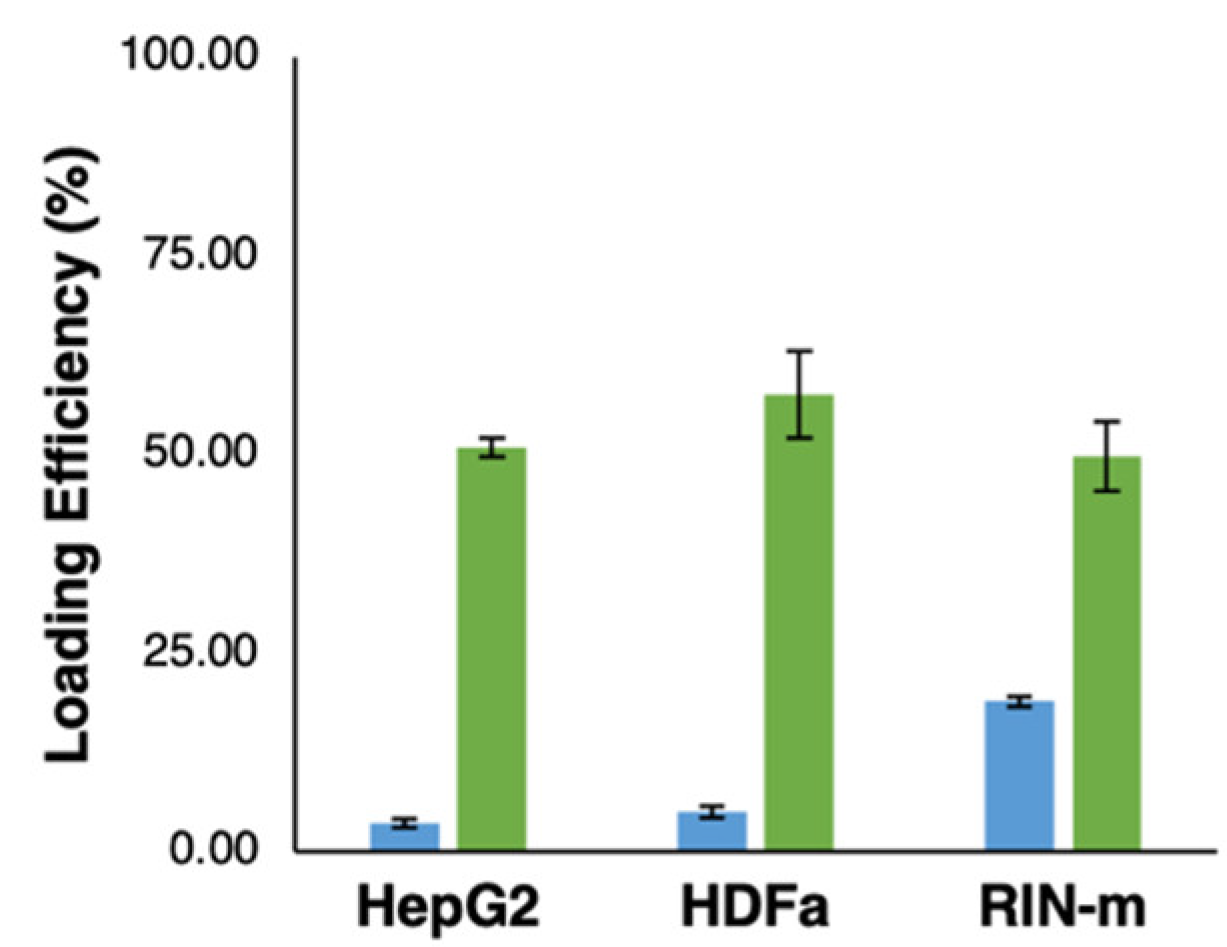
2.7. Evaluation of Exosome Loading Efficiency
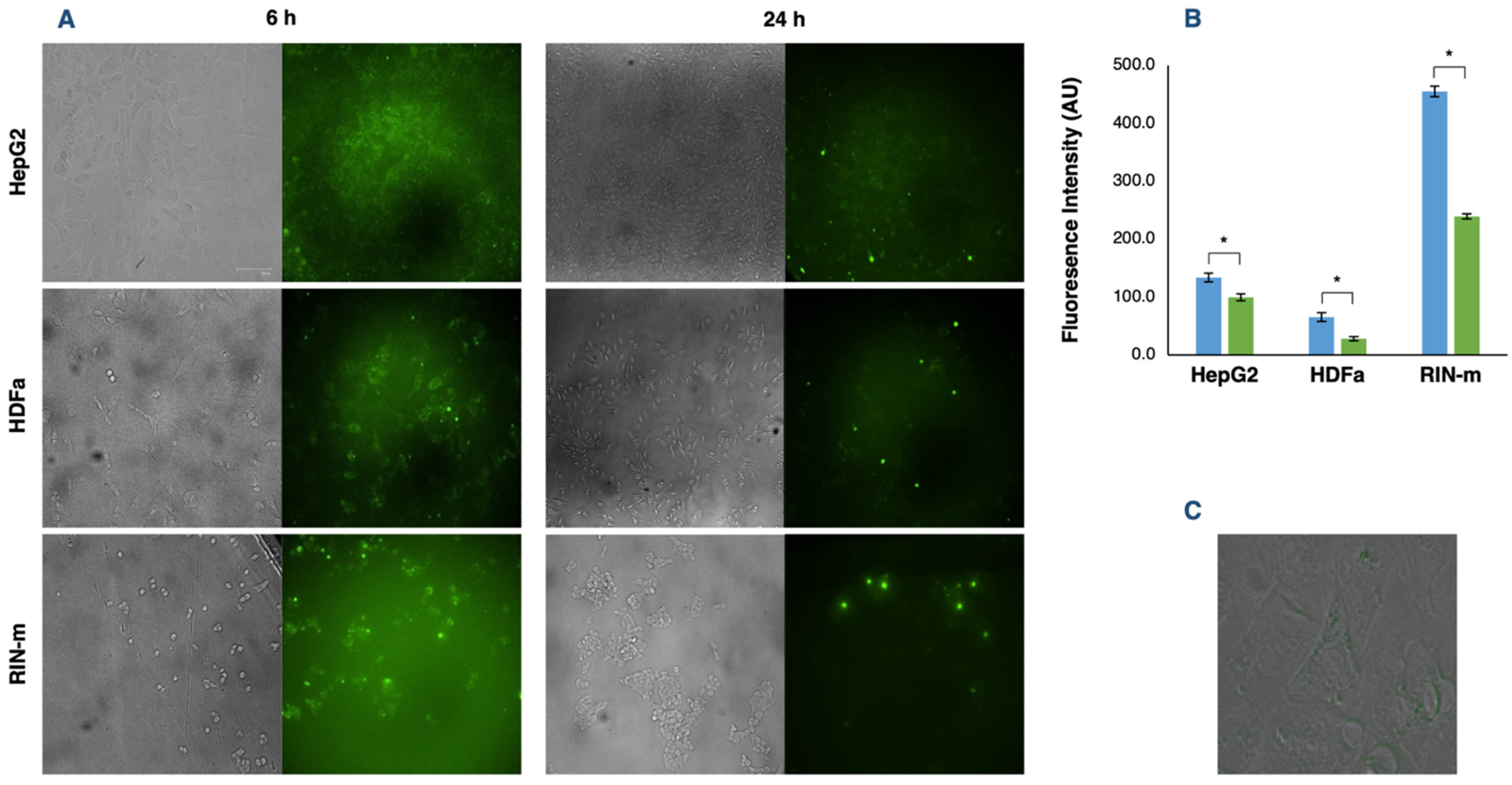
2.8. In Vitro Exosome Cellular Uptake and Evaluation of FITC–Insulin Cellular Internalization
2.9. In Vitro Evaluation of Glucose Regulation Levels of Exosome-Encapsulated Human Insulin
2.10. Data Analysis
3. Results and Discussion
3.1. Exosome Isolation and Characterization
3.2. Human Insulin Labeling and Detection
3.3. Evaluation of FITC–Insulin Loading Efficiency in Exosomes
3.4. In Vitro Evaluation of Cellular Exosome Uptake and Cellular Incorporation of FITC–Insulin
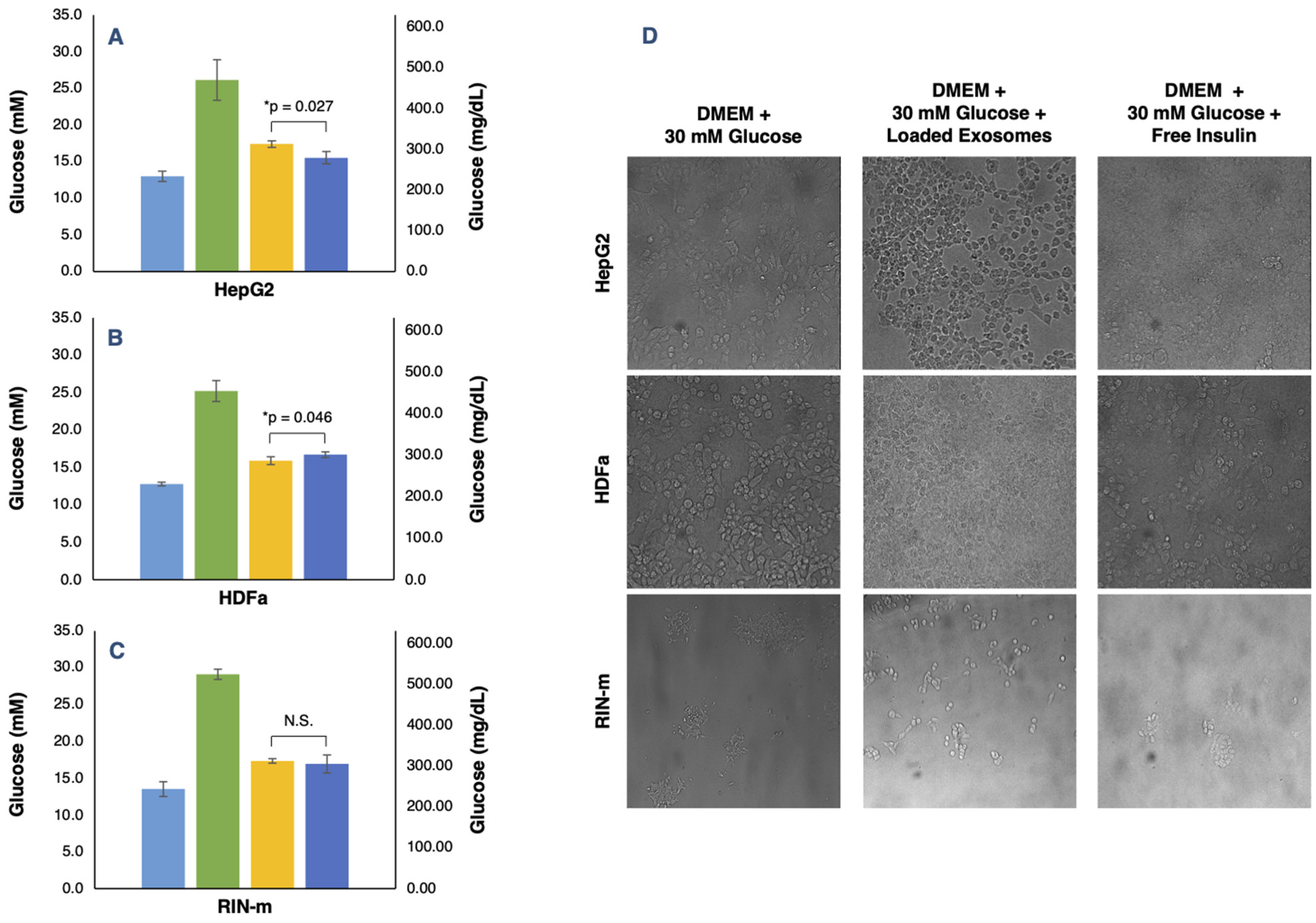
3.5. In Vitro Evaluation Glucose Regulation Levels of Exosome-Encapsulated Human Insulin
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Raposo, G. Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Lila, A.S.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemler, M.E. Tetraspanins, functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 2003, 19, 397–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Grizzle, W.E. Exosomes and cancer: A newly described pathway of immune suppression. Clin. Cancer Res. 2011, 17, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Familtseva, A.; Jeremic, N.; Tyagi, S.C. Exosomes: Cell-created drug delivery systems. Mol. Cell Biochem. 2019, 459, 1–6. [Google Scholar] [CrossRef]
- Tan, C.; Lai, R.; Wong, W.; Dan, Y.; Lim, S.-K.; Ho, H. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res. Ther. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamalden, T.A.; Macgregor-Das, A.M.; Kannan, S.M.; Dunkerly-Eyring, B.; Khaliddin, N.; Xu, Z.; Fusco, A.P.; Yazib, S.A.; Chow, R.C.; Duh, E.J.; et al. Exosomal microRNA-15a transfer from the pancreas augments diabetic complications by inducing oxidative stress. Antioxid. Redox Signal. 2017, 27, 913–930. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.W. Human insulin: Basic sciences to therapeutic uses. Drug Dev. Indus. Pharm. 1996, 22, 753–789. [Google Scholar] [CrossRef]
- Woodley, J.F. Enzymatic barriers for GI peptide and protein delivery. Crit Rev. Ther. Drug Carrier Syst. 1994, 11, 61–95. [Google Scholar] [PubMed]
- Park, S.J.; Jeong, U.H.; Lee, J.W.; Park, J.S. Preparation and characterization of bovine albumin-loaded cationic liposomes: Effect of hydration phase. J. Pharmceut. Invest. 2010, 40, 353–356. [Google Scholar]
- Aboubakar, M.; Puisieux, F.; Couvreur, P.; Deyme, D.; Vauthier, C. Study of the mechanism of insulin encapsulation in poly(isobutylcyanoacrylate) nanocapsules obtained by interfacial polymerization. J. Biomed. Mater. Res. 1999, 47, 568–576. [Google Scholar] [CrossRef]
- Whiteside, T.L. Tumour-derived exosomes or microvesicles: Another mechanism of tumour escape from the host immune system? Br. J. Cancer 2005, 92, 209–211. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Guo, Y.; Ocando, J.; Shao, J. FITC labeling of human insulin and transport of FITC-insulin conjugates through MDCK cell monolayer. J. Pharm. Anal. 2019, 9, 400–405. [Google Scholar] [CrossRef]
- Donoso-Quezada, J.; Guajardo-Flores, D.; González-Valdez, J. Enhanced exosome-mediated delivery of black bean phytochemicals (Phaseolus vulgaris L.) for cancer treatment applications. Biomed. Pharmacother. 2020, 131, 110771. [Google Scholar] [CrossRef]
- Miller, G.L. Use of dinitrosalicylicacid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Summer, J.; Graham, V. Dinitrosalicylic acid: A reagent for estimation of sugar in normal and diabetic urine. J. Biol. Chem. 1921, 47, 5–9. [Google Scholar] [CrossRef]
- Başkan, K.S.; Tütem, E.; Akyüz, E.; Özen, S.; Apak, R. Spectrophotometric total reducing sugars assay based on cupric reduction. Talanta 2016, 147, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Ludwig, A.K.; Hornung, S.; Rotan, O.; Horn, P.A.; Epple, M.; Giebel, B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces 2011, 87, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Kan, S.; Zhu, Y.; Feng, S.; Feng, W.; Gao, S. Engineered exosome-mediated delivery of functionally active miR-26a and its enhanced suppression effect in HepG2 cells. Int. J. Nanomed. 2018, 13, 585–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathappa, M.; Alder, N.N. Ionization properties of phospholipids determined by zeta potential measurements. Bio. Protoc. 2016, 6, e2030. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Ade, L.; Ntambi, J.M.; Ansari, I.-U.H.; Stoker, S.W. Characterization of Phospholipids in Insulin Secretory Granules and Mitochondria in Pancreatic Beta Cells and Their Changes with Glucose Stimulation. J. Biol. Chem. 2015, 290, 11075–11092. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.J. Zeta Potential in Colloid Science: Principles and Applications, 3rd ed.; NSW Academic Press: Sydney, Australia, 2013; pp. 59–124. [Google Scholar]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabro, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Fedreici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, 5219. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, Y.; Konishi, Y.; Kosaka, N.; Katsuda, T.; Kato, T.; Ochiya, T. Comparative marker analysis of extracellular vesicles in different human cancer types. J. Extracell. Vesicles 2013, 2, 20424. [Google Scholar] [CrossRef] [PubMed]
- Jacob, D.; Taylor, M.J.; Tomlins, P.; Sahota, T.S. Synthesis and identification of FITCinsulin conjugates produced using human insulin and insulin analogues for biomedical applications. J. Fluoresc. 2016, 26, 617–629. [Google Scholar] [CrossRef]
- Banks, P.R.; Paquette, D.M. Comparison of three common amine reactive fluorescent probes used for conjugation to biomolecules by capillary zone electrophoresis. Bioconjug. Chem. 1995, 6, 447–458. [Google Scholar] [CrossRef]
- Hentz, N.G.; Richardson, J.M.; Sportsman, J.R.; Daijo, J.; Sittampalam, G.S. Synthesis and characterization of insulin-fluorescein derivatives for bioanalytical applications. Anal. Chem. 1997, 69, 4994–5000. [Google Scholar] [CrossRef]
- Camenisch, G.; Alsenz, J.; Van de Waterbeemd, H.; Folkers, G. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs’ lipophilicity and molecular weight. Eur. J. Pharm. Sci. 1998, 6, 313–319. [Google Scholar] [CrossRef]
- Schroeder, N.E.; Macguidwin, A.E. Behavioral quiescence reduces the penetration and toxicity of exogenous compounds in second-stage juveniles of Heterodera glycines. Nematology 2010, 12, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Demetzos, C. Differential Scanning Calorimetry (DSC): A tool to study the thermal behavior of lipid bilayers and liposomal stability. J. Liposome Res. 2008, 18, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Bala, S.; Bukong, T.; Szabo, G. Exosome-mediated delivery of functionally active miRNA-155 inhibitor to macrophages. Nanomed. Nanotech. Biol. Med. 2014, 10, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Batrakova, E.V. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotech. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Murthy, S.N.; Zhao, Y.-L.; Marlan, K.; Hui, S.W.; Kazim, A.L.; Sen, A. Lipid and Electroosmosis Enhanced Transdermal Delivery of Insulin by Electroporation. J. Pharm. Sci. 2006, 95, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Satake, A.; Sumi, S.; Inoue, K.; Miyakoshi, J. An extremely low frequency magnetic field attenuates insulin secretion from the insulinoma cell line, RIN-m. Bioelectromagnetics 2004, 25, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Berthod, F.; Germain, L.; Tremblay, N.; Auger, F.A. Extracellular matrix deposition by fibroblasts is neccesarry to promote capillary-like tube formation in vitro. J. Cell Physiol. 2006, 207, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wang, Y.; Wang, H.; Zhu, Z.; Xiao, Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J. Cell Biochem. 2010, 111, 488–496. [Google Scholar] [CrossRef]
- Obregon, C.; Rothen-Rutishauser, B.; Gitahi, S.K.; Gehr, P.; Nicod, L.P. Active uptake of dendritic cell-derived exovesicles by epithelial cells induces the release of inflammatory mediators through a TNF-a-mediated pathway. Am. J. Pathol. 2009, 175, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denzer, K.; Van Eijk, M.; Kleijmeer, M.J.; Jakobson, E.; de Groot, C.; Geuze, H.J. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J. Immunol. 2000, 165, 1259–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef]
- Zöller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef]
- Stoorvogel, W.; Keijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef]
- Urban, S.; Zieseniss, S.; Werder, M.; Hauser, H.; Budzinski, R.; Engelmann, B. Scavenger receptor BI transfers major lipoprotein-associated phospholipids into the cells. J. Biol. Chem. 2000, 275, 33409–33415. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, S.; Gibertini, S.; Blasevich, F.; Bragato, C.; Ruggieri, A.; Saredi, S.; Fabbri, M.; Bernasconi, P.; Maggi, L.; Mantegazza, R.; et al. Exosomes and exosomal miRNAs from muscle-derived fibroblasts promote skeletal muscle fibrosis. Matrix Biol. 2018, 74, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Yamauchi, K.; Shigematsu, S.; Ikeo, S.; Komatsu, M.; Aizawa, T.; Hashizume, K. Selective attenuation of metabolic branch of insulin receptor down-signaling by high glucose in a hepatoma cell line, HepG2 cells. J. Biol. Chem. 2000, 275, 20880–20886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavin, J.R.; Roth, J.; Jen, P.; Freychet, P. Insulin receptors in human circulating cells and fibroblasts. Proc. Natl. Acad. Sci. USA 1972, 69, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Petruzziello, A.; Formisano, P.; Miele, C.; Di Finizio, B.; Riccardi, G.; Ferrara, A.; Beguinot, L.; Beguinot, F. Defective insulin action in fibroblasts from noninsulin-dependent diabetes mellitus patients with Gln”52 insulin receptor mutation. J. Clin. Endocrinol. Metab 1993, 77, 409–412. [Google Scholar] [PubMed]
- Rebelato, E.; Santos, L.R.; Carpinelli, A.R.; Rorsman, P.; Abdulkader, F. Short-term high glucose culture potentiates pancreatic beta cell function. Sci. Rep. 2018, 8, 13061. [Google Scholar] [CrossRef] [PubMed]
- Guerci, B.; Sauvanet, J.P. Subcutaneous insulin: Pharmacokinetic variability and glycemic variability. Diabetes Metab. 2005, 31, 4S7–4S24. [Google Scholar] [CrossRef]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Gupta, R. Exosomes for the Enhanced Tissue Bioavailability and Efficacy of Curcumin. AAPS J. 2017, 19, 1691–1702. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell-Derived Exosomes | Room Temperature (mg/mL) | Electroporation (mg/mL) |
|---|---|---|
| HepG2 | 0.07 ± 0.08 | 1.04 ± 0.99 |
| HDFa | 0.09 ± 0.05 | 1.10 ± 1.19 |
| RIN-m | 0.38 ± 0.12 | 0.91 ± 1.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Morales, B.; Antunes-Ricardo, M.; González-Valdez, J. Exosome-Mediated Insulin Delivery for the Potential Treatment of Diabetes Mellitus. Pharmaceutics 2021, 13, 1870. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111870
Rodríguez-Morales B, Antunes-Ricardo M, González-Valdez J. Exosome-Mediated Insulin Delivery for the Potential Treatment of Diabetes Mellitus. Pharmaceutics. 2021; 13(11):1870. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111870
Chicago/Turabian StyleRodríguez-Morales, Belén, Marilena Antunes-Ricardo, and José González-Valdez. 2021. "Exosome-Mediated Insulin Delivery for the Potential Treatment of Diabetes Mellitus" Pharmaceutics 13, no. 11: 1870. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13111870