
Nanotechnology-Based Drug Delivery Strategies to Repair the Mitochondrial Function in Neuroinflammatory and Neurodegenerative Diseases


Abstract
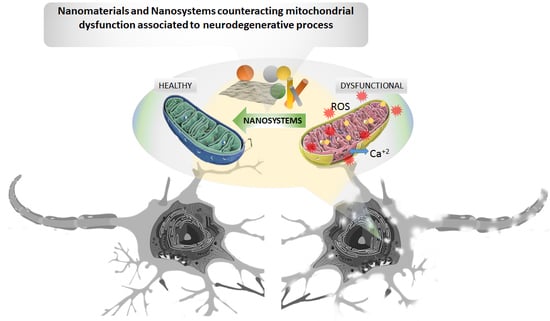
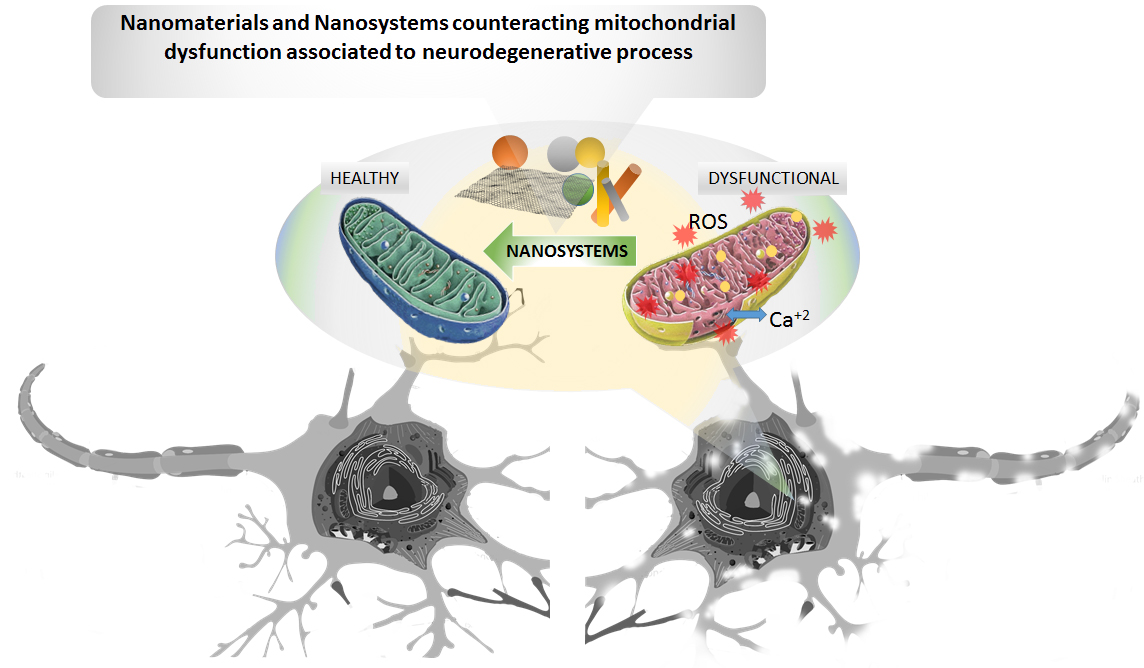
:
1. Introduction
2. Organization of Mitochondria
3. Mitochondrial Dynamics
4. Mitochondrial Alterations Associated with NI&NDDs
4.1. Proteinopathies and Alteration of Mitochondrial Biology in NI&NDDs
4.2. Alteration of Mitochondrial Dynamics in NI&NDDs
4.3. Energy Impairment Associated with Mitochondrial Dysfunction in NI&NDDs
4.4. Oxidative Stress Associated with Mitochondrial Dysfunction in NI&NDDs
5. Mitochondria-Targeted Drug Delivery Nanosystems to Repair Mitochondrial Function in NI&NDDs
5.1. Nanosystems to Counteract the Oxidative Stress Associated with Mitochondrial Dysfunction in NI&NDDs
5.2. Nanocarriers Improve CNS Bioavailability of Plant Compounds with Antioxidative and Neuroprotective Activity
5.3. Nanosystems Containing Antioxidative and Neuroprotective Plant Chemicals to Repair Mitochondrial Function in Neurodegeneration

{kind=link}
{kind=link}
| Nano-System | Drug | Disease Model | Effect on Mitochondria and Neurodegeneration | Refs | ||||
|---|---|---|---|---|---|---|---|---|
| Oxidative Stress | Cell Viability | Inflammation | Clinical Manifestations | Other Effects | ||||
| NLCs | Curcumin | AD mouse model | ↓cellular and mitochondrial oxidative stress markers | neuroprotection of hippocampal cells | ↓Aβ deposition in hippocampus | ↑learning and memory functions | ↑brain levels of curcumin, ↑ATP levels | [200] |
| NLCs-Gellan/ xanthan | Resveratrol | AD mouse model | - | - | - | ↑learning and memory functions | ↑permeation through nasal mucosa | [204] |
| SLN | Ferulic acid | Cellular model of AD | ↓cellular and mitochondrial oxidative stress markers | ↓death by apoptosis and ↑viability of neurons | ↓release of mitochondrial cytochrome c | - | ↑stabilization of mitochondrial membranes | [209] |
| Nano-emulsion | Osthole | Cellular and mouse models of AD | ↓oxidative stress markers, ↑activity of antioxidant enzymes (SOD and GSH-Px) | ↓death by apoptosis and ↑viability of neurons | - | ↑learning and memory functions | ↑acetylcholine activity in cortex and hippocampus | [213] |
| SPION | Quercetin | Rat model of AD | ↑expression levels of antioxidant enzymes (SOD, GSH-Px, CAT) | ↓apoptotic pathways | ↓expression of nitric oxide synthase and amyloid precursor protein | ↑learning and memory functions | ↑acetylcholine activity in hippocampus | [218] |
| Graphene oxide NPs | Dauricine | Cellular and mouse model of AD | ↓oxidative stress markers, ↑SOD activity | ↓apoptosis and ↑viability of neurons | ↓ glial activation | ↑learning and memory functions | ↑brain-derived neurotrophic factor (BDNF) | [222] |
| Grapehene oxide sheets/lactoferrin | Puerarin | Cellular and mouse model of PD | ↓oxidative stress markers, ↑GSH levels and SOD activity | ↓dopaminergic neuron loss | - | ↑cognitive and motor functions | ↑permeation through a BBB model, ↑brain accumulation of Puerarin, ↑dopamine levels | [198] |
| Liposomes/ Transferrin | Osthole | Cellular and mouse model of AD | ↓mitochondrial oxidative stress, ↓lipid oxidation, ↑SOD activity | ↑viability of neurons, ↓neuron apoptotic pathway | ↓inflammatory markers in brain tissue, ↓Aβ accumulation in hippocampus | ↑cognitive functions | ↑mitochondrial stability, ↑permeation through a BBB model, ↑brain accumulation of Osthole | [199] |
| SLN/RVG29/TPP | Genistein | Cellular and mouse model of AD | ↓mitochondrial oxidative stress | ↓neuronal apoptotic pathways, ↑cellular stability in hippocampus | ↓inflammatory markers and glial activation, ↓Aβ accumulation in hippocampus | ↑cognitive functions | ↑localization in neuronal mitochondria, ↑permeation through a BBB model, ↑RES evasion | [231] |
| Resveratrol | Cellular and mouse model of AD | ↓mitochondrial oxidative stress | ↓neuronal apoptotic pathways, | ↓glial activation, ↓Aβ accumulation in hippocampus | ↑cognitive function | ↑localization in neuronal mitochondria, ↓nanoparticle uptake by macrophages, ↑permeation through an in vitro BBB model | [232] | |
| Nanocrystal | Puerarin | Cellular and mouse model of PD | ↓oxidative stress markers, ↑SOD activity and GSH levels in brain | ↑dopaminergic neuronal viability | - | ↑cognitive and motor functions | ↑mitochondrial stabilization, ↑brain accumulation of Puerarin, ↑dopamine levels in the striatum | [225] |
| Paeoniflorin | Cellular model of PD | - | ↑viability of neural cells | - | - | ↑stability of mitochondrial membranes, ↑brain levels of Paeoniflorin, ↑ATP levels | [233] | |
| Resveratrol | PD rat model | ↓oxidative stress markers, ↑catalase activity and GSH levels in brain | ↑stability of neural cells | - | ↑cognitive and motor functions | ↑activity of ETC complexes | [234] | |
| Quercetin | PD mouse model | ↓lipid oxidation, ↑catalase and SOD activity, ↑GSH levels in hippocampus | - | - | ↑memory function, ↓anxious behavior | - | [235] | |
| Hesperetin | Cellular model of AD | ↓activity of cytochrome c as a peroxidase | - | - | - | ↑mitochondrial stabilization, ↑ATP levels, ↑activity of ETC complexes | [236, 237] | |
5.4. Nanocrystals of Natural Compounds as Antioxidative Agents to Repair Mitochondrial Function in Neurodegeneration
6. Restoring Production and Utilization of Mitochondrial Energy in NI&NDDs
7. Reducing the Impact of Proteinopathies on Mitochondria
7.1. Interfering with Protein Aggregation
7.2. Nanosystems Containing Natural Compounds as Mitochondrial Agents for the Treatment of Proteinopathies in Neurodegeneration
8. Therapeutic Nanomaterials for Mitochondrial Dysfunction in NI&NDDs
8.1. Photothermal Nanomaterials
8.2. Nanozymes: Nanomaterials with Enzymatic Activity
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial Dynamics—Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Panchal, K.; Tiwari, A.K. Mitochondrial Dynamics, a Key Executioner in Neurodegenerative Diseases. Mitochondrion 2019, 47, 151–173. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, R.P.; Layé, S. Polyunsaturated Fatty Acids and Their Metabolites in Brain Function and Disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 Reasons Why the Brain Is Susceptible To Oxidative Stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 Signalling Rescues Amyloid Pathology and Mitochondrial Dysfunction in Alzheimer’s Disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Liévens, J.C. PINK1-Induced Mitophagy Promotes Neuroprotection in Huntington’s Disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Rosenkranz, S.C.; Shaposhnykov, A.A.; Träger, S.; Engler, J.B.; Witte, M.E.; Roth, V.; Vieira, V.; Paauw, N.; Bauer, S.; Schwencke-Westphal, C.; et al. Enhancing Mitochondrial Activity in Neurons Protects against Neurodegeneration in a Mouse Model of Multiple Sclerosis. eLife 2021, 10, e61798. [Google Scholar] [CrossRef]
- Aman, Y.; Ryan, B.; Torsetnes, S.B.; Knapskog, A.-B.; Watne, L.O.; McEwan, W.A.; Fang, E.F. Enhancing mitophagy as a therapeutic approach for neurodegenerative diseases. In Metabolic and Bioenergetic Drivers of Neurodegenerative Disease: Treating Neurodegenerative Diseases as Metabolic Diseases, 1st ed.; Söderbom, G., Esterline, R., Oscarsson, J., Mattson, M.P., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 155, pp. 169–202. [Google Scholar]
- Debska-Vielhaber, G.; Miller, I.; Peeva, V.; Zuschratter, W.; Walczak, J.; Schreiber, S.; Petri, S.; Machts, J.; Vogt, S.; Szczepanowska, J.; et al. Impairment of Mitochondrial Oxidative Phosphorylation in Skin Fibroblasts of SALS and FALS Patients Is Rescued by in Vitro Treatment with ROS Scavengers. Exp. Neurol. 2021, 339, 113620. [Google Scholar] [CrossRef]
- Nesci, S. The Mitochondrial Permeability Transition Pore in Cell Death: A Promising Drug Binding Bioarchitecture. Med. Res. Rev. 2020, 40, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Cesura, A.M.; Pinard, E.; Schubenel, R.; Goetschy, V.; Friedlein, A.; Langen, H.; Polcic, P.; Forte, M.A.; Bernardi, P.; Kemp, J.A. The Voltage-Dependent Anion Channel Is the Target for a New Class of Inhibitors of the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2003, 278, 49812–49818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The Overlooked Oncogenome? BMC Biol. 2019, 17, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.; McFarland, R.; Taylor, R.W.; Alston, C.L. The Genetic Basis of Isolated Mitochondrial Complex II Deficiency. Mol. Genet. Metab. 2020, 131, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Madeira, V.M.C. Overview of Mitochondrial Bioenergetics. Methods Mol. Biol. 2018, 1782, 1–6. [Google Scholar] [CrossRef]
- Nath, S.; Villadsen, J. Oxidative Phosphorylation Revisited. Biotechnol. Bioeng. 2015, 112, 429–437. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial Production of Reactive Oxygen Species. Biochemistry 2013, 78, 1490–1511. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleier, L.; Dröse, S. Superoxide Generation by Complex III: From Mechanistic Rationales to Functional Consequences. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Woo, J.; Cho, H.; Seol, Y.; Kim, S.H.; Park, C.; Yousefian-Jazi, A.; Hyeon, S.J.; Lee, J.; Ryu, H. Power Failure of Mitochondria and Oxidative Stress in Neurodegeneration and Its Computational Models. Antioxidants 2021, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.K.; Kang, H.Y.; Lu, B. Altered ER-Mitochondria Contact Impacts Mitochondria Calcium Homeostasis and Contributes to Neurodegeneration in Vivo in Disease Models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK Regulates Energy Expenditure by Modulating NAD + Metabolism and SIRT1 Activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial Biogenesis in Neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Gureev, A.P.; Popov, V.N. Nrf2/ARE Pathway as a Therapeutic Target for the Treatment of Parkinson Diseases. Neurochem. Res. 2019, 44, 2273–2279. [Google Scholar] [CrossRef]
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional Activation of Antioxidant Gene Expression by Nrf2 Protects against Mitochondrial Dysfunction and Neuronal Death Associated with Acute and Chronic Neurodegeneration. Exp. Neurol. 2020, 328, 113247. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial Fission and Fusion: A Dynamic Role in Aging and Potential Target for Age-Related Disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial Fusion/Fission Dynamics in Neurodegeneration and Neuronal Plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Klionsky, D.J. Mitochondrial Fission Facilitates Mitophagy in Saccharomyces Cerevisiae. Autophagy 2013, 9, 1900–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Tanaka, K. The PINK1–Parkin Axis: An Overview. Neurosci. Res. 2020, 159, 9–15. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN Signalling in Neurodegeneration and Neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, H.; Tang, W.; Wang, S.; Tian, X.; Zhu, Y.; He, H. Mitochondrial Ca2+ Oscillation Induces Mitophagy Initiation through the PINK1-Parkin Pathway. Cell Death Dis. 2021, 12, 632. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Guan, J.S.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial Alterations near Amyloid Plaques in an Alzheimer’s Disease Mouse Model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial dysfunction in huntington’s disease. In Polyglutamine Disorders, 1st ed.; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer: Cham, Switzerland; New York, NY, USA, 2018; Volume 1049, pp. 59–83. [Google Scholar]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial Fission in Huntington’s Disease Mouse Striatum Disrupts ER-Mitochondria Contacts Leading to Disturbances in Ca2+ Efflux and Reactive Oxygen Species (ROS) Homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA Deletions and Neurodegeneration in Multiple Sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Simkins, T.J.; Duncan, G.J.; Bourdette, D. Chronic Demyelination and Axonal Degeneration in Multiple Sclerosis: Pathogenesis and Therapeutic Implications. Curr. Neurol. Neurosci. Rep. 2021, 21, 654284. [Google Scholar] [CrossRef]
- Yu, X.; Koczan, D.; Sulonen, A.M.; Akkad, D.A.; Kroner, A.; Comabella, M.; Costa, G.; Corongiu, D.; Goertsches, R.; Camina-Tato, M.; et al. MtDNA Nt13708A Variant Increases the Risk of Multiple Sclerosis. PLoS ONE 2008, 3, e1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andalib, S.; Emamhadi, M.; Yousefzadeh-Chabok, S.; Salari, A.; Sigaroudi, A.E.; Vafaee, M.S. MtDNA T4216C Variation in Multiple Sclerosis: A Systematic Review and Meta-Analysis. Acta Neurol. Belg. 2016, 116, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective Mitochondrial DNA Homeostasis in the Substantia Nigra in Parkinson Disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Gonzalo, H.; Nogueras, L.; Gil-Sánchez, A.; Hervás, J.V.; Valcheva, P.; González-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Damiano, M.; Diguet, E.; Malgorn, C.; D’Aurelio, M.; Galvan, L.; Petit, F.; Benhaim, L.; Guillermier, M.; Houitte, D.; Dufour, N.; et al. A Role of Mitochondrial Complex II Defects in Genetic Models of Huntington’s Disease Expressing N-Terminal Fragments of Mutant Huntingtin. Hum. Mol. Genet. 2013, 22, 3869–3882. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via MPTP to Activate CGAS/STING in ALS. Cell 2020, 183, 636-649.e18. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.; Vempati, U.D.; Diaz, F.; Peralta, S.; Moraes, C.T. Ablation of Cytochrome c in Adult Forebrain Neurons Impairs Oxidative Phosphorylation Without Detectable Apoptosis. Mol. Neurobiol. 2019, 56, 3722–3735. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.; Clark, J.B.; Sharpe, M. Cytochrome c Release from Rat Brain Mitochondria Is Proportional to the Mitochondrial Functional Deficit: Implications for Apoptosis and Neurodegenerative Disease. J. Neurochem. 2005, 92, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R.; et al. Mitochondrial Dysfunction Is an Important Cause of Neurological Deficits in an Inflammatory Model of Multiple Sclerosis. Sci. Rep. 2016, 6, 33249. [Google Scholar] [CrossRef]
- Sorbara, C.D.; Wagner, N.E.; Ladwig, A.; Nikić, I.; Merkler, D.; Kleele, T.; Marinković, P.; Naumann, R.; Godinho, L.; Bareyre, F.M.; et al. Pervasive Axonal Transport Deficits in Multiple Sclerosis Models. Neuron 2014, 84, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Licht-Mayer, S.; Campbell, G.R.; Canizares, M.; Mehta, A.R.; Gane, A.B.; McGill, K.; Ghosh, A.; Fullerton, A.; Menezes, N.; Dean, J.; et al. Enhanced Axonal Response of Mitochondria to Demyelination Offers Neuroprotection: Implications for Multiple Sclerosis. Acta Neuropathol. 2020, 140, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Panes, J.D.; Godoy, P.A.; Silva-Grecchi, T.; Celis, M.T.; Ramirez-Molina, O.; Gavilan, J.; Muñoz-Montecino, C.; Castro, P.A.; Moraga-Cid, G.; Yévenes, G.E.; et al. Changes in PGC-1α/SIRT1 Signaling Impact on Mitochondrial Homeostasis in Amyloid-Beta Peptide Toxicity Model. Front. Pharmacol. 2020, 11, 709. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Bae, J.E.; Jo, D.S.; Kim, J.B.; Park, N.Y.; Fang, J.; Jung, Y.K.; Jo, D.G.; Cho, D.H. Increased O-GlcNAcylation of Drp1 by Amyloid-Beta Promotes Mitochondrial Fission and Dysfunction in Neuronal Cells. Mol. Brain 2021, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated Tau Impairs Mitophagy by Inhibiting Parkin Translocation to Mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Burgess, J.D.; Faroqi, A.H.; Demeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-Synuclein-Induced Mitochondrial Dysfunction Is Mediated via a Sirtuin 3-Dependent Pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeve, A.K.; Ludtmann, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-Synuclein and Complex I Deficiency: Exploration of Their Relationship in Differentiated Neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Gao, C.; Arakawa, H.; Perry, G.; Wang, X. TDP-43 Inhibitory Peptide Alleviates Neurodegeneration and Memory Loss in an APP Transgenic Mouse Model for Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165580. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant Superoxide Dismutase 1 Forms Aggregates in the Brain Mitochondrial Matrix of Amyotrophic Lateral Sclerosis Mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn Superoxide Dismutase That Causes Motoneuron Degeneration Is Present in Mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (Huntingtin) Impairs Mitophagy in a Cellular Model of Huntington Disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s Interaction with Mitochondrial Protein Drp1 Impairs Mitochondrial Biogenesis and Causes Defective Axonal Transport and Synaptic Degeneration in Huntington’s Disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Manzanza, N.; de Oliveira Manzanza, N.; Sedlackova, L.; Kalaria, R.N. Alpha-Synuclein Post-Translational Modifications: Implications for Pathogenesis of Lewy Body Disorders. Front. Aging Neurosci. 2021, 13, 690293. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Zwaans, B.M.M. SIRT3: As Simple as It Seems? Gerontology 2014, 60, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Lieberknecht, V.; Junqueira, S.C.; Cunha, M.P.; Barbosa, T.A.; de Souza, L.F.; Coelho, I.S.; Santos, A.R.S.; Rodrigues, A.L.S.; Dafré, A.L.; Dutra, R.C. Pramipexole, a Dopamine D2/D3 Receptor-Preferring Agonist, Prevents Experimental Autoimmune Encephalomyelitis Development in Mice. Mol. Neurobiol. 2017, 54, 1033–1045. [Google Scholar] [CrossRef]
- Papadopoulos, D.; Ewans, L.; Pham-Dinh, D.; Knott, J.; Reynolds, R. Upregulation of α-Synuclein in Neurons and Glia in Inflammatory Demyelinating Disease. Mol. Cell. Neurosci. 2006, 31, 597–612. [Google Scholar] [CrossRef]
- Wang, H.; Wang, K.; Xu, W.; Wang, C.; Qiu, W.; Zhong, X.; Dai, Y.; Wu, A.; Hu, X. Cerebrospinal Fluid α-Synuclein Levels Are Elevated in Multiple Sclerosis and Neuromyelitis Optica Patients during Replase. J. Neurochem. 2012, 122, 19–23. [Google Scholar] [CrossRef]
- Sun, B.L.; Li, W.W.; Zhu, C.; Jin, W.S.; Zeng, F.; Liu, Y.H.; Bu, X.L.; Zhu, J.; Yao, X.Q.; Wang, Y.J. Clinical Research on Alzheimer’s Disease: Progress and Perspectives. Neurosci. Bull. 2018, 34, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Sivanesan, S.; Chang, E.; Howell, M.D.; Rajadas, J. Amyloid Protein Aggregates: New Clients for Mitochondrial Energy Production in the Brain? FEBS J. 2020, 287, 3386–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of VDAC1 with Amyloid Beta and Phosphorylated Tau Causes Mitochondrial Dysfunction in Alzheimer’s Disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal Interaction between the Mitochondrial Fission Protein Drp1 and Hyperphosphorylated Tau in Alzheimer’s Disease Neurons: Implications for Mitochondrial Dysfunction and Neuronal Damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired Mitochondrial Dynamics and Abnormal Interaction of Amyloid Beta with Mitochondrial Protein Drp1 in Neurons from Patients with Alzheimer’s Disease: Implications for Neuronal Damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Özdinler, P. Mitochondria, ER, and Nuclear Membrane Defects Reveal Early Mechanisms for Upper Motor Neuron Vulnerability with Respect to TDP-43 Pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, S.; Jing, L.; Huang, L.; Chen, L.; Zhao, X.; Yang, W.; Pan, Y.; Yin, P.; Qin, Z.S.; et al. Truncation of Mutant Huntingtin in Knock-in Mice Demonstrates Exon1 Huntingtin Is a Key Pathogenic Form. Nat. Commun. 2020, 11, 2582. [Google Scholar] [CrossRef] [PubMed]
- Genç, B.; Gautam, M.; Gözütok, Ö.; Dervishi, I.; Sanchez, S.; Goshu, G.M.; Koçak, N.; Xie, E.; Silverman, R.B.; Özdinler, P.H. Improving Mitochondria and ER Stability Helps Eliminate Upper Motor Neuron Degeneration That Occurs Due to MSOD1 Toxicity and TDP-43 Pathology. Clin. Transl. Med. 2021, 11, e336. [Google Scholar] [CrossRef]
- Gilmozzi, V.; Gentile, G.; Castelo Rueda, M.P.; Hicks, A.A.; Pramstaller, P.P.; Zanon, A.; Lévesque, M.; Pichler, I. Interaction of Alpha-Synuclein With Lipids: Mitochondrial Cardiolipin as a Critical Player in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 2020, 14, 578993. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant Huntingtin Binds the Mitochondrial Fission GTPase Dynamin-Related Protein-1 and Increases Its Enzymatic Activity. Nat. Med. 2011, 17, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Velde, C.V.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-Linked Mutant Superoxide Dismutase 1 (SOD1) Alters Mitochondrial Protein Composition and Decreases Protein Import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons With TDP-43 Pathology Take, Very Early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filichia, E.; Hoffer, B.; Qi, X.; Luo, Y. Inhibition of Drp1 Mitochondrial Translocation Provides Neural Protection in Dopaminergic System in a Parkinson’s Disease Model Induced by MPTP. Sci. Rep. 2016, 6, 32656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of Mitochondrial Fragmentation Diminishes Huntington’s Disease-Associated Neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; De Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 Inhibition Attenuates Neurotoxicity and Dopamine Release Deficits in Vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef]
- Zhao, F.; Austria, Q.; Wang, W.; Zhu, X. Mfn2 Overexpression Attenuates Mptp Neurotoxicity in Vivo. Int. J. Mol. Sci. 2021, 22, 601. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; García, E.; Simón, D.; Avila, J.; García-Escudero, V. Mitophagy Failure in APP and Tau Overexpression Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent Advances in the Genetics of Parkinson’s Disease. Annu. Rev. Genomics Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Fu, Y.H.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, W.; Wang, C.; Chen, Y.; Liu, P.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. Silibinin Attenuates Motor Dysfunction in a Mouse Model of Parkinson’s Disease by Suppression of Oxidative Stress and Neuroinflammation along with Promotion of Mitophagy. Physiol. Behav. 2021, 239, 113510. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J.; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 Function Promotes Mitophagy through Effects on Oxidative Stress and Mitochondrial Fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial Transmitophagy and Parkinson’s Disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef] [PubMed]
- Choong, C.J.; Okuno, T.; Ikenaka, K.; Baba, K.; Hayakawa, H.; Koike, M.; Yokota, M.; Doi, J.; Kakuda, K.; Takeuchi, T.; et al. Alternative Mitochondrial Quality Control Mediated by Extracellular Release. Autophagy 2021, 17, 2962–2974. [Google Scholar] [CrossRef] [PubMed]
- Araujo, B.G.; Souza e Silva, L.F.; de Barros Torresi, J.L.; Siena, A.; Valerio, B.C.O.; Brito, M.D.; Rosenstock, T.R. Decreased Mitochondrial Function, Biogenesis, and Degradation in Peripheral Blood Mononuclear Cells from Amyotrophic Lateral Sclerosis Patients as a Potential Tool for Biomarker Research. Mol. Neurobiol. 2020, 57, 5084–5102. [Google Scholar] [CrossRef] [PubMed]
- Drabik, K.; Malińska, D.; Piecyk, K.; Dębska-Vielhaber, G.; Vielhaber, S.; Duszyński, J.; Szczepanowska, J. Effect of Chronic Stress Present in Fibroblasts Derived from Patients with a Sporadic Form of AD on Mitochondrial Function and Mitochondrial Turnover. Antioxidants 2021, 10, 938. [Google Scholar] [CrossRef]
- Bennett, J.P.; Keeney, P.M. Alzheimer’s and Parkinson’s Brain Tissues Have Reduced Expression of Genes for MtDNA OXPHOS Proteins, Mitobiogenesis Regulator PGC-1α Protein and MtRNA Stabilizing Protein LRPPRC (LRP130). Mitochondrion 2020, 53, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.R.; Gerritsen, W.; Geerts, D.; Van Het Hof, B.; Reijerkerk, A.; De Vries, H.E.; Van Der Valk, P.; Van Horssen, J. Reduced Expression of PGC-1α Partly Underlies Mitochondrial Changes and Correlates with Neuronal Loss in Multiple Sclerosis Cortex. Acta Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Terni, B.; Boada, J.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Mitochondrial ATP-Synthase in the Entorhinal Cortex Is a Target of Oxidative Stress at Stages I/II of Alzheimer’s Disease Pathology. Brain Pathol. 2010, 20, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Cell Bioenergetics: Increasingly Recognized Components and a Possible Etiologic Cause of Alzheimer’s Disease. Antioxidants Redox Signal. 2012, 16, 1434–1455. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, F.; Ballarini, T.; Neumann, J.; Schroeter, M.L. FDG-PET Hypometabolism Is More Sensitive than MRI Atrophy in Parkinson’s Disease: A Whole-Brain Multimodal Imaging Meta-Analysis. NeuroImage Clin. 2019, 21, 101594. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Valette, J.; Pépin, J.; Flament, J.; Brouillet, E. Energy Defects in Huntington’s Disease: Why “in Vivo” Evidence Matters. Biochem. Biophys. Res. Commun. 2017, 483, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Tondo, G.; Iaccarino, L.; Caminiti, S.P.; Presotto, L.; Santangelo, R.; Iannaccone, S.; Magnani, G.; Perani, D. The Combined Effects of Microglia Activation and Brain Glucose Hypometabolism in Early-Onset Alzheimer’s Disease. Alzheimer’s Res. Ther. 2020, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-Scale Proteomic Analysis of Alzheimer’s Disease Brain and Cerebrospinal Fluid Reveals Early Changes in Energy Metabolism Associated with Microglia and Astrocyte Activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS Glucose Metabolism in Amyotrophic Lateral Sclerosis: A Therapeutic Target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.S.; Liu, T.H.; Wang, K.L.; Han, C.L.; Liu, Y.P.; Michitomo, S.; Zhang, J.G.; Fang, T.; Meng, F.G. The Metabolic Activity of Caudate and Prefrontal Cortex Negatively Correlates with the Severity of Idiopathic Parkinson’s Disease. Aging Dis. 2019, 10, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, D.C.; Lerman, H.; Lukic, A.; Andrews, R.D.; Mirelman, A.; Wernick, M.N.; Giladi, N.; Strother, S.C.; Evans, K.C.; Cedarbaum, J.M.; et al. FDG PET Parkinson’s Disease-Related Pattern as a Biomarker for Clinical Trials in Early Stage Disease. NeuroImage Clin. 2018, 20, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid Beta Oligomers Induce Impairment of Neuronal Insulin Receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s Disease a Type 3 Diabetes? A Critical Appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Aghanoori, M.R.; Smith, D.R.; Roy Chowdhury, S.; Sabbir, M.G.; Calcutt, N.A.; Fernyhough, P. Insulin Prevents Aberrant Mitochondrial Phenotype in Sensory Neurons of Type 1 Diabetic Rats. Exp. Neurol. 2017, 297, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.T.; Chen, K.Y.; Wang, W.; Chiu, J.Y.; Wu, D.; Chao, T.Y.; Hu, C.J.; Chau, K.Y.D.; Bamodu, O.A. Insulin Resistance Promotes Parkinson’s Disease through Aberrant Expression of α-Synuclein, Mitochondrial Dysfunction, and Deregulation of the Polo-Like Kinase 2 Signaling. Cells 2020, 9, 740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sintini, I.; Schwarz, C.G.; Martin, P.R.; Graff-Radford, J.; Machulda, M.M.; Senjem, M.L.; Reid, R.I.; Spychalla, A.J.; Drubach, D.A.; Lowe, V.J.; et al. Regional Multimodal Relationships between Tau, Hypometabolism, Atrophy, and Fractional Anisotropy in Atypical Alzheimer’s Disease. Hum. Brain Mapp. 2019, 40, 1618–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain Energy Rescue: An Emerging Therapeutic Concept for Neurodegenerative Disorders of Ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.C.; Zimmer, E.R.; Rosa-Neto, P.; Yoon, S.O. Consequences of Metabolic Disruption in Alzheimer’s Disease Pathology. Neurotherapeutics 2019, 16, 600–610. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial Dysfunction as a Cause of Axonal Degeneration in Multiple Sclerosis Patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon Proteinopathy Drives Neurodegeneration in Multiple Sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef]
- Hariharan, A.; Shetty, S.; Shirole, T.; Jagtap, A.G. Potential of Protease Inhibitor in 3-Nitropropionic Acid Induced Huntington’s Disease like Symptoms: Mitochondrial Dysfunction and Neurodegeneration. Neurotoxicology 2014, 45, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A. Protective Effect of Hesperidin and Naringin against 3-Nitropropionic Acid Induced Huntington’s like Symptoms in Rats: Possible Role of Nitric Oxide. Behav. Brain Res. 2010, 206, 38–46. [Google Scholar] [CrossRef]
- Maegawa, H.; Niwa, H. Generation of mitochondrial toxin rodent models of Parkinson’s disease using 6-OHDA, MPTP, and rotenone. In Experimental Models of Parkinson’s Disease, 1st ed.; Imai, Y., Ed.; Springer: New York, NY, USA, 2021; Volume 2322, pp. 95–110. [Google Scholar]
- Prasad, E.M.; Hung, S.Y. Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease. Antioxidants 2020, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Teil, M.; Arotcarena, M.L.; Dehay, B. A New Rise of Non-Human Primate Models of Synucleinopathies. Biomedicines 2021, 9, 272. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; del Rey, N.L.G.; Obeso, J.A. The Use of Nonhuman Primate Models to Understand Processes in Parkinson’s Disease. J. Neural Transm. 2018, 125, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered Glucose Catabolism in the Presynaptic and Perisynaptic Compartments of SOD1G93A Mouse Spinal Cord and Motor Cortex Indicates That Mitochondria Are the Site of Bioenergetic Imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef]
- Li, W.; Feng, J.; Gao, C.; Wu, M.; Du, Q.; Tsoi, B.; Wang, Q.; Yang, D.; Shen, J. Nitration of Drp1 Provokes Mitophagy Activation Mediating Neuronal Injury in Experimental Autoimmune Encephalomyelitis. Free Radic. Biol. Med. 2019, 143, 70–83. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal Escape Pathways for Delivery of Biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Dowding, J.M.; Song, W.; Bossy, K.; Karakoti, A.; Kumar, A.; Kim, A.; Bossy, B.; Seal, S.; Ellisman, M.H.; Perkins, G.; et al. Cerium Oxide Nanoparticles Protect against Aβ-Induced Mitochondrial Fragmentation and Neuronal Cell Death. Cell Death Differ. 2014, 21, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Song, S.J.; Bae, Y.; Choi, J.S. Self-Assembled Nanoparticles Composed of Glycol Chitosan-Dequalinium for Mitochondria-Targeted Drug Delivery. Int. J. Biol. Macromol. 2019, 132, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Mursaleen, L.; Noble, B.; Chan, S.H.Y.; Somavarapu, S.; Zariwala, M.G. N-Acetylcysteine Nanocarriers Protect against Oxidative Stress in a Cellular Model of Parkinson’s Disease. Antioxidants 2020, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Mendiola, M.R.; Nguyen, H.X.; Yambao, F.; Yu, N.; Weissig, V. Conjugation of triphenylphosphonium cation to hydrophobic moieties to prepare mitochondria-targeting nanocarriers. In Pharmaceutical Nanotechnology, 1st ed.; Weissig, V., Elbayoumi, T., Eds.; Springer: New York, NY, USA, 2019; Volume 2000, pp. 183–189. [Google Scholar]
- Teixeira, J.; Basit, F.; Willems, P.H.G.M.; Wagenaars, J.A.; van de Westerlo, E.; Amorim, R.; Cagide, F.; Benfeito, S.; Oliveira, C.; Borges, F.; et al. Mitochondria-Targeted Phenolic Antioxidants Induce ROS-Protective Pathways in Primary Human Skin Fibroblasts. Free Radic. Biol. Med. 2021, 163, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, D.; Da Silva, O.; Benfeito, S.; Barreiro, S.; Garrido, J.; Cagide, F.; Soares, P.; Remião, F.; Brazzolotto, X.; Nachon, F.; et al. Fine-tuning the Biological Profile of Multitarget Mitochondriotropic Antioxidants for Neurodegenerative Diseases. Antioxidants 2021, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Teixeira, J.; Simões, R.F.; Cagide, F.; Benfeito, S.; Borges, F.; Raimundo, N.; Oliveira, P.J. A Mitochondria-Targeted Caffeic Acid Derivative Reverts Cellular and Mitochondrial Defects in Human Skin Fibroblasts from Male Sporadic Parkinson’s Disease Patients. Redox Biol. 2021, 45, 102037. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The Mitochondria-Targeted Antioxidant MitoQ Extends Lifespan and Improves Healthspan of a Transgenic Caenorhabditis Elegans Model of Alzheimer Disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benfeito, S.; Fernandes, C.; Vilar, S.; Remião, F.; Uriarte, E.; Borges, F. Exploring the Multi-Target Performance of Mitochondriotropic Antioxidants against the Pivotal Alzheimer’s Disease Pathophysiological Hallmarks. Molecules 2020, 25, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxidants Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef]
- Kwon, H.J.; Cha, M.Y.; Kim, D.; Kim, D.K.; Soh, M.; Shin, K.; Hyeon, T.; Mook-Jung, I. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer’s Disease. ACS Nano 2016, 10, 2860–2870. [Google Scholar] [CrossRef]
- Kwon, H.J.; Kim, D.; Seo, K.; Kim, Y.G.; Han, S.I.; Kang, T.; Soh, M.; Hyeon, T. Ceria Nanoparticle Systems for Selective Scavenging of Mitochondrial, Intracellular, and Extracellular Reactive Oxygen Species in Parkinson’s Disease. Angew. Chemie Int. Ed. 2018, 57, 9408–9412. [Google Scholar] [CrossRef] [PubMed]
- Hasan, W.; Kori, R.K.; Thakre, K.; Yadav, R.S.; Jat, D. Synthesis, Characterization and Efficacy of Mitochondrial Targeted Delivery of TPP-Curcumin in Rotenone-Induced Toxicity. DARU J. Pharm. Sci. 2019, 27, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Kotha, R.R.; Luthria, D.L. Curcumin: Biological, Pharmaceutical, Nutraceutical, and Analytical Aspects. Molecules 2019, 24, 2930. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of Curcumin: Problems and Promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Tsai, Y.M.; Chien, C.F.; Lin, L.C.; Tsai, T.H. Curcumin and Its Nano-Formulation: The Kinetics of Tissue Distribution and Blood-Brain Barrier Penetration. Int. J. Pharm. 2011, 416, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. Engineering of Blended Nanoparticle Platform for Delivery of Mitochondria-Acting Therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Wang, Y.; Sun, J.; Han, Y.; Gong, W.; Li, Y.; Feng, Y.; Wang, H.; Yang, M.; Li, Z.; et al. Neuronal Mitochondria-Targeted Delivery of Curcumin by Biomimetic Engineered Nanosystems in Alzheimer’s Disease Mice. Acta Biomater. 2020, 108, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A.A.G.; et al. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenza, T.M.; Ghaisas, S.; Ramirez, J.E.V.; Harischandra, D.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G.; Narasimhan, B. Neuronal Protection against Oxidative Insult by Polyanhydride Nanoparticle-Based Mitochondria-Targeted Antioxidant Therapy. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonruamkaew, P.; Chonpathompikunlert, P.; Vong, L.B.; Sakaue, S.; Tomidokoro, Y.; Ishii, K.; Tamaoka, A.; Nagasaki, Y. Chronic Treatment with a Smart Antioxidative Nanoparticle for Inhibition of Amyloid Plaque Propagation in Tg2576 Mouse Model of Alzheimer’s Disease. Sci. Rep. 2017, 7, 3785. [Google Scholar] [CrossRef] [Green Version]
- Pichla, M.; Bartosz, G.; Stefaniuk, I.; Sadowska-Bartosz, I. Ph-Responsive Redox Nanoparticles Protect Sh-Sy5y Cells at Lowered Ph in a Cellular Model of Parkinson’s Disease. Molecules 2021, 26, 543. [Google Scholar] [CrossRef]
- Pichla, M.; Pulaski, Ł.; Kania, K.D.; Stefaniuk, I.; Cieniek, B.; Pieńkowska, N.; Bartosz, G.; Sadowska-Bartosz, I. Nitroxide Radical-Containing Redox Nanoparticles Protect Neuroblastoma SH-SY5Y Cells against 6-Hydroxydopamine Toxicity. Oxid. Med. Cell. Longev. 2020, 2020, 9260748. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxid. Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulikova, O.I.; Berezhnoy, D.S.; Stvolinsky, S.L.; Lopachev, A.V.; Orlova, V.S.; Fedorova, T.N. Neuroprotective Effect of the Carnosine–α-Lipoic Acid Nanomicellar Complex in a Model of Early-Stage Parkinson’s Disease. Regul. Toxicol. Pharmacol. 2018, 95, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Piersimoni, M.E.; Teng, X.; Cass, A.E.G.; Ying, L. Antioxidant Lipoic Acid Ligand-Shell Gold Nanoconjugates against Oxidative Stress Caused by α-Synuclein Aggregates. Nanoscale Adv. 2020, 2, 5666–5681. [Google Scholar] [CrossRef]
- Asha Devi, S.; Chamoli, A. Polyphenols as an effective therapeutic intervention against cognitive decline during normal and pathological brain aging. In Reviews on New Drug Targets in Age-Related Disorders, 1st ed.; Guest, P.C., Ed.; Springer: Cham, Switzerland; New York, NY, USA, 2020; Volume 1260, pp. 159–174. [Google Scholar]
- Fukutomi, R.; Ohishi, T.; Koyama, Y.; Pervin, M.; Nakamura, Y.; Isemura, M. Beneficial Effects of Epigallocatechin-3-o-Gallate, Chlorogenic Acid, Resveratrol, and Curcumin on Neurodegenerative Diseases. Molecules 2021, 26, 415. [Google Scholar] [CrossRef] [PubMed]
- Burkon, A.; Somoza, V. Quantification of Free and Protein-Bound Trans-Resveratrol Metabolites and Identification of Trans-Resveratrol-C/O-Conjugated Diglucuronides - Two Novel Resveratrol Metabolites in Human Plasma. Mol. Nutr. Food Res. 2008, 52, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.; Prasain, J.; D’Alessandro, T.; Arabshahi, A.; Botting, N.; Lila, M.A.; Jackson, G.; Janle, E.M.; Weaver, C.M. The Metabolism and Analysis of Isoflavones and Other Dietary Polyphenols in Foods and Biological Systems. Food Funct. 2011, 2, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Franciosoa, A.; Mastromarino, P.; Masci, A.; D’Erme, M.; Mosca, L. Chemistry, Stability and Bioavailability of Resveratrol. Med. Chem. 2014, 10, 237–245. [Google Scholar] [CrossRef]
- Faria, A.; Meireles, M.; Fernandes, I.; Santos-Buelga, C.; Gonzalez-Manzano, S.; Dueñas, M.; De Freitas, V.; Mateus, N.; Calhau, C. Flavonoid Metabolites Transport across a Human BBB Model. Food Chem. 2014, 149, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood Brain Barrier Permeability of (−)-Epigallocatechin Gallate, Its Proliferation-Enhancing Activity of Human Neuroblastoma SH-SY5Y Cells, and Its Preventive Effect on Age-Related Cognitive Dysfunction in Mice. Biochem. Biophys. Reports 2017, 9, 180–186. [Google Scholar] [CrossRef]
- Shimazu, R.; Anada, M.; Miyaguchi, A.; Nomi, Y.; Matsumoto, H. Evaluation of Blood-Brain Barrier Permeability of Polyphenols, Anthocyanins, and Their Metabolites. J. Agric. Food Chem. 2021, 69, 11676–11686. [Google Scholar] [CrossRef]
- Figueira, I.; Garcia, G.; Pimpão, R.C.; Terrasso, A.P.; Costa, I.; Almeida, A.F.; Tavares, L.; Pais, T.F.; Pinto, P.; Ventura, M.R.; et al. Polyphenols Journey through Blood-Brain Barrier towards Neuronal Protection. Sci. Rep. 2017, 7, 11456. [Google Scholar] [CrossRef]
- Faria, A.; Pestana, D.; Teixeira, D.; Azevedo, J.; De Freitas, V.; Mateus, N.; Calhau, C. Flavonoid Transport across RBE4 Cells: A Blood-Brain Barrier Model. Cell. Mol. Biol. Lett. 2010, 15, 234–241. [Google Scholar] [CrossRef]
- Carecho, R.; Carregosa, D.; dos Santos, C.N. Low Molecular Weight (Poly)Phenol Metabolites Across the Blood-Brain Barrier: The Underexplored Journey. Brain Plast. 2020, 6, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticulate Systems for Brain Delivery of Drugs. Adv. Drug Deliv. Rev. 2012, 64, 213–222. [Google Scholar] [CrossRef]
- Cai, Q.; Wang, L.; Deng, G.; Liu, J.; Chen, Q.; Chen, Z. Systemic Delivery to Central Nervous System by Engineered PLGA Nanoparticles. Am. J. Transl. Res. 2016, 8, 749–764. [Google Scholar] [PubMed]
- Gelperina, S.; Maksimenko, O.; Khalansky, A.; Vanchugova, L.; Shipulo, E.; Abbasova, K.; Berdiev, R.; Wohlfart, S.; Chepurnova, N.; Kreuter, J. Drug Delivery to the Brain Using Surfactant-Coated Poly(Lactide-Co-Glycolide) Nanoparticles: Influence of the Formulation Parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Simo, G.M.; Gao, T.; Alhindi, N.; Xu, N.; Graham, D.J.; Gamble, L.J.; Nance, E. Surfactants Influence Polymer Nanoparticle Fate within the Brain. Biomaterials 2021, 277, 121086. [Google Scholar] [CrossRef]
- Pasche, S.; Vörös, J.; Griesser, H.J.; Spencer, N.D.; Textor, M. Effects of Ionic Strength and Surface Charge on Protein Adsorption at PEGylated Surfaces. J. Phys. Chem. B 2005, 109, 17545–17552. [Google Scholar] [CrossRef] [PubMed]
- Nance, E.A.; Woodworth, G.F.; Sailor, K.A.; Shih, T.Y.; Xu, Q.; Swaminathan, G.; Xiang, D.; Eberhart, C.; Hanes, J. A Dense Poly(Ethylene Glycol) Coating Improves Penetration of Large Polymeric Nanoparticles within Brain Tissue. Sci. Transl. Med. 2012, 4, 149ra119. [Google Scholar] [CrossRef] [Green Version]
- De Luca, M.A.; Lai, F.; Corrias, F.; Caboni, P.; Bimpisidis, Z.; Maccioni, E.; Fadda, A.M.; Di Chiara, G. Lactoferrin- and Antitransferrin-Modified Liposomes for Brain Targeting of the NK3 Receptor Agonist Senktide: Preparation and in Vivo Evaluation. Int. J. Pharm. 2015, 479, 129–137. [Google Scholar] [CrossRef]
- Yu, A.; Pang, Z.; Lu, W.; Yin, Q.; Gao, H.; Jiang, X. Self-Assembled Polymersomes Conjugated with Lactoferrin as Novel Drug Carrier for Brain Delivery. Pharm. Res. 2012, 29, 83–96. [Google Scholar] [CrossRef]
- Xiong, S.; Li, Z.; Liu, Y.; Wang, Q.; Luo, J.; Chen, X.; Xie, Z.; Zhang, Y.; Zhang, H.; Chen, T. Brain-Targeted Delivery Shuttled by Black Phosphorus Nanostructure to Treat Parkinson’s Disease. Biomaterials 2020, 260, 120339. [Google Scholar] [CrossRef]
- Xiong, S.; Luo, J.; Wang, Q.; Li, Z.; Li, J.; Liu, Q.; Gao, L.; Fang, S.; Li, Y.; Pan, H.; et al. Targeted Graphene Oxide for Drug Delivery as a Therapeutic Nanoplatform against Parkinson’s Disease. Biomater. Sci. 2021, 9, 1705–1715. [Google Scholar] [CrossRef]
- Kong, L.; Li, X.T.; Ni, Y.N.; Xiao, H.H.; Yao, Y.J.; Wang, Y.Y.; Ju, R.J.; Li, H.Y.; Liu, J.J.; Fu, M.; et al. Transferrin-Modified Osthole PEGylated Liposomes Travel the Blood-Brain Barrier and Mitigate Alzheimer’s Disease-Related Pathology in APP/PS-1 Mice. Int. J. Nanomed. 2020, 15, 2841–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvajerd, S.S.; Izadi, Z.; Azadi, A.; Kurd, M.; Derakhshankhah, H.; Zadeh, M.S.; Javar, H.A.; Hamidi, M. Neuroprotective Potential of Curcumin-Loaded Nanostructured Lipid Carrier in an Animal Model of Alzheimer’s Disease: Behavioral and Biochemical Evidence. J. Alzheimer’s Dis. 2019, 69, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Juan, M.E.; Maijó, M.; Planas, J.M. Quantification of Trans-Resveratrol and Its Metabolites in Rat Plasma and Tissues by HPLC. J. Pharm. Biomed. Anal. 2010, 51, 391–398. [Google Scholar] [CrossRef]
- Wang, P.; Sang, S. Metabolism and Pharmacokinetics of Resveratrol and Pterostilbene. BioFactors 2018, 44, 16–25. [Google Scholar] [CrossRef]
- Rajput, A.; Bariya, A.; Allam, A.; Othman, S.; Butani, S.B. In Situ Nanostructured Hydrogel of Resveratrol for Brain Targeting: In Vitro-in Vivo Characterization. Drug Deliv. Transl. Res. 2018, 8, 1460–1470. [Google Scholar] [CrossRef]
- Sgarbossa, A.; Giacomazza, D.; Di Carlo, M. Ferulic Acid: A Hope for Alzheimer’s Disease Therapy from Plants. Nutrients 2015, 7, 5764–5782. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.P.; Rai, H.; Singh, G.; Singh, G.K.; Mishra, S.; Kumar, S.; Srikrishna, S.; Modi, G. A Review on Ferulic Acid and Analogs Based Scaffolds for the Management of Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 215, 113278. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.P.; Tej, G.N.V.C.; Pandey, A.; Priya, K.; Pandey, P.; Shankar, G.; Nayak, P.K.; Rai, G.; Chittiboyina, A.G.; Doerksen, R.J.; et al. Design, Synthesis and Biological Evaluation of Novel Naturally-Inspired Multifunctional Molecules for the Management of Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 198, 112257. [Google Scholar] [CrossRef]
- Anis, E.; Zafeer, M.F.; Firdaus, F.; Islam, S.N.; Anees Khan, A.; Ali, A.; Hossain, M.M. Ferulic Acid Reinstates Mitochondrial Dynamics through PGC1α Expression Modulation in 6-Hydroxydopamine Lesioned Rats. Phyther. Res. 2020, 34, 214–226. [Google Scholar] [CrossRef]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic Acid Inhibits Oxidative Stress and Cell Death Induced by Ab Oligomers: Improved Delivery by Solid Lipid Nanoparticles. Free Radic. Res. 2009, 43, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.B.; Zhou, J.; Qu, Y.; Li, X.; Lu, C.T.; Xie, K.L.; Sun, X.L.; Fei, Z. Neuroprotective Effect of Osthole on MPP+-Induced Cytotoxicity in PC12 Cells via Inhibition of Mitochondrial Dysfunction and ROS Production. Neurochem. Int. 2010, 57, 206–215. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Li, L.; Feng, F.; Yuan, H.; Gao, D.K.; Fu, L.A.; Fei, Z. Osthole Attenuates Spinal Cord Ischemia-Reperfusion Injury through Mitochondrial Biogenesis-Independent Inhibition of Mitochondrial Dysfunction in Rats. J. Surg. Res. 2013, 185, 805–814. [Google Scholar] [CrossRef]
- Yang, Y.F.; Xu, W.; Song, W.; Ye, M.; Yang, X.W. Transport of Twelve Coumarins from Angelicae Pubescentis Radix across a MDCK-PHaMDR Cell Monolayer - An in Vitro Model for Blood-Brain Barrier Permeability. Molecules 2015, 20, 11719–11732. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, X.; Wang, X.; Wang, J.; Hao, Q.; Hao, J.; Hou, X. Osthole-Loaded Nanoemulsion Enhances Brain Target in the Treatment of Alzheimer’s Disease via Intranasal Administration. Oxid. Med. Cell. Longev. 2021, 2021, 8844455. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Madathil, S.K.; Pandey, M.; Haobam, R.; Rajamma, U.; Mohanakumar, K.P. Quercetin Up-Regulates Mitochondrial Complex-I Activity to Protect against Programmed Cell Death in Rotenone Model of Parkinson’s Disease in Rats. Neuroscience 2013, 236, 136–148. [Google Scholar] [CrossRef]
- Sandhir, R.; Mehrotra, A. Quercetin Supplementation Is Effective in Improving Mitochondrial Dysfunctions Induced by 3-Nitropropionic Acid: Implications in Huntington’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Saber Batiha, G.; Beshbishy, A.M.; Ikram, M.; Mulla, Z.S.; Abd El-Hack, M.E.; Taha, A.E.; Algammal, A.M.; Ali Elewa, Y.H. The Pharmacological Activity, Biochemical Properties, and Pharmacokinetics of the Major Natural Polyphenolic Flavonoid: Quercetin. Foods 2020, 9, 374. [Google Scholar] [CrossRef] [Green Version]
- Dabeek, W.M.; Marra, M.V. Dietary Quercetin and Kaempferol: Bioavailability and Potential Cardiovascular-Related Bioactivity in Humans. Nutrients 2019, 11, 2288. [Google Scholar] [CrossRef] [Green Version]
- Amanzadeh Jajin, E.; Esmaeili, A.; Rahgozar, S.; Noorbakhshnia, M. Quercetin-Conjugated Superparamagnetic Iron Oxide Nanoparticles Protect AlCl3-Induced Neurotoxicity in a Rat Model of Alzheimer’s Disease via Antioxidant Genes, APP Gene, and MiRNA-101. Front. Neurosci. 2021, 14, 598617. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, X.; Zhou, H.; Wang, L.; Zhang, Z.; Ren, X.; Zhu, F.; Guo, Y.; Huang, X.; Liu, J.; et al. The Isoquinoline Alkaloid Dauricine Targets Multiple Molecular Pathways to Ameliorate Alzheimer-like Pathological Changes in Vitro. Oxid. Med. Cell. Longev. 2018, 2018, 2025914. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Pu, Z.; Li, M.; Wang, K.; Deng, L.; Chen, W. Antioxidative and Antiapoptosis: Neuroprotective Effects of Dauricine in Alzheimer’s Disease Models. Life Sci. 2020, 243, 117237. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, P.; Wang, J.; Yu, H.; Zhang, Z.; Liu, J.; Chen, X.; Zhu, F.; Yang, X. Dauricine Attenuates Spatial Memory Impairment and Alzheimer-Like Pathologies by Enhancing Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 8, 624339. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, L.; Chen, L.; Peng, C.; Luo, B.; Mo, J.; Chen, W. Intranasal Administration of Dauricine Loaded on Graphene Oxide: Multi-Target Therapy for Alzheimer’s Disease. Drug Deliv. 2021, 28, 580–593. [Google Scholar] [CrossRef]
- Chen, X.; Yi, L.; Song, S.; Wang, L.; Liang, Q.; Wang, Y.; Wu, Y.; Gao, Q. Puerarin Attenuates Palmitate-Induced Mitochondrial Dysfunction, Impaired Mitophagy and Inflammation in L6 Myotubes. Life Sci. 2018, 206, 84–92. [Google Scholar] [CrossRef]
- Song, X.; Bai, X.; Liu, S.; Dong, L.; Deng, H.; Wang, C. A Novel Microspheres Formulation of Puerarin: Pharmacokinetics Study and in Vivo Pharmacodynamics Evaluations. Evid.-Based Complement. Altern. Med. 2016, 2016. [Google Scholar] [CrossRef]
- Xiong, S.; Liu, W.; Li, D.; Chen, X.; Liu, F.; Yuan, D.; Pan, H.; Wang, Q.; Fang, S.; Chen, T. Oral Delivery of Puerarin Nanocrystals to Improve Brain Accumulation and Anti-Parkinsonian Efficacy. Mol. Pharm. 2019, 16, 1444–1455. [Google Scholar] [CrossRef]
- Siddique, Y.H.; Naz, F.; Jyoti, S.; Ali, F. Rahul Effect of Genistein on the Transgenic Drosophila Model of Parkinson’s Disease. J. Diet. Suppl. 2019, 16, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Magalingam, K.B.; Radhakrishnan, A.K.; Haleagrahara, N. Protective Mechanisms of Flavonoids in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 314560. [Google Scholar] [CrossRef] [Green Version]
- Ferramosca, A.; Lorenzetti, S.; Di Giacomo, M.; Lunetti, P.; Murrieri, F.; Capobianco, L.; Dolce, V.; Coppola, L.; Zara, V. Modulation of Human Sperm Mitochondrial Respiration Efficiency by Plant Polyphenols. Antioxidants 2021, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.J.; Cherng, Y.G.; Chen, J.T.; Chang, C.C.; Liu, S.H.; Chen, R.M. Genistein Triggers Translocation of Estrogen Receptor-Alpha in Mitochondria to Induce Expressions of ATP Synthesis-Associated Genes and Improves Energy Production and Osteoblast Maturation. Am. J. Chin. Med. 2021, 49, 901–923. [Google Scholar] [CrossRef]
- Zhou, S.; Hu, Y.; Zhang, B.; Teng, Z.; Gan, H.; Yang, Z.; Wang, Q.; Huan, M.; Mei, Q. Dose-Dependent Absorption, Metabolism, and Excretion of Genistein in Rats. J. Agric. Food Chem. 2008, 56, 8354–8359. [Google Scholar] [CrossRef]
- Han, Y.; Gao, C.; Wang, H.; Sun, J.; Liang, M.; Feng, Y.; Liu, Q.; Fu, S.; Cui, L.; Gao, C.; et al. Macrophage Membrane-Coated Nanocarriers Co-Modified by RVG29 and TPP Improve Brain Neuronal Mitochondria-Targeting and Therapeutic Efficacy in Alzheimer’s Disease Mice. Bioact. Mater. 2021, 6, 529–542. [Google Scholar] [CrossRef]
- Han, Y.; Chu, X.; Cui, L.; Fu, S.; Gao, C.; Li, Y.; Sun, B. Neuronal Mitochondria-Targeted Therapy for Alzheimer’s Disease by Systemic Delivery of Resveratrol Using Dual-Modified Novel Biomimetic Nanosystems. Drug Deliv. 2020, 27, 502–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Li, B.; Zhang, Y.; Chen, T.; Chen, C.; Jiang, W.; Wang, Q.; Chen, T. Intranasal Delivery of Paeoniflorin Nanocrystals for Brain Targeting. Asian J. Pharm. Sci. 2020, 15, 326–335. [Google Scholar] [CrossRef]
- Palle, S.; Neerati, P. Improved Neuroprotective Effect of Resveratrol Nanoparticles as Evinced by Abrogation of Rotenone-Induced Behavioral Deficits and Oxidative and Mitochondrial Dysfunctions in Rat Model of Parkinson’s Disease. Naunyn. Schmiedebergs. Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef]
- Ghaffari, F.; Moghaddam, A.H.; Zare, M. Neuroprotective Effect of Quercetin Nanocrystal in a 6-Hydroxydopamine Model of Parkinson Disease: Biochemical and Behavioral Evidence. Basic Clin. Neurosci. 2018, 9, 317–324. [Google Scholar] [CrossRef]
- Babylon, L.; Grewal, R.; Stahr, P.L.; Eckert, R.W.; Keck, C.M.; Eckert, G.P. Hesperetin Nanocrystals Improve Mitochondrial Function in a Cell Model of Early Alzheimer Disease. Antioxidants 2021, 10, 1003. [Google Scholar] [CrossRef]
- Stahr, P.L.; Grewal, R.; Eckert, G.P.; Keck, C.M. Investigating Hesperetin Nanocrystals with Tailor-Made Sizes for the Prevention and Treatment of Alzheimer’s Disease. Drug Deliv. Transl. Res. 2021, 11, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Figueiredo, P.; Zhang, P.; Hirvonen, J.T.; Liu, D.; Santos, H.A. Production of Pure Drug Nanocrystals and Nano Co-Crystals by Confinement Methods. Adv. Drug Deliv. Rev. 2018, 131, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Shen, C.; Zhu, W.; Yuan, H. The Contribution of Absorption of Integral Nanocrystals to Enhancement of Oral Bioavailability of Quercetin. Acta Pharm. Sin. B 2021, 11, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Biesemann, N.; Ried, J.S.; Ding-Pfennigdorff, D.; Dietrich, A.; Rudolph, C.; Hahn, S.; Hennerici, W.; Asbrand, C.; Leeuw, T.; Strübing, C. High Throughput Screening of Mitochondrial Bioenergetics in Human Differentiated Myotubes Identifies Novel Enhancers of Muscle Performance in Aged Mice. Sci. Rep. 2018, 8, 9408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, L.; Zhu, X.; Wu, W.; Wang, Y. Hesperidin Alleviates Cognitive Impairment, Mitochondrial Dysfunction and Oxidative Stress in a Mouse Model of Alzheimer’s Disease. Cell. Mol. Neurobiol. 2014, 34, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Stokes, K.; Mahngar, K.; Domazet-Damjanov, D.; Sikorska, M.; Pandey, S. Inhibition of Stress Induced Premature Senescence in Presenilin-1 Mutated Cells with Water Soluble Coenzyme Q10. Mitochondrion 2014, 17, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mukherjee, A.; Das, N.; Swarnakar, S. Protective Roles of Nanomelatonin in Cerebral Ischemia-Reperfusion of Aged Brain: Matrixmetalloproteinases as Regulators. Exp. Gerontol. 2017, 92, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Sandhir, R.; Yadav, A.; Mehrotra, A.; Sunkaria, A.; Singh, A.; Sharma, S. Curcumin Nanoparticles Attenuate Neurochemical and Neurobehavioral Deficits in Experimental Model of Huntington’s Disease. NeuroMolecular Med. 2014, 16, 106–118. [Google Scholar] [CrossRef]
- Ferreira, A.; Rodrigues, M.; Fortuna, A.; Falcão, A.; Alves, G. Huperzine A from Huperzia Serrata: A Review of Its Sources, Chemistry, Pharmacology and Toxicology. Phytochem. Rev. 2016, 15, 51–85. [Google Scholar] [CrossRef]
- Gao, X.; Xi, C.T. Huperzine A Attenuates Mitochondrial Dysfunction in β-Amyloid-Treated PC12 Cells by Reducing Oxygen Free Radicals Accumulation and Improving Mitochondrial Energy Metabolism. J. Neurosci. Res. 2006, 83, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zheng, C.Y.; Yang, L.; Tang, X.C.; Zhang, H.Y. Huperzine A Protects Isolated Rat Brain Mitochondria against β-Amyloid Peptide. Free Radic. Biol. Med. 2009, 46, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Wang, A.; Hua, H.; Jiang, Y.; Wang, Y.; Mu, H.; Wu, Z.; Sun, K. Intranasal Delivery of Huperzine A to the Brain Using Lactoferrin-Conjugated N-Trimethylated Chitosan Surface-Modified PLGA Nanoparticles for Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2018, 13, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, C.; Zhai, W.; Zhuang, N.; Han, T.; Ding, Z. The Optimization Design of Lactoferrin Loaded Hupa Nanoemulsion for Targeted Drug Transport via Intranasal Route. Int. J. Nanomed. 2019, 14, 9217–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Han, L.; Qin, J.; Lu, W.; Wang, J. The Use of Borneol as an Enhancer for Targeting Aprotinin-Conjugated PEG-PLGA Nanoparticles to the Brain. Pharm. Res. 2013, 30, 2560–2572. [Google Scholar] [CrossRef]
- Talbot, K. Brain Insulin Resistance in Alzheimer’s Disease and Its Potential Treatment with GLP-1 Analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, A.; Sah, S.P. Insulin Signaling Pathway and Related Molecules: Role in Neurodegeneration and Alzheimer’s Disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Hölscher, C. Brain Insulin Resistance: Role in Neurodegenerative Disease and Potential for Targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef]
- Pharaoh, G.; Owen, D.; Yeganeh, A.; Premkumar, P.; Farley, J.; Bhaskaran, S.; Ashpole, N.; Kinter, M.; Van Remmen, H.; Logan, S. Disparate Central and Peripheral Effects of Circulating IGF-1 Deficiency on Tissue Mitochondrial Function. Mol. Neurobiol. 2020, 57, 1317–1331. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Cui, C.; Duan, W.; Wang, W.; Wang, Y.; Liu, Y.; Li, Z.; Li, C. The Role of Insulin-like Growth Factor 1 in ALS Cell and Mouse Models: A Mitochondrial Protector. Brain Res. Bull. 2019, 144, 1–13. [Google Scholar] [CrossRef]
- Qi, Z.; Guo, W.; Zheng, S.; Fu, C.; Ma, Y.; Pan, S.; Liu, Q.; Yang, X. Enhancement of Neural Stem Cell Survival, Proliferation and Differentiation by IGF-1 Delivery in Graphene Oxide-Incorporated PLGA Electrospun Nanofibrous Mats. RSC Adv. 2019, 9, 8315–8325. [Google Scholar] [CrossRef] [Green Version]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diab. Rep. 2019, 19, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, A.; Kumar, A. Role of Nuclear Receptor on Regulation of BDNF and Neuroinflammation in Hippocampus of β-Amyloid Animal Model of Alzheimer’s Disease. Neurotox. Res. 2014, 25, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Wong, L.R.; Pee, H.N.; Yang, S.; Ho, P.C.L. Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Agonist. Mol. Neurobiol. 2019, 56, 7267–7283. [Google Scholar] [CrossRef] [PubMed]
- Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Lecrux, C.; Fernandes, P.; Rosa-Neto, P.; Tong, X.K.; Hamel, E. Complete Rescue of Cerebrovascular Function in Aged Alzheimer’s Disease Transgenic Mice by Antioxidants and Pioglitazone, a Peroxisome Proliferator-Activated Receptor γ Agonist. J. Neurosci. 2008, 28, 9287–9296. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.R.; Wong, P.; Ho, P.C.L. Metabolic Profiling of Female Tg2576 Mouse Brains Provides Novel Evidence Supporting Intranasal Low-Dose Pioglitazone for Long-Term Treatment at an Early Stage of Alzheimer’s Disease. Biomedicines 2020, 8, 589. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Dey, P.N.; Chen, C.; Monteiro, O.; et al. Metformin Reverses Early Cortical Network Dysfunction and Behavior Changes in Huntington’s Disease. eLife 2018, 7, e38744. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin Treatment Prevents Amyloid Plaque Deposition and Memory Impairment in APP/PS1 Mice. Brain. Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, N. Sirtuin 1-Mediated Cellular Metabolic Memory of High Glucose via the LKB1/AMPK/ROS Pathway and Therapeutic Effects of Metformin. Diabetes 2012, 61, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.M.; Lee, W.K.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. Metformin Restores Parkin-Mediated Mitophagy, Suppressed by Cytosolic P53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511-1523.e5. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [Green Version]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Sikora, A.; Zielonka, J.; Dwinell, M. Mitochondria-Targeted Metformins: Antitumour and Redox Signalling Mechanisms. Interface Focus 2017, 7, 20160109. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the Basics of Repurposing Mitochondria-Targeted Drugs: From Parkinson’s Disease to Cancer and Back to Parkinson’s Disease. Redox Biol. 2020, 36, 101665. [Google Scholar] [CrossRef] [PubMed]
- Girges, C.; Vijiaratnam, N.; Athauda, D.; Auld, G.; Gandhi, S.; Foltynie, T. The Future of Incretin-Based Approaches for Neurodegenerative Diseases in Older Adults: Which to Choose? A Review of Their Potential Efficacy and Suitability. Drugs Aging 2021, 38, 355–373. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, J.; Li, S.; Li, H.; Zhou, Y.; Zheng, W.; Zhang, M.; Liu, L.; Chen, Z. GLP-1 Improves the Neuronal Supportive Ability of Astrocytes in Alzheimer’s Disease by Regulating Mitochondrial Dysfunction via the CAMP/PKA Pathway. Biochem. Pharmacol. 2021, 188, 114578. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Watson, K.T.; Wroolie, T.E.; Tong, G.; Foland-Ross, L.C.; Frangou, S.; Singh, M.; McIntyre, R.S.; Roat-Shumway, S.; Myoraku, A.; Reiss, A.L.; et al. Neural Correlates of Liraglutide Effects in Persons at Risk for Alzheimer’s Disease. Behav. Brain Res. 2019, 356, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.; Liu, W.; Li, Y.; Li, L.; Hölscher, C. Neuroprotective Effects of a Triple GLP-1/GIP/Glucagon Receptor Agonist in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Brain Res. 2018, 1678, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.J.; Xue, G.F.; Cheng, H.F.; Meng, P.F.; Lian, X.; Hölscher, C.; Li, D.F. The GLP-1/GIP Dual-Receptor Agonist DA5-CH Inhibits the NF-ΚB Inflammatory Pathway in the MPTP Mouse Model of Parkinson’s Disease More Effectively than the GLP-1 Single-Receptor Agonist NLY01. Brain Behav. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, L.; Shi, M.; Yu, Z.; Shao, S.; Yuan, Y.; Zhang, Z.; Hölscher, C. Neuroprotective Effects of a GLP-2 Analogue in the MPTP Parkinson’s Disease Mouse Model. J. Parkinson’s Dis. 2021, 11, 529–543. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Hölscher, C. GIP Has Neuroprotective Effects in Alzheimer and Parkinson’s Disease Models. Peptides 2020, 125, 170184. [Google Scholar] [CrossRef]
- Gharagozloo, M.; Smith, M.D.; Sotirchos, E.S.; Jin, J.; Meyers, K.; Taylor, M.; Garton, T.; Bannon, R.; Lord, H.-N.; Dawson, T.M.; et al. Therapeutic Potential of a Novel Glucagon-like Peptide-1 Receptor Agonist, NLY01, in Experimental Autoimmune Encephalomyelitis. Neurotherapeutics 2021, 18, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Jeon, S.J.; Cho, K.S.; Moon, E.; Sapkota, A.; Jun, H.S.; Ryu, J.H.; Choi, J.W. Activation of Glucagon-Like Peptide-1 Receptor Promotes Neuroprotection in Experimental Autoimmune Encephalomyelitis by Reducing Neuroinflammatory Responses. Mol. Neurobiol. 2018, 55, 3007–3020. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.B.; Park, M.R.; Song, S.C. Sustained Release of Exendin 4 Using Injectable and Ionic-Nano-Complex Forming Polymer Hydrogel System for Long-Term Treatment of Type 2 Diabetes Mellitus. ACS Appl. Mater. Interfaces 2019, 11, 15201–15211. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Lim, S.M.; Chae, S.Y.; Kim, K.; Park, E.J.; Lee, K.C.; Na, D.H. Mono-Lithocholated Exendin-4-Loaded Glycol Chitosan Nanoparticles with Prolonged Antidiabetic Effects. Int. J. Pharm. 2015, 495, 81–86. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Hu, Y.; Gui, Z.; Zhou, Y.; Nie, T.; Zhu, J.; Liu, Z.; Chen, K.; Liu, L.; Leong, K.W.; et al. Sustained Release of Exendin-4 from Tannic Acid/Fe (III) Nanoparticles Prolongs Blood Glycemic Control in a Mouse Model of Type II Diabetes. J. Control. Release 2019, 301, 119–128. [Google Scholar] [CrossRef]
- Bao, X.; Qian, K.; Yao, P. Oral Delivery of Exenatide-Loaded Hybrid Zein Nanoparticles for Stable Blood Glucose Control and β-Cell Repair of Type 2 Diabetes Mice. J. Nanobiotechnol. 2020, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Ismail, R.; Phan, T.N.Q.; Laffleur, F.; Csóka, I.; Bernkop-Schnürch, A. Hydrophobic Ion Pairing of a GLP-1 Analogue for Incorporating into Lipid Nanocarriers Designed for Oral Delivery. Eur. J. Pharm. Biopharm. 2020, 152, 10–17. [Google Scholar] [CrossRef]
- Ren, T.; Zheng, X.; Bai, R.; Yang, Y.; Jian, L. Utilization of PLGA Nanoparticles in Yeast Cell Wall Particle System for Oral Targeted Delivery of Exenatide to Improve Its Hypoglycemic Efficacy. Int. J. Pharm. 2021, 601, 120583. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Nie, T.; Hu, Y.; Zhou, Y.; Zhu, J.; Liu, Z.; Liu, L.; Leong, K.W.; Chen, Y.; Mao, H.Q. A Polyphenol-Metal Nanoparticle Platform for Tunable Release of Liraglutide to Improve Blood Glycemic Control and Reduce Cardiovascular Complications in a Mouse Model of Type II Diabetes. J. Control. Release 2020, 318, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Tramontin, N.; da Silva, S.; Arruda, R.; Ugioni, K.S.; Canteiro, P.B.; de Bem Silveira, G.; Mendes, C.; Silveira, P.C.L.; Muller, A.P. Gold Nanoparticles Treatment Reverses Brain Damage in Alzheimer’s Disease Model. Mol. Neurobiol. 2020, 57, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, M.; Dolui, S.; Chaudhuri, S.; Paul, U.; Bhattacharjee, G.; Ghosal, M.; Maiti, N.C.; Mukhopadhyay, D.; Senapati, D. Impact of Porous Nanomaterials on Inhibiting Protein Aggregation Behaviour. RSC Adv. 2021, 11, 3354–3362. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, L.P.; Dai, W.; Dong, H.; Wen, Y.; Zhang, X. Graphene Quantum Dots for the Inhibition of β Amyloid Aggregation. Nanoscale 2015, 7, 19060–19065. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Sun, Y.; Lei, J.; Wei, G. Mechanistic Insights into the Inhibition and Size Effects of Graphene Oxide Nanosheets on the Aggregation of an Amyloid-β Peptide Fragment. Nanoscale 2018, 10, 8989–8997. [Google Scholar] [CrossRef]
- Liu, C.; Huang, H.; Ma, L.; Fang, X.; Wang, C.; Yang, Y. Modulation of β-Amyloid Aggregation by Graphene Quantum Dots. R. Soc. Open Sci. 2019, 6, 190271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaeidamini, M.; Bernson, D.; Sasanian, N.; Kumar, R. Graphene Oxide Sheets and Quantum Dots Inhibit A-Synuclein Amyloid Formation by Different Mechanisms. Nanoscale 2020, 12, 19450–19460. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Yoo, J.M.; Hwang, H.; Lee, J.; Lee, S.H.; Yun, S.P.; Park, M.J.; Lee, M.J.; Choi, S.; Kwon, S.H.; et al. Graphene Quantum Dots Prevent α-Synucleinopathy in Parkinson’s Disease. Nat. Nanotechnol. 2018, 13, 812–818. [Google Scholar] [CrossRef]
- Joshi, A.S.; Singh, V.; Gahane, A.; Thakur, A.K. Biodegradable Nanoparticles Containing Mechanism Based Peptide Inhibitors Reduce Polyglutamine Aggregation in Cell Models and Alleviate Motor Symptoms in a Drosophila Model of Huntington’s Disease. ACS Chem. Neurosci. 2019, 10, 1603–1614. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, S.; Jin, P.; Huang, Y.; Dai, Q.; Zhu, Q.; Wei, P.; Yang, Z.; Zhang, L.; Liu, H.; et al. Graphene Oxide Improves Postoperative Cognitive Dysfunction by Maximally Alleviating Amyloid Beta Burden in Mice. Theranostics 2020, 10, 11908–11920. [Google Scholar] [CrossRef] [PubMed]
- Sardoiwala, M.N.; Srivastava, A.K.; Kaundal, B.; Karmakar, S.; Choudhury, S.R. Recuperative Effect of Metformin Loaded Polydopamine Nanoformulation Promoting EZH2 Mediated Proteasomal Degradation of Phospho-α-Synuclein in Parkinson’s Disease Model. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102088. [Google Scholar] [CrossRef] [PubMed]
- Sardoiwala, M.N.; Karmakar, S.; Choudhury, S.R. Chitosan Nanocarrier for FTY720 Enhanced Delivery Retards Parkinson’s Disease via PP2A-EzH2 Signaling in Vitro and Ex Vivo. Carbohydr. Polym. 2021, 254, 117435. [Google Scholar] [CrossRef]
- Rojas-Prats, E.; Tosat-Bitrián, C.; Martínez-González, L.; Nozal, V.; Pérez, D.I.; Martínez, A. Increasing Brain Permeability of PHA-767491, a Cell Division Cycle 7 Kinase Inhibitor, with Biodegradable Polymeric Nanoparticles. Pharmaceutics 2021, 13, 180. [Google Scholar] [CrossRef]
- Singh, N.A.; Bhardwaj, V.; Ravi, C.; Ramesh, N.; Mandal, A.K.A.; Khan, Z.A. EGCG Nanoparticles Attenuate Aluminum Chloride Induced Neurobehavioral Deficits, Beta Amyloid and Tau Pathology in a Rat Model of Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Chen, Z.X.; Lu, Z.G.; Yang, Q.H.; Liu, L.Y.; Jiang, Z.T.; Zhang, L.Q.; Zhang, X.; Qing, H. “Cell-Addictive” Dual-Target Traceable Nanodrug for Parkinson’s Disease Treatment via Flotillins Pathway. Theranostics 2018, 8, 5469–5481. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-Drug Loaded Nanoparticles of Epigallocatechin-3-Gallate (EGCG)/Ascorbic Acid Enhance Therapeutic Efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s Disease Mice Model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Yi, P.; He, T.; Song, X.; Liu, Y.; Li, Q.; Zheng, J.; Song, R.; Liu, C.; Zhang, Z.; et al. Quercetin-Loaded Selenium Nanoparticles Inhibit Amyloid-β Aggregation and Exhibit Antioxidant Activity. Colloids Surfaces A Physicochem. Eng. Asp. 2020, 602, 125058. [Google Scholar] [CrossRef]
- Yang, L.; Wang, N.; Zheng, G. Enhanced Effect of Combining Chlorogenic Acid on Selenium Nanoparticles in Inhibiting Amyloid β Aggregation and Reactive Oxygen Species Formation In Vitro. Nanoscale Res. Lett. 2018, 13, 303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Yan, F.; Liang, X.; Wu, M.; Shen, Y.; Chen, M.; Xu, Y.; Zou, G.; Jiang, P.; Tang, C.; et al. Localized Delivery of Curcumin into Brain with Polysorbate 80-Modified Cerasomes by Ultrasound-Targeted Microbubble Destruction for Improved Parkinson’s Disease Therapy. Theranostics 2018, 8, 2264–2277. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, C.; Zhang, J.; Zhang, Y.; Liu, K.; Song, J.X.; Sreenivasmurthy, S.G.; Wang, Z.; Shi, Y.; Chu, C.; et al. A Self-Assembled α-Synuclein Nanoscavenger for Parkinson’s Disease. ACS Nano 2020, 14, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Aliakbari, F.; Mohammad-Beigi, H.; Abbasi, S.; Rezaei-Ghaleh, N.; Lermyte, F.; Parsafar, S.; Becker, S.; Tafreshi, A.P.; O’Connor, P.B.; Collingwood, J.F.; et al. Multiple Protective Roles of Nanoliposome-Incorporated Baicalein against Alpha-Synuclein Aggregates. Adv. Funct. Mater. 2021, 31, 2007765. [Google Scholar] [CrossRef]
- Saleh, S.R.; Abady, M.M.; Nofal, M.; Yassa, N.W.; Abdel-Latif, M.S.; Nounou, M.I.; Ghareeb, D.A.; Abdel-Monaem, N. Berberine Nanoencapsulation Attenuates Hallmarks of Scoplomine Induced Alzheimer’s-like Disease in Rats. Curr. Rev. Clin. Exp. Pharmacol. 2021, 16, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, N.; Zeng, Z.; Huang, J.; Xiang, Z.; Guan, Y.Q. Neuroprotective Effect of Chitosan Nanoparticle Gene Delivery System Grafted with Acteoside (ACT) in Parkinson’s Disease Models. J. Mater. Sci. Technol. 2020, 43, 197–207. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Stuani, C.; Guarnaccia, C.; Romano, V.; De Conti, L.; Baralle, F.E. Cellular Model of TAR DNA-Binding Protein 43 (TDP-43) Aggregation Based on Its C-Terminal Gln/Asn-Rich Region. J. Biol. Chem. 2012, 287, 7512–7525. [Google Scholar] [CrossRef] [Green Version]
- Mompeán, M.; Buratti, E.; Guarnaccia, C.; Brito, R.M.M.; Chakrabartty, A.; Baralle, F.E.; Laurents, D.V. Structural Characterization of the Minimal Segment of TDP-43 Competent for Aggregation. Arch. Biochem. Biophys. 2014, 545, 53–62. [Google Scholar] [CrossRef]
- Mompeán, M.; Ramírez de Mingo, D.; Hervás, R.; Fernández-Ramírez, M. del C.; Carrión-Vázquez, M.; Laurents, D. V. Molecular Mechanism of the Inhibition of TDP-43 Amyloidogenesis by QBP1. Arch. Biochem. Biophys. 2019, 675, 108113. [Google Scholar] [CrossRef]
- Hervás, R.; Oroz, J.; Galera-Prat, A.; Goñi, O.; Valbuena, A.; Vera, A.M.; Gómez-Sicilia, À.; Losada-Urzáiz, F.; Uversky, V.N.; Menéndez, M.; et al. Common Features at the Start of the Neurodegeneration Cascade. PLoS Biol. 2012, 10, e1001335. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Tucker, T.; Ren, H.; Kenan, D.J.; Henderson, B.S.; Keene, J.D.; Strittmatter, W.J.; Burke, J.R. Inhibition of Polyglutamine Protein Aggregation and Cell Death by Novel Peptides Identified by Phage Display Screening. J. Biol. Chem. 2000, 275, 10437–10442. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.R.; Feng, T.; Zhang, Q.; Chan, H.Y.E.; Chau, Y. Co-Encapsulation and Co-Delivery of Peptide Drugs via Polymeric Nanoparticles. Polymers 2019, 11, 288. [Google Scholar] [CrossRef] [Green Version]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 2–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef] [PubMed]
- Eck, R.J.; Kraemer, B.C.; Liachko, N.F. Regulation of TDP-43 Phosphorylation in Aging and Disease. GeroScience 2021, 43, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef]
- Vidal-Martinez, G.; Najera, K.; Miranda, J.D.; Gil-Tommee, C.; Yang, B.; Vargas-Medrano, J.; Diaz-Pacheco, V.; Perez, R.G. FTY720 Improves Behavior, Increases Brain Derived Neurotrophic Factor Levels and Reduces α-Synuclein Pathology in Parkinsonian GM2 +/− Mice. Neuroscience 2019, 411, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 Aggregation Induced by Oxidative Stress Causes Global Mitochondrial Imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, Y.; Katsuno, M.; Takagi, S.; Ishigaki, S.; Niwa, J.; Hasegawa, M.; Tanaka, F.; Sobue, G. Oxidative Stress Induced by Glutathione Depletion Reproduces Pathological Modifications of TDP-43 Linked to TDP-43 Proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective Effect of Chlorogenic Acid on Mitochondrial Dysfunction-Mediated Apoptotic Death of Da Neurons in a Parkinsonian Mouse Model. Oxid. Med. Cell. Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef]
- Zhao, N.; Yang, X.; Calvelli, H.R.; Cao, Y.; Francis, N.L.; Chmielowski, R.A.; Joseph, L.B.; Pang, Z.P.; Uhrich, K.E.; Baum, J.; et al. Antioxidant Nanoparticles for Concerted Inhibition of α-Synuclein Fibrillization, and Attenuation of Microglial Intracellular Aggregation and Activation. Front. Bioeng. Biotechnol. 2020, 8, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, H.; Meeran, M.F.N.; Azimullah, S.; Adem, A.; Sadek, B.; Ojha, S.K. Plant Extracts and Phytochemicals Targeting α-Synuclein Aggregation in Parkinson’s Disease Models. Front. Pharmacol. 2019, 9, 1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardah, M.T.; Ghanem, S.S.; Abdulla, S.A.; Lv, G.; Emara, M.M.; Paleologou, K.E.; Vaikath, N.N.; Lu, J.H.; Li, M.; Vekrellis, K.; et al. Inhibition of Alpha-Synuclein Seeded Fibril Formation and Toxicity by Herbal Medicinal Extracts. BMC Complement. Med. Ther. 2020, 20, 73. [Google Scholar] [CrossRef]
- Rehman, H.; Krishnasamy, Y.; Haque, K.; Thurman, R.G.; Lemasters, J.J.; Schnellmann, R.G.; Zhong, Z. Green Tea Polyphenols Stimulate Mitochondrial Biogenesis and Improve Renal Function after Chronic Cyclosporin A Treatment in Rats. PLoS ONE 2013, 8, e65029. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-Gallate Prevents Oxidative Phosphorylation Deficit and Promotes Mitochondrial Biogenesis in Human Cells from Subjects with Down’s Syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Godoi, G.L.; De Oliveira Porciúncula, L.; Schulz, J.F.; Kaufmann, F.N.; Da Rocha, J.B.; De Souza, D.O.G.; Ghisleni, G.; De Almeida, H.L. Selenium Compounds Prevent Amyloid β-Peptide Neurotoxicity in Rat Primary Hippocampal Neurons. Neurochem. Res. 2013, 38, 2359–2363. [Google Scholar] [CrossRef] [PubMed]
- Kuršvietienė, L.; Mongirdienė, A.; Bernatonienė, J.; Šulinskienė, J.; Stanevičienė, I. Selenium Anticancer Properties and Impact on Cellular Redox Status. Antioxidants 2020, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M.Y. Baicalein Attenuates α-Synuclein Aggregation, Inflammasome Activation and Autophagy in the MPP+-Treated Nigrostriatal Dopaminergic System in Vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhuang, X.; Lu, J. Neuroprotective Effects of Baicalein in Animal Models of Parkinson’s Disease: A Systematic Review of Experimental Studies. Phytomedicine 2019, 55, 302–309. [Google Scholar] [CrossRef]
- Zhang, X.; Du, L.; Zhang, W.; Yang, Y.; Zhou, Q.; Du, G. Therapeutic Effects of Baicalein on Rotenone-Induced Parkinson’s Disease through Protecting Mitochondrial Function and Biogenesis. Sci. Rep. 2017, 7, 9968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, N.N.; Cai, C.Z.; Wu, M.Y.; Su, H.X.; Li, M.; Lu, J.H. Neuroprotective Effects of Berberine in Animal Models of Alzheimer’s Disease: A Systematic Review of Pre-Clinical Studies. BMC Complement. Altern. Med. 2019, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Jia, Y.; Pan, D.; Ma, Z.G. Berberine Alleviates Rotenone-Induced Cytotoxicity by Antioxidation and Activation of PI3K/Akt Signaling Pathway in SH-SY5Y Cells. Neuroreport 2020, 31, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Su, P.; Lv, C.; Guo, L.; Cao, G.; Qin, C.; Zhang, W. Berberine Alleviates Amyloid β-Induced Mitochondrial Dysfunction and Synaptic Loss. Oxid. Med. Cell. Longev. 2019, 2019, 7593608. [Google Scholar] [CrossRef] [Green Version]
- Rajasekhar, K.; Samanta, S.; Bagoband, V.; Murugan, N.A.; Govindaraju, T. Antioxidant Berberine-Derivative Inhibits Multifaceted Amyloid Toxicity. iScience 2020, 23, 101005. [Google Scholar] [CrossRef]
- Kurisu, M.; Miyamae, Y.; Murakami, K.; Han, J.; Isoda, H.; Irie, K.; Shigemori, H. Inhibition of Amyloid β Aggregation by Acteoside, a Phenylethanoid Glycoside. Biosci. Biotechnol. Biochem. 2013, 77, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, Y.; Yan, J.; Zhao, X.; Sun, X.; Zhang, Y.; Guo, J.; Zhu, C. Acteoside Protects Human Neuroblastoma SH-SY5Y Cells against β-Amyloid-Induced Cell Injury. Brain Res. 2009, 1283, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cai, X.; Wang, R.; Zhai, S.; Zhang, Y.; Hu, W.; Zhang, Y.; Wang, D. Neuroprotective Effects of Verbascoside against Alzheimer’s Disease via the Relief of Endoplasmic Reticulum Stress in Aβ-Exposed U251 Cells and APP/PS1 Mice. J. Neuroinflammation 2020, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, D.; Luo, J.; Chen, X.; Gao, L.; Liu, W.; Chen, T. NIR-II-Activated Yolk-Shell Nanostructures as an Intelligent Platform for Parkinsonian Therapy. ACS Appl. Bio Mater. 2020, 3, 6876–6887. [Google Scholar] [CrossRef]
- Chen, W.; Ouyang, J.; Yi, X.; Xu, Y.; Niu, C.; Zhang, W.; Wang, L.; Sheng, J.; Deng, L.; Liu, Y.N.; et al. Black Phosphorus Nanosheets as a Neuroprotective Nanomedicine for Neurodegenerative Disorder Therapy. Adv. Mater. 2018, 30, 1703458. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Dong, K.; Zhao, A.; Sun, H.; Wang, Y.; Ren, J.; Qu, X. Polyoxometalate-Based Nanozyme: Design of a Multifunctional Enzyme for Multi-Faceted Treatment of Alzheimer’s Disease. Nano Res. 2016, 9, 1079–1090. [Google Scholar] [CrossRef]
- Chaudhary, H.; Iashchishyn, I.A.; Romanova, N.V.; Rambaran, M.A.; Musteikyte, G.; Smirnovas, V.; Holmboe, M.; Ohlin, C.A.; Svedružić, Ž.M.; Morozova-Roche, L.A. Polyoxometalates as Effective Nano-Inhibitors of Amyloid Aggregation of Pro-Inflammatory S100A9 Protein Involved in Neurodegenerative Diseases. ACS Appl. Mater. Interfaces 2021, 13, 26721–26734. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Mao, Y.; Xu, E.; Jia, H.; Zhang, S.; Dawson, V.L.; Dawson, T.M.; Li, Y.M.; Zheng, Z.; He, W.; et al. Nanozyme Scavenging ROS for Prevention of Pathologic α-Synuclein Transmission in Parkinson’s Disease. Nano Today 2021, 36, 101027. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, P.; Yue, C.; Jin, Z.; Liu, Q.; Du, X.; He, Q. Sustained Release of Bioactive Hydrogen by Pd Hydride Nanoparticles Overcomes Alzheimer’s Disease. Biomaterials 2019, 197, 393–404. [Google Scholar] [CrossRef]
- Robinson, A.P.; Zhang, J.Z.; Titus, H.E.; Karl, M.; Merzliakov, M.; Dorfman, A.R.; Karlik, S.; Stewart, M.G.; Watt, R.K.; Facer, B.D.; et al. Nanocatalytic Activity of Clean-Surfaced, Faceted Nanocrystalline Gold Enhances Remyelination in Animal Models of Multiple Sclerosis. Sci. Rep. 2020, 10, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, A.; Mondal, S.; Das, M.; Biswas, P.; Pal, U.; Darbar, S.; Bhattacharya, S.S.; Pal, D.; Saha-Dasgupta, T.; Das, A.K.; et al. Incorporation of a Biocompatible Nanozyme in Cellular Antioxidant Enzyme Cascade Reverses Huntington’s Like Disorder in Preclinical Model. Adv. Healthc. Mater. 2021, 10, 2001736. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Savanur, M.A.; Srivastava, S.; D’Silva, P.; Mugesh, G. A Redox Modulatory Mn3O4 Nanozyme with Multi-Enzyme Activity Provides Efficient Cytoprotection to Human Cells in a Parkinson’s Disease Model. Angew. Chemie Int. Ed. 2017, 56, 14267–14271. [Google Scholar] [CrossRef]
- DeCoteau, W.; Heckman, K.L.; Estevez, A.Y.; Reed, K.J.; Costanzo, W.; Sandford, D.; Studlack, P.; Clauss, J.; Nichols, E.; Lipps, J.; et al. Cerium Oxide Nanoparticles with Antioxidant Properties Ameliorate Strength and Prolong Life in Mouse Model of Amyotrophic Lateral Sclerosis. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, R.; De Giorgio, G.; Minato, I.; Bianchi, M.G.; Bussolati, O.; Marmiroli, N. Cerium Oxide Nanoparticles Rescue α-Synuclein-Induced Toxicity in a Yeast Model of Parkinson’s Disease. Nanomaterials 2020, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Li, M.; Dong, K.; Gao, N.; Ren, J.; Zheng, Y.; Qu, X. Ceria/POMs Hybrid Nanoparticles as a Mimicking Metallopeptidase for Treatment of Neurotoxicity of Amyloid-β Peptide. Biomaterials 2016, 98, 92–102. [Google Scholar] [CrossRef]
- Singh, N.; NaveenKumar, S.K.; Geethika, M.; Mugesh, G. A Cerium Vanadate Nanozyme with Specific Superoxide Dismutase Activity Regulates Mitochondrial Function and ATP Synthesis in Neuronal Cells. Angew. Chemie Int. Ed. 2021, 60, 3121–3130. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Li, D.; Zhou, Q.; Hu, X. Mitochondria-Targeted TPP-MoS2 with Dual Enzyme Activity Provides Efficient Neuroprotection through M1/M2 Microglial Polarization in an Alzheimer’s Disease Model. Biomaterials 2020, 232, 119752. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Qu, A.; Xu, L.; Sun, M.; Zhang, H.; Xu, C.; Kuang, H. Chiral Molecule-Mediated Porous Cu x O Nanoparticle Clusters with Antioxidation Activity for Ameliorating Parkinson’s Disease. J. Am. Chem. Soc. 2019, 141, 1091–1099. [Google Scholar] [CrossRef]
- Ren, C.; Hu, X.; Zhou, Q. Graphene Oxide Quantum Dots Reduce Oxidative Stress and Inhibit Neurotoxicity In Vitro and In Vivo through Catalase-Like Activity and Metabolic Regulation. Adv. Sci. 2018, 5, 1700595. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Zhou, T.; Cheng, K.; Chen, M.; Wang, Y.; Jiang, Y.; Yang, P. Carboxylic Acid Fullerene (C60) Derivatives Attenuated Neuroinflammatory Responses by Modulating Mitochondrial Dynamics. Nanoscale Res. Lett. 2015, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.S.; Hardt, J.I.; Quick, K.L.; Sook Kim-Han, J.; Erlanger, B.F.; Huang, T.T.; Epstein, C.J.; Dugan, L.L. A Biologically Effective Fullerene (C 60) Derivative with Superoxide Dismutase Mimetic Properties. Free Radic. Biol. Med. 2004, 37, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Gonchar, O.O.; Maznychenko, A.V.; Klyuchko, O.M.; Mankovska, I.M.; Butowska, K.; Borowik, A.; Piosik, J.; Sokolowska, I. C60 Fullerene Reduces 3-Nitropropionic Acid-Induced Oxidative Stress Disorders and Mitochondrial Dysfunction in Rats by Modulation of P53, Bcl-2 and Nrf2 Targeted Proteins. Int. J. Mol. Sci. 2021, 22, 5444. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Roy Choudhury, S.; Karmakar, S. Near-Infrared Responsive Dopamine/Melatonin-Derived Nanocomposites Abrogating in Situ Amyloid β Nucleation, Propagation, and Ameliorate Neuronal Functions. ACS Appl. Mater. Interfaces 2020, 12, 5658–5670. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gong, Y.; Liu, Y.; Huang, A.; Zhu, X.; Liu, J.; Yuan, G.; Zhang, L.; Wei, J.; Liu, J. Intelligently Thermoresponsive Flower-like Hollow Nano-Ruthenium System for Sustained Release of Nerve Growth Factor to Inhibit Hyperphosphorylation of Tau and Neuronal Damage for the Treatment of Alzheimer’s Disease. Biomaterials 2020, 237, 119822. [Google Scholar] [CrossRef] [PubMed]
- Gui, R.; Jin, H.; Wang, Z.; Li, J. Black Phosphorus Quantum Dots: Synthesis, Properties, Functionalized Modification and Applications. Chem. Soc. Rev. 2018, 47, 6795–6823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huynh, T.; Xiu, P.; Zhou, B.; Ye, C.; Luan, B.; Zhou, R. Revealing the Importance of Surface Morphology of Nanomaterials to Biological Responses: Adsorption of the Villin Headpiece onto Graphene and Phosphorene. Carbon N. Y. 2015, 94, 895–902. [Google Scholar] [CrossRef]
- Liang, M.; Yan, X. Nanozymes: From New Concepts, Mechanisms, and Standards to Applications. Acc. Chem. Res. 2019, 52, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.T.S.; Jarvet, J.; Zhao, L.; Jia, X.; Shankar, S.K.; Olofsson, A.; Brännström, T.; et al. The Role of Pro-Inflammatory S100A9 in Alzheimer’s Disease Amyloid-Neuroinflammatory Cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, I.; Iashchishyn, I.A.; Moskalenko, R.A.; Wang, C.; Wärmländer, S.K.T.S.; Wallin, C.; Gräslund, A.; Kovacs, G.G.; Morozova-Roche, L.A. Co-Aggregation of pro-Inflammatory S100A9 with α-Synuclein in Parkinson’s Disease: Ex Vivo and in Vitro Studies. J. Neuroinflamm. 2018, 15, 172. [Google Scholar] [CrossRef]
- Ha, T.Y.; Chang, K.A.; Kim, J.A.; Kim, H.S.; Kim, S.; Chong, Y.H.; Suh, Y.H. S100a9 Knockdown Decreases the Memory Impairment and the Neuropathology in Tg2576 Mice, AD Animal Model. PLoS ONE 2010, 5, e8840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braidy, N.; Lim, C.K.; Grant, R.; Brew, B.J.; Guillemin, G.J. Serum Nicotinamide Adenine Dinucleotide Levels through Disease Course in Multiple Sclerosis. Brain Res. 2013, 1537, 267–272. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C.; Menon, P.; Huynh, W.; Rynders, A.; Ho, K.S.; Glanzman, R.; Hotchkin, M.T. Study Protocol of RESCUE-ALS: A Phase 2, Randomised, Double-Blind, Placebo-Controlled Study in Early Symptomatic Amyotrophic Lateral Sclerosis Patients to Assess Bioenergetic Catalysis with CNM-Au8 as a Mechanism to Slow Disease Progression. BMJ Open 2021, 11, e041479. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Cheng, Y.; Zhou, M.; Zhao, S.; Lin, S.; Wang, X.; Wu, J.; Li, S.; Wei, H. ROS Scavenging Mn3O4 Nanozymes for: In Vivo Anti-Inflammation. Chem. Sci. 2018, 9, 2927–2933. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.C.; Johnson, M.E.; Walker, M.L.; Riley, K.R.; Sims, C.M. Antioxidant Cerium Oxide Nanoparticles in Biology and Medicine. Antioxidants 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Du, Y.; Zhang, K.; Liang, Z.; Li, J.; Yu, H.; Ren, R.; Feng, J.; Jin, Z.; Li, F.; et al. Tau-Targeted Multifunctional Nanocomposite for Combinational Therapy of Alzheimer’s Disease. ACS Nano 2018, 12, 1321–1338. [Google Scholar] [CrossRef]
- Heckman, K.L.; Decoteau, W.; Estevez, A.; Reed, K.J.; Costanzo, W.; Sanford, D.; Leiter, J.C.; Clauss, J.; Knapp, K.; Gomez, C.; et al. Custom Cerium Oxide Nanoparticles Protect against a Free Radical Mediated Autoimmune Degenerative Disease in the Brain. ACS Nano 2013, 7, 10582–10596. [Google Scholar] [CrossRef]
- Zand, Z.; Khaki, P.A.; Salihi, A.; Sharifi, M.; Nanakali, N.M.Q.; Alasady, A.A.B.; Aziz, F.M.; Shahpasand, K.; Hasan, A.; Falahati, M. Cerium Oxide NPs Mitigate the Amyloid Formation of α-Synuclein and Associated Cytotoxicity. Int. J. Nanomed. 2019, 14, 6989–7000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikheev, I.V.; Sozarukova, M.M.; Izmailov, D.Y.; Kareev, I.E.; Proskurnina, E.V.; Proskurnin, M.A. Antioxidant Potential of Aqueous Dispersions of Fullerenes C60, C70, and Gd@c82. Int. J. Mol. Sci. 2021, 22, 5838. [Google Scholar] [CrossRef] [PubMed]
- Chirico, F.; Fumelli, C.; Marconi, A.; Tinari, A.; Straface, E.; Malorni, W.; Pellicciari, R.; Pincelli, C. Carboxyfullerenes Localize within Mitochondria and Prevent the UVB-Induced Intrinsic Apoptotic Pathway. Exp. Dermatol. 2007, 16, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Quick, K.L.; Ali, S.S.; Arch, R.; Xiong, C.; Wozniak, D.; Dugan, L.L. A Carboxyfullerene SOD Mimetic Improves Cognition and Extends the Lifespan of Mice. Neurobiol. Aging 2008, 29, 117–128. [Google Scholar] [CrossRef]
- Dugan, L.L.; Turetsky, D.M.; Du, C.; Lobner, D.; Wheeler, M.; Almli, C.R.; Shen, C.K.F.; Luh, T.Y.; Choi, D.W.; Lin, T.S. Carboxyfullerenes as Neuroprotective Agents. Proc. Natl. Acad. Sci. USA 1997, 94, 9434–9439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugan, L.L.; Tian, L.L.; Quick, K.L.; Hardt, J.I.; Karimi, M.; Brown, C.; Loftin, S.; Flores, H.; Moerlein, S.M.; Polich, J.; et al. Carboxyfullerene Neuroprotection Postinjury in Parkinsonian Nonhuman Primates. Ann. Neurol. 2014, 76, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Protein Aggregates | Disease Model | Effect on Mitochondria | Refs |
|---|---|---|---|
| α-synuclein | PD | ↑mitochondrial ROS levels, ↓ ETC activity, ↓stability of mitochondrial membranes, ↑mPTP opening, ↓mitochondria-ER contacts, ↑DRP1 and ↓mitochondrial SIRT3 levels (a protective molecule of mitochondrial integrity and energetic function [84]) | [72,73,74,83,100] |
| Amyloid b | AD | ↑mitochondrial ROS levels, ↑mitochondrial fission (↓Mfn1, ↑DRP1 levels and ↑O-GlcNAcylation of DRP1) | [68,69,91, 92,94] |
| Tau | AD | ↑microtubule dissociation, ↑mitochondrial ROS levels, ↓ATP production, ↑mitochondrial fission and ↓mitophagy (interaction with DRP1 and PARKIN proteins) | [70,71,93] |
| Transactive response DNA-binding protein of 43 kDa (TDP-43) | AD, ALS | ↑mitochondrial ROS levels, ↓stability of mitochondrial structure, ↑mPTP opening | [61,75,95,96,97] |
| Huntingtin | HD | ↑mitochondrial ROS levels, ↓stability of mitochondrial structure, ↑mPTP opening, ↑mitochondrial fission by activation of DRP1, ↓mitophagy, ↑disruption Ca+2 flux between ER and mitochondria | [49,79, 80,101] |
| Superoxide dismutase | ALS | ↓mitophagy (by arresting optineurin protein), ↓stability of mitochondria structure, ↓flux of protein from and to the mitochondria | [102,103,104,105] |
| Nanosystem | Drug | Disease Model | Effect on Mitochondria and Neurodegeneration | Refs | ||||
|---|---|---|---|---|---|---|---|---|
| ROS Levels | Selective Localization of Drug | Cell Viability | Inflammation | Other Effects | ||||
| DQAsome/ P68 | NAC/ DFO | Cellular model of PD | ↓ (cellular and mitochondrial) | - | ↓neuronal death | - | ↑cellular antioxidant activity, ↓total non-ferritin-bound iron | [153] |
| CeO2 | Cerium | Cellular model of AD | ↓ (cellular and mitochondrial) | ↑ (mitochondrial) | ↓neuronal death | - | ↓DRP1 activation, ↓mitochondrial fission, ↓tyrosine nitration | [151] |
| CeO2-TPP | Cerium | Cellular model of AD | ↓ (cellular and mitochondrial) | ↑ (mitochondrial) | ↓neuronal death | ↓glial cell activation, ↓lipid peroxidation | ↑mitochondrial stabilization | [162] |
| CeO2-TPP | Cerium | PD mouse model | ↓ (cellular and mitochondrial) | ↑ (mitochondrial) | - | ↓glial cell activation, ↓lipid peroxidation | ↑mitochondrial stabilization, ↑brain tyrosine hydroxylase levels (enzyme that converts L-tyrosine into levodopa, a precursor of dopamine) | [163] |
| PLGA-bPEG-TPP | Curcumin | Cellular model of AD | - | ↑ (mitochondrial) | ↑neuronal viability | - | ↑endosomal and lysosomal escape | [168] |
| HSA/RBC/ t807/-TPP | Curcumin | Cellular and mouse model of AD | ↓ (cellular and mitochondrial) | ↑ (neuronal and mitochondrial) | ↑neuronal survival in hippocampus | ↓glial cell activation | ↑long-term circulation, ↑learning and memory function | [169] |
| Nanomicelles | TEMPO (4-amino-2,2,6,6-tetramethylpiperidinyloxy) | AD mouse model | ↓ (cellular) | - | - | ↓lipid peroxidation, ↓brain Aβ accumulation | ↓oxidative stress, ↑learning and memory function | [172] |
| Cellular model of PD | ↓ (cellular and mitochondrial) | - | ↑neuronal viability at pH 6,5 | - | ↑mitochondrial mass, ↑GSH content, ↑mitochondrial stabilization, ↑ATP levels | [173] | ||
| Nanomicelles | Carnosine/ lipoic acid | PD mouse model | - | - | - | ↓lipid peroxidation | ↑antioxidant activity in the brain tissue, ↑levels of dopamine and serotonin | [176] |
| Gold nanoparticles | Lipoic acid | Cellular model of PD | ↓ (cellular) | - | ↑neuronal viability | ↓lipid peroxidation | ↑ATP levels and mitochondrial respiratory activity, ↑cell membrane and microtubule stabilization | [177] |
| Nanosystem | Drug | Disease Model | Effect on Mitochondria and Neurodegeneration | Refs | ||
|---|---|---|---|---|---|---|
| Oxidative Stress | Energetic Function | Other Effects | ||||
| Nanomicellar CoQ10 | Ubiquinone | Fibroblasts from AD patients | ↓cellular and mitochondrial ROS | ↑ATP levels | delays the onset of premature senescence | [243] |
| PLGA NPs | Melatonin | Ischemia–reperfusion injury | ↓mitochondrial oxidative stress, restoration of SOD, CAT and GSH-Px activity | ↑activity of ETC complexes | ↑stability of mitochondrial membranes, ↑stability and survival of pyramidal neurons | [244] |
| SLN | Curcumin | HD rat model | ↓cellular ROS, lipid peroxidation and protein oxidation. ↑mitochondrial GSH and SOD activity | ↑activity of ETC complexes and cytochrome levels | ↓mitochondrial swelling, ↑neuromotor coordination | [245] |
| PLGA-PEG NPs | Huperzine A | AD rat model | - | - | ↑memory and cognitive recovery, ↑bioavailability | [251] |
| PLGA NPs | Pioglitazone | Cellular model of AD | - | ↑mitochondrial respiratory activity, ↑ATP levels | ↑brain bioavailability. Modulation of locomotor activity and brain energetic metabolism | [262] |
| Role of Nanosystem | Nanosystem Composition | Disease Model | Effect on Proteinopathies, Mitochondria and Neurodegeneration | Refs | |||
|---|---|---|---|---|---|---|---|
| Oxidative Stress | Protein Aggregates | Cell Viability | Other Effects | ||||
| Dissolution or inhibition of protein aggregation | Inorganic, Gold NPs | AD rat model | ↓oxidative markers, ↑antioxidant status (SOD and catalase activity and GSH levels) | ↓phosphorylation and Tau levels | - | ↓inflammatory markers, ↑ETC complex activity, ↑ATP levels, ↑cognitive functions | [288] |
| Cellular model of HD | - | ↓mHTT aggregation | - | ↓amyloid aggregation of insulin | [289] | ||
| Inorganic, porous silica NPs | Cellular model of HD | - | ↓aggregation and accumulation of mHTT | - | ↓amount of aggregated amyloid of insulin and ↓insulin amyloid fibrillation | [289] | |
| Organic, graphene nanosystem (GQD, GO) | Cellular AD model | - | ↓Aβ aggregation, | ↑cell viability from Aβ-mediated cellular toxicity | ↑preservation of mitochondrial function | [290,291,292] | |
| Cellular and mouse models of PD | ↓cellular and mitochondrial ROS induced by α-syn in neurons | ↓α-syn aggregation, ↑dissociation of α-syn fibrils | ↓loss of dopaminergic neurons induced by α-syn | ↓mitochondrial damage in α-syn-treated primary cortical neurons, ↑preservation of mitochondrial function, ↓glial activation, ↓motor deficits | [293, 294] | ||
| Organic, PLGA/polysorbate80- QBP1 | Cellular and fly models of HD | - | ↓mHTT aggregation | - | ↑motor performance in Drosophila model of HD | [295] | |
| Degradation of toxic proteins | Organic, GO nanosheet | Cellular model of AD | - | ↓accumulation of hippocampal Aβ aggregates, ↑Aβ delivery to lysosomes for degradation | - | ↓β-cleavage of amyloid precursor protein (APP) by BACE1 activity | [296] |
| Organic, Polydopamine NPs/ metformin | Cellular model of PD | ↓cellular ROS levels | ↑proteasomal degradation of phosphorylated α-syn | ↓neuron loss induced by rotenone | ↓inflammatory markers, ↑stabilization of mitochondrial membranes | [297] | |
| Inhibition of protein phosphorylation | Chitosan NPs/ Fingolimod | Cellular model of PD | ↓cellular ROS levels | ↓phosphorylation and accumulation of α-syn | ↑cell viability of rotenone-treated neurons | ↑stability of mitochondrial membranes | [298] |
| PLGA NPs/PHA-767491 | Cellular model of ALS | ↓cellular and mitochondrial ROS levels | ↓phosphorylation and accumulation of TDP-43 | ↓loss of dopaminergic neurons | ↑permeability through BBB in a cellular model | [299] | |
| Therapeutics against protein aggregation based on natural compounds | PLA-PEG NPs/EGCG | AD rat model | ↓cellular ROS levels | ↓accumulation of Aβ aggregates in the hippocampus | - | ↑locomotor and cognitive abilities | [300] |
| Micellar SPIONS/ EGCG | PD mouse model | EGCG release upon oxidative conditions | ↓α-syn aggregation | ↑dopaminergic neurons | ↑permeation through a BBB model, ↑motor and cognitive abilities | [301] | |
| PLGA-PEG NPs/EGCG | AD mouse model | - | ↓levels of soluble and insoluble Aβ peptide | - | ↑permeation through a BBB model, ↓glial activation in cortex and hippocampus, ↑learning and memory abilities | [302] | |
| SeNPs/CGA or QRC | Cellular model of AD | ↓ROS levels | ↓Aβ aggregation | ↓cell death induced by Aβ | ↑mitochondrial stability, ↓release of mitochondrial lactate dehydrogenase | [303, 304] | |
| Liposomal-polysorbate80/ curcumin | PD mouse model | - | ↓α-syn accumulation in brain tissue | - | ↑curcumin brain accumulation and circulation lifetime, ↑dopamine levels, ↑motor function | [305] | |
| PEG- carbonyl curcumin | Cellular and mouse model of PD | - | ↑α-syn lysosomal degradation in vitro, ↓α-syn accumulation in brain tissue | ↓cell death of dopaminergic neurons | ↑memory and motor function | [306] | |
| PEG- cholesterol-Nanoliposomes/ baicalein | Cellular and mouse model of PD | ↓ROS levels in cellular model of PD | ↓α-syn fibrillation, ↑disaggregation of α-syn fibrils | ↑neurite growth, ↑dopamine levels and stability of dopaminergic neurons | ↑motor function in PD mouse model | [307] | |
| Chitosan NPs/berberine | AD rat model | ↓oxidative stress in brain tissue | ↓levels of Aβ and Tau | - | ↑learning and memory abilities | [308] | |
| Chitosan-PLA-PEG-NGF NPs/ Acteoside | PD mouse model | - | ↓α-syn aggregation in cells and brain of PD mice | ↑stability of dopaminergic neurons | ↑dopamine levels of dopaminergic neurons, ↑motor function | [309] | |
| Nanomaterial | Nanosystem Composition | Disease Model | Effect on Mitochondria and Neurodegeneration | Refs | ||||
|---|---|---|---|---|---|---|---|---|
| Oxidative Stress | Mitochondrial Function | Cell Viability | Protein Aggregates | Other Effects | ||||
| Photothermal | Gold nanorods coated by mesoporous silica and associated to quercetin | Cellular and mouse models of PD | - | ↑ATP levels, ↑stability of mitochondrial membranes | ↑preservation of dopaminergic neurons | - | ↑permeability through a BBB model, ↑brain accumulation, ↑striatal dopamine levels, ↑motor performance | [342] |
| Black Phosphorous | Cellular and mouse models of Cu toxicity | ↓cellular oxidative stress and damage | ↑stability of mitochondrial membranes | ↑neural cell viability under oxidative stress conditions | - | ↑permeability through a BBB model, ↑brain accumulation | [343] | |
| Cellular and mouse models of PD | ↑antioxidant status (↓lipid oxidation, ↑SOD activity and GSH levels) | ↑mitochondrial localization | ↑preservation of dopaminergic neurons | - | ↑permeability through a BBB model, ↑brain accumulation, ↑dopamine levels, ↑motor function | [197] | ||
| Metallic nanozymes | Polyoxometalates | Cellular and cell-free models of AD | ↓cell ROS levels induced by Aβ, ↑ability to scavenge oxidant Cu ions, ↑SOD-like activity | - | ↑cell viability under Aβ toxicity | ↑degradation of Aβ fibrils and aggregates, ↓assembly of amyloid S100A9 protein | ↑permeability through a BBB model | [344, 345] |
| Platinum-copper/ polyvinyl pyrrolidone | Cellular and mouse models of PD | ↓cell ROS levels, ↑activity of antioxidant-like enzymes | - | ↑cellular neuroprotection by expression of NeuN protein | ↓α-syn aggregation and spreading | - | [346] | |
| Palladium hydride | AD mouse model | ↓cellular ROS levels | ↑expression of ETC complex IV, ↑mitochondrial respiratory activity, ↓levels of fission protein DRP1, ↑expression of fusion protein Mfn2 | ↓apoptotic pathway | ↓Aβ aggregation | ↑cognitive function | [347] | |
| Nanocrystals of gold (CNM-Au8) | Cellular and mouse models of MS | - | ↑ATP and NAD+ levels in oligodendrocyte precursor cells | ↑differentiation of oligodendrocytes from precursor cells | - | ↑remyelinating activity, ↑expression of myelin synthesis-related genes, ↑motor functions | [348] | |
| Metal oxide nanozyme | Flower-like Mn3O4 | Cellular model of PD and mouse model of HD | ↓mitochondrial ROS and lipid oxidation, ↑GSH peroxidase-like activity and brain antioxidant activity | ↑brain activity of ETC complexes and ATP levels, ↓mitochondrial swelling and mPTP opening, ↑stability of mitochondrial membranes | ↑preservation of brain cell structure, ↓focal degeneration of neural cells | - | ↑motor and cognitive functions, ↓anxious behavior | [349, 350] |
| Cerium oxide NPs | Mouse model of ALS | ↑catalase-like activity in cell-free system, ↓oxidative stress in brain tissue | - | ↑cell viability under oxidative conditions | - | ↑muscle strength, motor function and mouse lifespan | [351] | |
| Yeast model of PD | ↓ROS levels | ↓mitochondrial fission | ↑cell viability under α-syn damage | ↓α-syn aggregation and accumulation | - | [352] | ||
| Cellular model of AD | ↓oxidative stress and formation of peroxynitrite | ↓mitochondrial fission, ↓DRP1 activation | ↓neuronal cell death under oxidative stress and Aβ treatment | - | ↑localization in mitochondria, ↑protein tyrosine nitration | [151] | ||
| Cerium oxide polyoxometalates | Cell-free system and cellular model of AD | ↑SOD-like activity, ↓cellular ROS | - | ↑cell viability under Aβ cytotoxicity | ↑degradation of Aβ monomers and disaggregation of Aβ fibrils, ↓Aβ aggregation | ↓microglial activation | [353] | |
| Nanorods of CeVO4 | Cellular model of ALS | ↑SOD-like activity, ↓cellular and mitochondrial ROS | ↑ATP levels, ↑stability of mitochondrial membranes | ↑cell viability and ↓apoptotic pathway | - | - | [354] | |
| MoS2-TPP | Cellular and mouse models of AD | ↑Activity of antioxidant-like enzymes, ↓cellular and mitochondrial ROS | ↑mitochondrial localization, ↓structural alteration and degradation of mitochondria, ↑mitophagy | ↑levels of neuroprotective protein NEuN | ↓Aβ aggregation and deposition in hippocampus | ↑permeation through a BBB model, ↑microglial transition from pro- inflammatory M1 to anti- inflammatory M2 state | [355] | |
| CuxO NPs | Cellular and mouse models of PD | ↑Activity of antioxidant-like enzymes, ↓ROS and lipid oxidation levels | - | ↑cell viability in PD cells model, ↑viability of dopaminergic neurons in PD mice, ↓apoptotic pathway | - | ↑dopamine levels, ↑memory function | [356] | |
| Carbon-based nanozymes | GQD and GOQD | Cellular and animal models of PD | ↓ROS levels, ↑activity of antioxidant-like enzymes | ↑stability of mitochondrial structure in brain cells | ↓dopaminergic neuron loss, ↓apoptotic pathway | ↓α-syn and Aβ aggregation, ↑dissociation of α-syn fibrils | ↑locomotor function | [294, 357] |
| Carboxyfullerene (C60) | Cellular neuroinflammatory and animal models of PD and HD | ↓ROS levels, ↑ROS scavenging and GSH levels in mitochondria from HD rats | ↑Activity of ETC complexes, ↑stability of mitochondrial membranes, ↓mitochondrial fragmentation and DRP1 activity | ↑stability and preservation of dopaminergic neurons, ↓apoptotic pathway | - | ↓inflammatory markers, ↑mouse lifespan, ↑dopamine levels and motor function in a PD primate model | [358,359,360] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, L.F.; Bevilacqua, L.E.; Naves, R. Nanotechnology-Based Drug Delivery Strategies to Repair the Mitochondrial Function in Neuroinflammatory and Neurodegenerative Diseases. Pharmaceutics 2021, 13, 2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122055
González LF, Bevilacqua LE, Naves R. Nanotechnology-Based Drug Delivery Strategies to Repair the Mitochondrial Function in Neuroinflammatory and Neurodegenerative Diseases. Pharmaceutics. 2021; 13(12):2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122055
Chicago/Turabian StyleGonzález, Luis F., Lorenzo E. Bevilacqua, and Rodrigo Naves. 2021. "Nanotechnology-Based Drug Delivery Strategies to Repair the Mitochondrial Function in Neuroinflammatory and Neurodegenerative Diseases" Pharmaceutics 13, no. 12: 2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122055