
Identification of Phytoconstituents as Potent Inhibitors of Casein Kinase-1 Alpha Using Virtual Screening and Molecular Dynamics Simulations


 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computer Environment and Web Resources
2.2. Receptor Preparation and Library Preparation
2.3. Molecular Docking-Based Virtual Screening
2.4. ADMET Prediction
2.5. PASS Evaluation
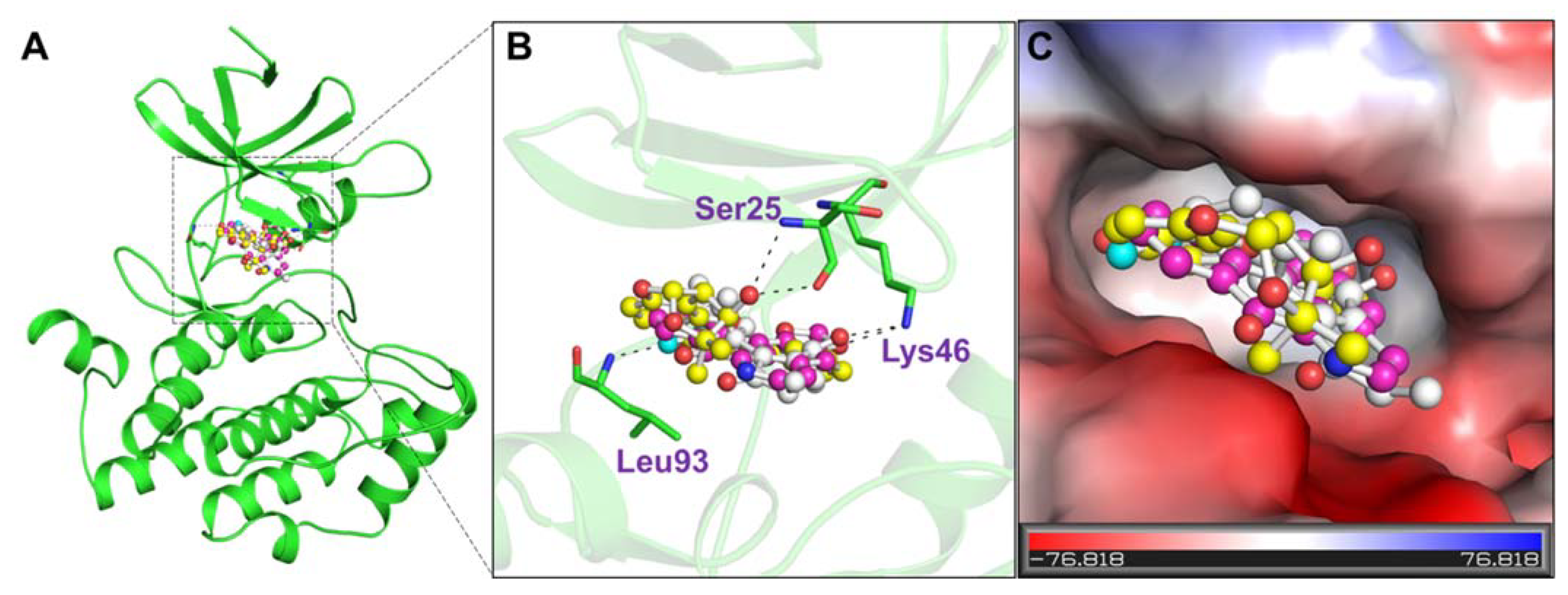
2.6. Interaction Analysis
2.7. MD Simulations
2.7.1. Systems Preparation and Simulation Protocol
2.7.2. Dynamical Cross-Correlation Matrix
2.7.3. Principal Component Analysis
2.7.4. MM-PBSA Calculations
3. Results and Discussion
3.1. Molecular Docking-Based Virtual Screening
3.2. ADMET Properties
3.3. PASS Evaluation
3.4. Interaction Analysis
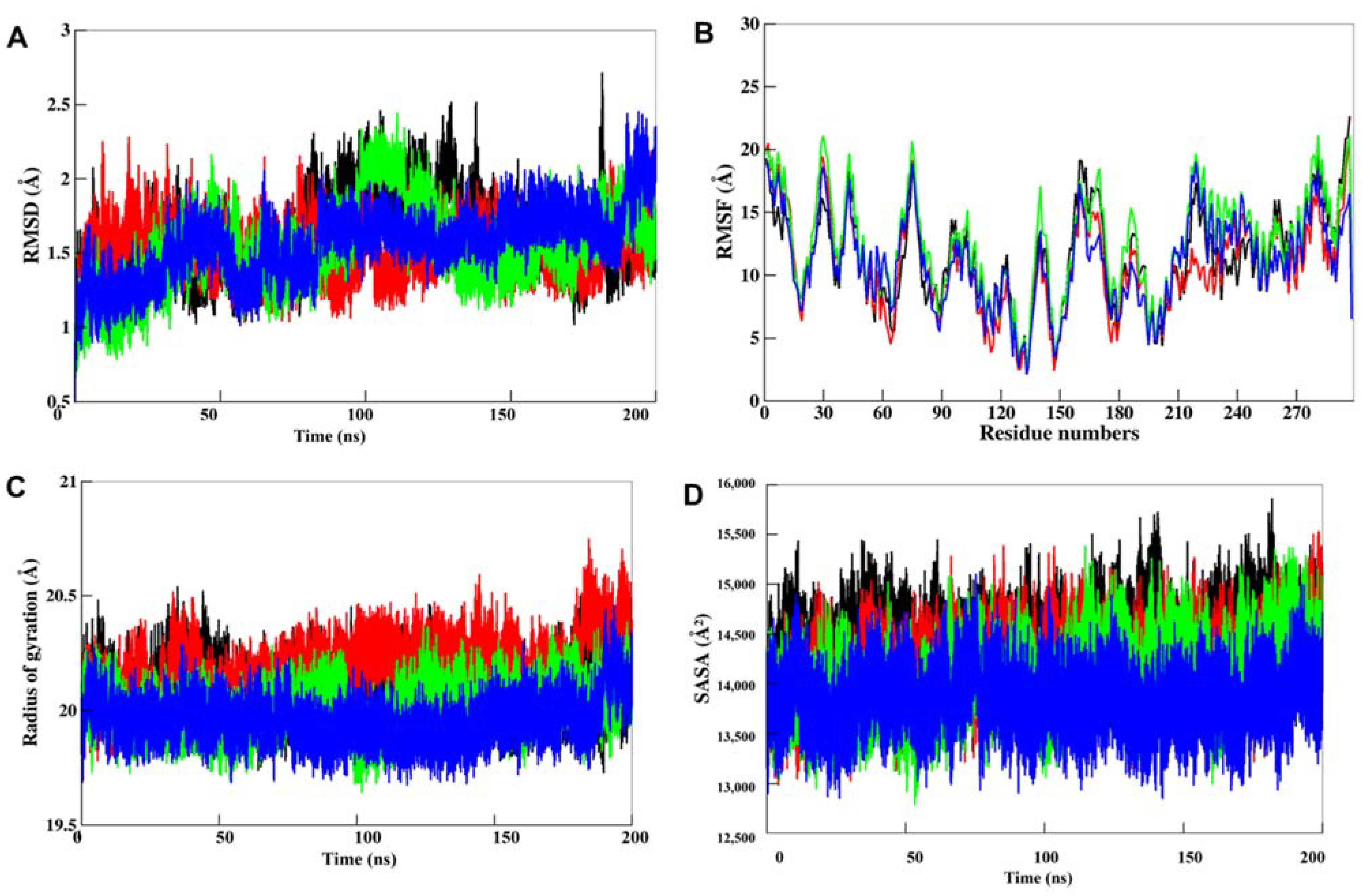
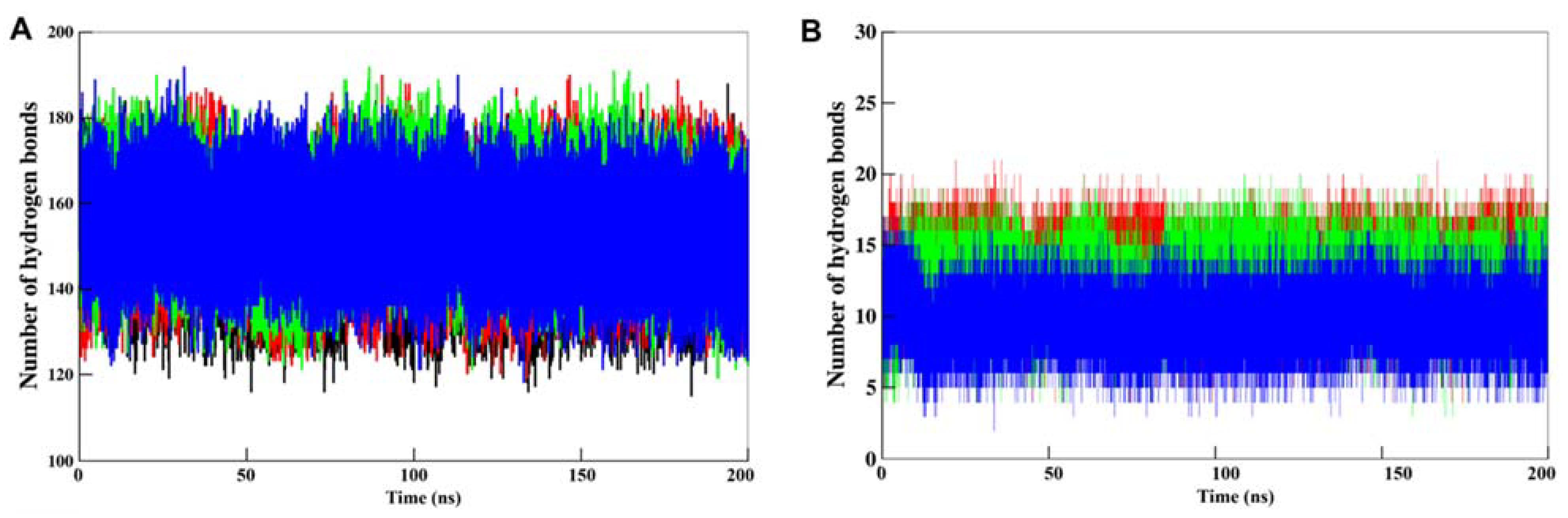
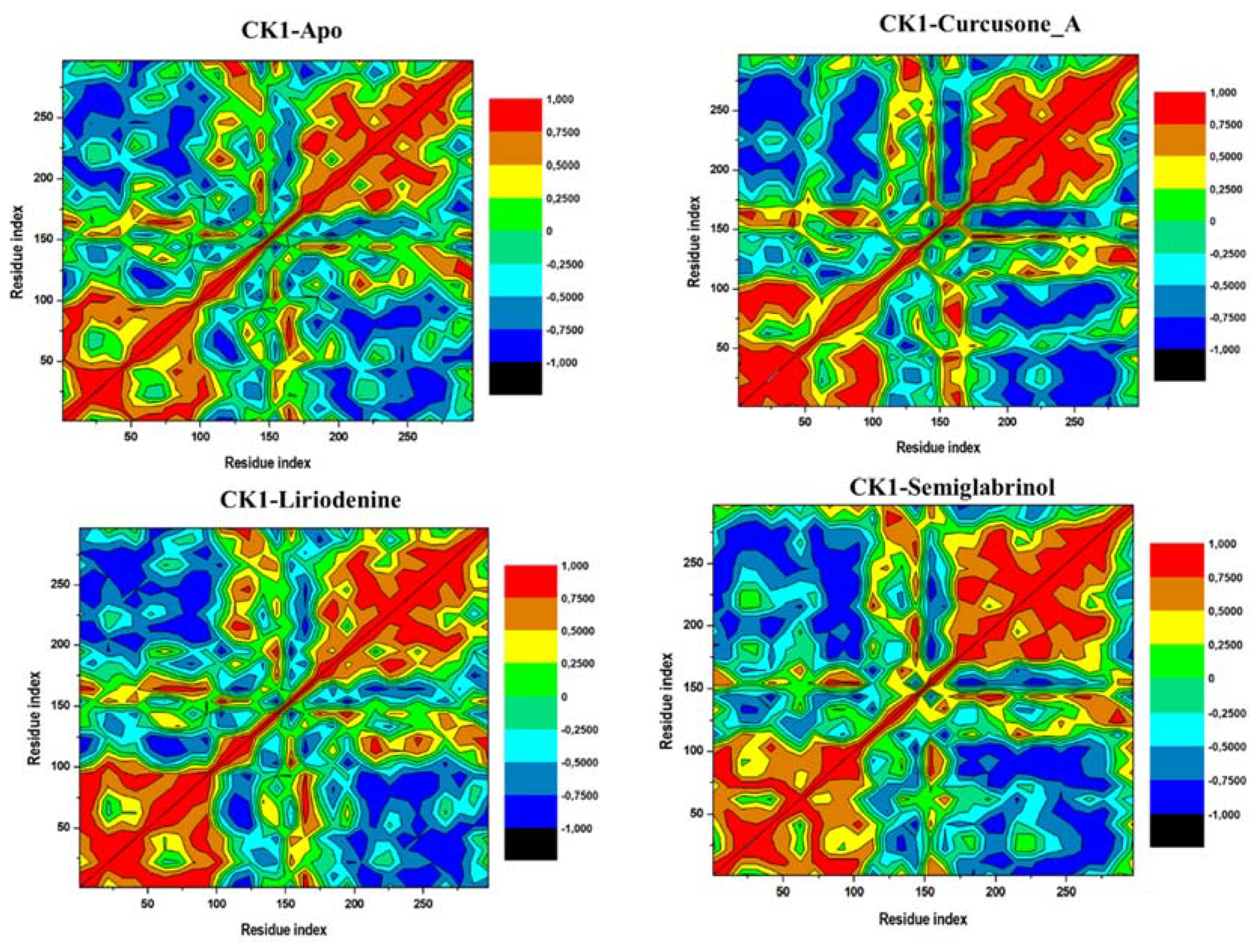
3.5. MD Simulations
3.5.1. Structural Deviations in CK1α
3.5.2. Dynamics of Hydrogen Bonds
3.5.3. Secondary Structure Dynamics
3.5.4. DCCM
3.5.5. PCA
3.5.6. MMPBSA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Byun, K.; Kim, J.Y.; Bayarsaikhan, E.; Kim, D.; Jeong, G.-B.; Na Yun, K.; Min, H.K.; Kim, S.U.; Yoo, J.S.; Lee, B. Quantitative proteomic analysis reveals that lipopolysaccharide induces mitogen-activated protein kinase-dependent activation in human microglial cells. Electrophoresis 2012, 33, 3756–3763. [Google Scholar] [CrossRef]
- Chun, K.; Cho, S.; Lee, J.; Seo, J.H.; Kim, K.; Lee, S. Protein kinase C-δ interacts with and phosphorylates ARD1. J. Cell. Physiol. 2021, 236, 379–391. [Google Scholar] [CrossRef]
- Goh, Y.-M.; Cinghu, S.; Hong, E.T.H.; Lee, Y.-S.; Kim, J.-H.; Jang, J.-W.; Li, Y.-H.; Chi, X.-Z.; Lee, K.-S.; Wee, H.; et al. Src Kinase Phosphorylates RUNX3 at Tyrosine Residues and Localizes the Protein in the Cytoplasm. J. Biol. Chem. 2010, 285, 10122–10129. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-H.; Hwang, J.H.; Kim, K.-S.; Noh, J.-R.; Gang, G.-T.; Oh, W.K.; Jeong, K.-H.; Kwak, T.H.; Choi, H.-S.; Lee, I.-K.; et al. Enhanced activation of NAD(P)H. J. Hypertens. 2014, 32, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, D.-S.; Kim, C.-E.; Shin, M.-S.; Seo, C.-S.; Shin, H.-K.; Hwang, G.S.; An, J.M.; Kim, S.-N.; Kang, K.S. Effects of fermented black ginseng on wound healing mediated by angiogenesis through the mitogen-activated protein kinase pathway in human umbilical vein endothelial cells. J. Ginseng Res. 2018, 42, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, H.-H.; Jung, Y.-S.; Kang, S.-J.; Cheong, H.-K.; Song, H.-K.; Lee, B.-J. Backbone resonances assignment of 19 kDa CD1 domain of human mitotic checkpoint serine/threonine-protein kinase, Bub1. Biomol. NMR Assign. 2011, 6, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M. Casein kinase 1 and Wnt/β-catenin signaling. Curr. Opin. Cell Biol. 2014, 31, 46–55. [Google Scholar] [CrossRef]
- Choi, J.-S.; Bae, W.-Y.; Nam, S.; Jeong, J.-W. New Targets for Parkinson’s Disease: Adhesion G Protein-Coupled Receptor B1 is Downregulated by AMP-Activated Protein Kinase Activation. OMICS A J. Integr. Biol. 2018, 22, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Baek, S.Y.; Jang, E.J.; Ku, S.K.; Kim, K.M.; Ki, S.H.; Kim, C.-E.; Park, K.I.; Kim, S.C.; Kim, Y.W. Oxyresveratrol ameliorates nonalcoholic fatty liver disease by regulating hepatic lipogenesis and fatty acid oxidation through liver kinase B1 and AMP-activated protein kinase. Chem. Interact. 2018, 289, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Choi, A.Y.; Oh, Y.T.; Choe, W.; Yeo, E.-J.; Ha, J.; Kang, I. Corrigendum to “AMP-activated protein kinase mediates T cell activation-induced expression of FasL and COX-2 via protein kinase C theta-dependent pathway in human Jurkat T leukemia cells”. Cell. Signal. 2018, 52, 163. [Google Scholar] [CrossRef]
- Lee, S.-H.; Ko, S.-C.; Kang, M.-C.; Lee, D.H.; Jeon, Y.-J. Octaphlorethol A, a marine algae product, exhibits antidiabetic effects in type 2 diabetic mice by activating AMP-activated protein kinase and upregulating the expression of glucose transporter 4. Food Chem. Toxicol. 2016, 91, 58–64. [Google Scholar] [CrossRef]
- Son, E.S.; Kyung, S.Y.; Lee, S.P.; Jeong, S.H.; Shin, J.Y.; Ohba, M.; Yeo, E.J.; Park, J.W. Role of protein kinase C-η in cigarette smoke extract-induced apoptosis in MRC-5-cells. Hum. Exp. Toxicol. 2015, 34, 869–877. [Google Scholar] [CrossRef]
- Son, K.H.; Park, C.H.; Park, K.Y.; Choi, C.H. Which Variables Should be Considered as Confounders of p38-Mitogen Activated Protein Kinase Activation Measurements? Ann. Thorac. Surg. 2016, 102, 1764–1765. [Google Scholar] [CrossRef] [PubMed]
- Lantermann, A.B.; Chen, D.; McCutcheon, K.; Hoffman, G.; Frias, E.; Ruddy, D.; Rakiec, D.; Korn, J.; McAllister, G.; Stegmeier, F. Inhibition of casein kinase 1 alpha prevents acquired drug resistance to Erlotinib in EGFR-mutant non–small cell lung cancer. Cancer Res. 2015, 75, 4937–4948. [Google Scholar] [CrossRef] [Green Version]
- Janovská, P.; Normant, E.; Miskin, H.; Bryja, V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int. J. Mol. Sci. 2020, 21, 9026. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Wolff, S.; Giamas, G.; Brockschmidt, C.; Wittau, M.; Würl, P.U.; Eismann, T.; Stöter, M. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Oncol. Res. Treat. 2005, 28, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, M.; Sun, J.; Yang, X. Casein kinase 1α: Biological mechanisms and theranostic potential. Cell Commun. Signal. 2018, 16, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Kim, D.Y.; Cheon, H.G. Clomiphene promotes browning of white adipocytes via casein kinase-2 inhibition. Eur. J. Pharmacol. 2019, 861, 172596. [Google Scholar] [CrossRef]
- Tran, N.; Chun, K.-H. ROCK2-Specific Inhibitor KD025 Suppresses Adipocyte Differentiation by Inhibiting Casein Kinase 2. Molecules 2021, 26, 4747. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-M.; Carmel, G.; Sweet, R.M.; Kuret, J.; Cheng, X. Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J. 1995, 14, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A. Small molecules co-targeting CKIα and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 2018, 175, 171–185. e125. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.M.; Yang, E.H.; Quan, W.; Nam, E.H.; Cheon, H.G. Discovery of a novel fibroblast activation protein (FAP) inhibitor, BR103354, with anti-diabetic and anti-steatotic effects. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-M.; Kim, M.-S.; Hayat, F.; Shin, D. Recent advances in the discovery of novel antiprotozoal agents. Molecules 2019, 24, 3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.; Ahn, J.; Lee, T.; Jang, G.; Park, C.; Yoon, Y. Drug voyager: A computational platform for exploring unintended drug action. BMC Bioinform. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jairajpuri, D.S.; Mohammad, T.; Adhikari, K.; Gupta, P.; Hasan, G.M.; Alajmi, M.F.; Rehman, M.T.; Hussain, A.; Hassan, M.I. Identification of sphingosine kinase-1 inhibitors from bioactive natural products targeting cancer therapy. ACS Omega 2020, 5, 14720–14729. [Google Scholar] [CrossRef]
- Mohammad, T.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification and evaluation of bioactive natural products as potential inhibitors of human microtubule affinity-regulating kinase 4 (MARK4). J. Biomol. Struct. Dyn. 2019, 37, 1813–1829. [Google Scholar] [CrossRef]
- Alam, M.; Ali, S.; Mohammad, T.; Hasan, G.M.; Yadav, D.K.; Hassan, M. B Cell Lymphoma 2: A Potential Therapeutic Target for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 10442. [Google Scholar] [CrossRef]
- Choi, J.; Choi, K.-E.; Park, S.J.; Kim, S.Y.; Jee, J.-G. Ensemble-Based Virtual Screening Led to the Discovery of New Classes of Potent Tyrosinase Inhibitors. J. Chem. Inf. Model. 2016, 56, 354–367. [Google Scholar] [CrossRef]
- Jang, C.; Yadav, D.K.; Subedi, L.; Venkatesan, R.; Venkanna, A.; Afzal, S.; Lee, E.; Yoo, J.; Ji, E.; Kim, S.Y.; et al. Identification of novel acetylcholinesterase inhibitors designed by pharmacophore-based virtual screening, molecular docking and bioassay. Sci. Rep. 2018, 8, 14921. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Cho, S.J.; Kim, M.-H. Discovery of CNS-Like D3R-Selective Antagonists Using 3D Pharmacophore Guided Virtual screening. Molecules 2018, 23, 2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoichet, B.K. Virtual screening of chemical libraries. Nat. Cell Biol. 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.K.; Kumar, S.; Yadav, D.K.; Kim, M.H. In silico identification of prolyl hydroxylase inhibitor by per-residue energy decomposition-based pharmacophore approach. J. Cell. Biochem. 2021, 122, 1098–1112. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Choi, E.-H.; Chaudhary, S.; Kim, M.-H. Computational modeling on aquaporin-3 as skin cancer target: A virtual screening study. Front. Chem. 2020, 8, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseem, R.; Anwar, S.; Khan, S.; Shamsi, A.; Hassan, M.; Anjum, F.; Shafie, A.; Islam, A.; Yadav, D.K. MAP/Microtubule Affinity Regulating Kinase 4 Inhibitory Potential of Irisin: A New Therapeutic Strategy to Combat Cancer and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10986. [Google Scholar] [CrossRef]
- Teli, M.K.; Kumar, S.; Yadav, D.K.; Kim, M.-H. In silico identification of hydantoin derivatives: A novel natural prolyl hydroxylase inhibitor. J. Biomol. Struct. Dyn. 2021, 39, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Koparde, A.A.; Doijad, R.C.; Magdum, C.S. Natural products in drug discovery. In Pharmacognosy-Medicinal Plants; IntechOpen: London, UK, 2019. [Google Scholar]
- Reddy, L.; Odhav, B.; Bhoola, K. Natural products for cancer prevention: A global perspective. Pharmacol. Ther. 2003, 99, 1–13. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.11–8.14.40. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Mathur, Y.; Hassan, M.I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Brief Bioinform. 2021, 22, bbaa279. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Dassault Systemes BIOVIA. Discovery Studio, Accelrys [2.1]; Dassault Systemes BIOVIA: San Diego, CA, USA, 2008. [Google Scholar]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.B.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of I ndian M edicinal P lants, P hytochemistry A nd T herapeutics. Sci. Rep. 2018, 8, 1–17. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Rai, R.; Dutta, R.K.; Singh, S.; Yadav, D.K.; Kumari, S.; Singh, H.; Gupta, R.D.; Pratap, R. Synthesis, biological evaluation and molecular docking study of 1-amino-2-aroylnaphthalenes against prostate cancer. Bioorganic Med. Chem. Lett. 2018, 28, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Poornima, B.; Siva, B.; Venkanna, A.; Shankaraiah, G.; Jain, N.; Yadav, D.K.; Misra, S.; Babu, K.S. Novel Gomisin B analogues as potential cytotoxic agents: Design, synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 2017, 139, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Mohammad, T.; Prasad, K.; Hasan, G.M.; Kumar, V.; Dohare, R.; Islam, A.; Ahmad, F.; Imtaiyaz Hassan, M. Virtual high-throughput screening of natural compounds in-search of potential inhibitors for protection of telomeres 1 (POT1). J. Biomol. Struct. Dyn. 2020, 38, 4625–4634. [Google Scholar] [CrossRef]
- Mohammad, T.; Siddiqui, S.; Shamsi, A.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M. Virtual screening approach to identify high-affinity inhibitors of serum and glucocorticoid-regulated kinase 1 among bioactive natural products: Combined molecular docking and simulation studies. Molecules 2020, 25, 823. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Jaiswal, P.K.; Kumar, S.; Mathur, M.; Swami, A.K.; Yadav, D.K.; Chaudhary, S. Discovery of Aporphine Analogues as Potential Antiplatelet and Antioxidant Agents: Design, Synthesis, Structure-Activity Relationships, Biological Evaluations, and in silico Molecular Docking Studies. ChemMedChem 2018, 13, 1817–1832. [Google Scholar] [CrossRef]
- Gimeno, A.; Montes, M.J.O.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, M.K.; Coghi, P.; Agrawal, P.; Shyamlal, B.R.K.; Yang, L.J.; Yadav, L.; Peng, Y.; Sharma, R.; Yadav, D.K.; Sahal, D. Design, Synthesis, Structure-Activity Relationship and Docking Studies of Novel Functionalized Arylvinyl-1, 2, 4-Trioxanes as Potent Antiplasmodial as well as Anticancer Agents. ChemMedChem 2020, 15, 1216–1228. [Google Scholar] [CrossRef]
- Ahmed, N.; Anwar, S.; Thet Htar, T. Docking based 3D-QSAR study of tricyclic guanidine analogues of batzelladine K as anti-malarial agents. Front. Chem. 2017, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Kalani, K.; Yadav, D.K.; Alam, S.; Khan, F.; Kashyap, M.P.; Srivastava, S.K.; Pant, A.B. In-silico Studies and Wet-Lab Validation of Camptothecin Derivatives for Anti-Cancer Activity Against Liver (HepG2) and Lung (A549) Cancer Cell Lines. Curr. Top. Med. Chem. 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Kim, H.-H.; Hyun, J.-S.; Choi, J.; Choi, K.-E.; Jee, J.-G.; Park, S.J. Structural ensemble-based docking simulation and biophysical studies discovered new inhibitors of Hsp90 N-terminal domain. Sci. Rep. 2018, 8, 1–13. [Google Scholar]
- Guleria, V.; Pal, T.; Sharma, B.; Chauhan, S.; Jaiswal, V. Pharmacokinetic and molecular docking studies to design antimalarial compounds targeting Actin I. Int. J. Health Sci. 2021, 15, 4–13. [Google Scholar]
- Yadav, D.K.; Kumar, S.; Teli, M.K.; Kim, M.H. Ligand-based pharmacophore modeling and docking studies on vitamin D receptor inhibitors. J. Cell. Biochem. 2020, 121, 3570–3583. [Google Scholar] [CrossRef]
- Hirte, M.; Meese, N.; Mertz, M.; Fuchs, M.; Brück, T.B. Insights into the bifunctional aphidicolan-16-ß-ol synthase through rapid biomolecular modeling approaches. Front. Chem. 2018, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, R.; Sharmila, G.; Elumalai, P.; Senthilkumar, K.; Banudevi, S.; Gunadharini, D.; Benson, C.; Daisy, P.; Arunakaran, J. Effect of diallyl disulfide on insulin-like growth factor signaling molecules involved in cell survival and proliferation of human prostate cancer cells in vitro and in silico approach through docking analysis. Phytomedicine 2012, 19, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham III, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- He, X.; Liu, S.; Lee, T.-S.; Ji, B.; Man, V.H.; York, D.M.; Wang, J. Fast, accurate, and reliable protocols for routine calculations of protein–ligand binding affinities in drug design projects using AMBER GPU-TI with ff14SB/GAFF. ACS Omega 2020, 5, 4611–4619. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Fataftah, H.; Karain, W. Detecting protein atom correlations using correlation of probability of recurrence. Proteins: Struct. Funct. Bioinform. 2014, 82, 2180–2189. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. In Protein Dynamics; Humana Press: Totowa, NJ, USA, 2014; pp. 193–226. [Google Scholar]
- Amir, M.; Mohammad, T.; Kumar, V.; AlAjmi, M.; Rehman, M.T.; Hussain, A.; Alam, P.; Dohare, R.; Islam, A.; Ahmad, F. Structural analysis and conformational dynamics of STN1 gene mutations involved in coat plus syndrome. Front. Mol. Biosci. 2019, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Jairajpuri, D.S.; Hussain, A.; Nasreen, K.; Mohammad, T.; Anjum, F.; Rehman, T.; Hasan, G.M.; Alajmi, M.F.; Hassan, I. Identification of natural compounds as potent inhibitors of SARS-CoV-2 main protease using combined docking and molecular dynamics simulations. Saudi J. Biol. Sci. 2021, 28, 2423–2431. [Google Scholar] [CrossRef]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Razia, S.; Park, H.; Shin, E.; Shim, K.-S.; Cho, E.; Kim, S.-Y. Effects of Aloe vera Flower Extract and Its Active Constituent Isoorientin on Skin Moisturization via Regulating Involucrin Expression: In Vitro and Molecular Docking Studies. Molecules 2021, 26, 2626. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Saloni, S.; Singh, H.; Kim, M.-H.; Sharma, P.; Misra, S.; Khan, F. Molecular docking, QSAR and ADMET studies of withanolide analogs against breast cancer. Drug Des. Dev. Ther. 2017, ume 11, 1859–1870. [Google Scholar] [CrossRef] [Green Version]
- Anjum, F.; Mohammad, T.; Almalki, A.A.; Akhtar, O.; Abdullaev, B.; Hassan, M.I. Phytoconstituents and Medicinal Plants for Anticancer Drug Discovery: Computational Identification of Potent Inhibitors of PIM1 Kinase. OMICS A J. Integr. Biol. 2021, 25, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Gadhe, C.G.; Balupuri, A.; Cho, S.J. In silicocharacterization of binding mode of CCR8 inhibitor: Homology modeling, docking and membrane based MD simulation study. J. Biomol. Struct. Dyn. 2015, 33, 2491–2510. [Google Scholar] [CrossRef]
- Lee, K.-Y.; Choi, H.-S.; Choi, H.-S.; Chung, K.Y.; Lee, B.-J.; Maeng, H.-J.; Seo, M.-D. Quercetin Directly Interacts with Vitamin D Receptor (VDR): Structural Implication of VDR Activation by Quercetin. Biomol. Ther. 2016, 24, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Rocha, S.F.D.S.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M. Virtual Screening Techniques in Drug Discovery: Review and Recent Applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Choi, E.-H.; Sharma, P.; Misra, S.; Kim, M.-H. Insight Into the Molecular Dynamic Simulation Studies of Reactive Oxygen Species in Native Skin Membrane. Front. Pharmacol. 2018, 9, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. eLS 2010. [Google Scholar] [CrossRef]
- Xie, M.; Schowen, R.L. Secondary structure and protein deamidation. J. Pharm. Sci. 1999, 88, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choi, M.; Sim, J.; Viji, M.; Li, S.; Lee, Y.H.; Kim, Y.; Seo, S.-Y.; Zhou, Y.; Lee, K.; et al. Synthesis and biological evaluation of caffeic acid derivatives as potent inhibitors of α-MSH-stimulated melanogenesis. Bioorganic Med. Chem. Lett. 2017, 27, 3374–3377. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-H.; Park, S.J.; Han, J.-H.; Pathak, C.; Cheong, H.-K.; Lee, B.-J. Structural insight into the interaction between the Hox and HMGB1 and understanding of the HMGB1-enhancing effect of Hox-DNA binding. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2015, 1854, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Son, W.S.; Lee, B.-J. Structural Analysis of Hypothetical Proteins from Helicobacter pylori: An Approach to Estimate Functions of Unknown or Hypothetical Proteins. Int. J. Mol. Sci. 2012, 13, 7109–7137. [Google Scholar] [CrossRef] [PubMed]
- Grimaldo, M.; Roosen-Runge, F.; Zhang, F.; Schreiber, F.; Seydel, T. Dynamics of proteins in solution. Q. Rev. Biophys. 2019, 52, 52. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Jernigan, R.L. Protein dynamic communities from elastic network models align closely to the communities defined by molecular dynamics. PLoS ONE 2018, 13, e0199225. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Points of significance: Principal component analysis. Nat. Methods 2017, 14, 641–643. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Mohammad, T.; Jairajpuri, D.S.; Rehman, M.T.; Hussain, A.; Samim, M.; Ahmad, F.J.; Alajmi, M.F.; Hassan, M.I. Identification and evaluation of glutathione conjugate gamma-l-glutamyl-l-cysteine for improved drug delivery to the brain. J. Biomol. Struct. Dyn. 2020, 38, 3610–3620. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Khan, F.I.; Mohammad, T.; Lan, D.; Hassan, M.; Wang, Y. Identification and evaluation of inhibitors of lipase from Malassezia restricta using virtual high-throughput screening and molecular dynamics studies. Int. J. Mol. Sci. 2019, 20, 884. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, R.; Mohammad, T.; Roy, S.; Anwar, S.; Gupta, P.; Haque, A.; Khan, P.; Kazim, S.N.; Islam, A.; Ahmad, F. Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy. Int. J. Biol. Macromol. 2019, 136, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mohammad, T.; Shamsi, A.; Hussain, A.; Alajmi, M.F.; Husain, S.A.; Iqbal, M.A.; Hassan, M.I. Identification of plant-based hexokinase 2 inhibitors: Combined molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef]
- Limongelli, V. Ligand binding free energy and kinetics calculation in 2020. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1455. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound ID | Phytochemical Name | Source | Binding Affinity (kcal/mol) | pKi | * Ligand Efficiency |
|---|---|---|---|---|---|---|
| 1. | 443716 | Hydroxysanguinarine | Papaver somniferum | −10.1 | 7.41 | 0.33 |
| 2. | 94577 | Cepharadione A | Piper nigrum | −10.0 | 7.33 | 0.33 |
| 3. | 11035494 | Semiglabrinol | Tephrosia purpurea | −9.9 | 7.26 | 0.30 |
| 4. | 124069 | Dihydrosanguinarine | Fumaria indica | −9.8 | 7.19 | 0.33 |
| 5. | 175941 | Curcusone A | Jatropha curcas | −9.8 | 7.19 | 0.39 |
| 6. | 10144 | Liriodenine | Annona squamosa | −9.8 | 7.19 | 0.38 |
| 7. | 147329 | Corysamine | Meconopsis aculeata | −9.7 | 7.11 | 0.33 |
| 8. | 197018 | Ushinsunine | Michelia champaca | −9.7 | 7.11 | 0.31 |
| 9. | 2754650 | Irenolone | Musa paradisiaca | −9.7 | 7.11 | 0.35 |
| 10. | 442851 | Crinasiatine | Crinum asiaticum | −9.7 | 7.11 | 0.33 |
| S. No. | Compound | Absorption | Distribution | Metabolism | Excretion | Toxicity | |
|---|---|---|---|---|---|---|---|
| GI Absorption | Water Solubility | BBB Permeation | CYP2D6 Substrate/Inhibitor | OCT2 Substrate | AMES/Hepatotoxicity | ||
| 1. | Hydroxysanguinarine | High | Moderate | Yes | No | No | Yes |
| 2. | Cepharadione A | High | Moderate | Yes | No | No | Yes |
| 3. | Semiglabrinol | High | Moderate | Yes | No | No | No |
| 4. | Dihydrosanguinarine | High | Moderate | Yes | No | No | Yes |
| 5. | Curcusone A | High | Moderate | Yes | No | Yes | No |
| 6. | Liriodenine | High | Moderate | Yes | Yes | No | No |
| 7. | Corysamine | High | Moderate | Yes | Yes | Yes | Yes |
| 8. | Ushinsunine | High | High | Yes | Yes | No | Yes |
| 9. | Irenolone | High | Moderate | Yes | Yes | No | Yes |
| 10. | Crinasiatine | High | Moderate | Yes | Yes | No | Yes |
| Compound ID | Pa | Pi | Biological Activity |
| Semiglabrino | 0.808 | 0.005 | Kinase inhibitor |
| 0.793 | 0.011 | Membrane permeability inhibitor | |
| 0.783 | 0.014 | Antineoplastic | |
| 0.653 | 0.036 | TP53 expression enhancer | |
| 0.612 | 0.033 | Oxidoreductase inhibitor | |
| Curcusone_A | 0.889 | 0.005 | Antineoplastic |
| 0.819 | 0.015 | Antieczematic | |
| 0.803 | 0.004 | Carminative | |
| 0.744 | 0.004 | Transcription factor NF kappa B stimulant | |
| 0.702 | 0.015 | Apoptosis agonist | |
| Liriodenine | 0.784 | 0.014 | Antineoplastic |
| 0.763 | 0.008 | Caspase 3 stimulant | |
| 0.710 | 0.015 | Alkane 1-monooxygenase inhibitor | |
| 0.680 | 0.014 | Kinase inhibitor | |
| 0.629 | 0.005 | Caspase 8 stimulant |
| Complex | α | β | 310-Helix | Turn | Bend | Other |
|---|---|---|---|---|---|---|
| CK1α-Apo | 24 | 23 | 3 | 9 | 11 | 19 |
| CK1α-Curcusone_A | 21 | 19 | 5 | 10 | 8 | 21 |
| CK1α-Liriodenine | 27 | 23 | 6 | 12 | 10 | 18 |
| CK1α-Semiglabrinol | 28 | 26 | 7 | 13 | 14 | 24 |
| Complex | ∆EvdW | ∆Eele | ∆Ggas | ∆Gpolar | ∆Gnonpolar | ∆Gsol | ∆Gbind |
|---|---|---|---|---|---|---|---|
| CK1α-Curcusone_A | −45.23 | −8.27 | −53.50 | 20.77 | −5.74 | 15.03 | −38.47 |
| CK1α-Liriodenine | −38.60 | −9.31 | −47.91 | 20.77 | −4.24 | 16.53 | −31.38 |
| CK1α-Semiglabrinol | −43.58 | −1.98 | −45.56 | 9.54 | −5.06 | 4.47 | −41.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafie, A.; Khan, S.; Zehra; Mohammad, T.; Anjum, F.; Hasan, G.M.; Yadav, D.K.; Hassan, M.I. Identification of Phytoconstituents as Potent Inhibitors of Casein Kinase-1 Alpha Using Virtual Screening and Molecular Dynamics Simulations. Pharmaceutics 2021, 13, 2157. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122157
Shafie A, Khan S, Zehra, Mohammad T, Anjum F, Hasan GM, Yadav DK, Hassan MI. Identification of Phytoconstituents as Potent Inhibitors of Casein Kinase-1 Alpha Using Virtual Screening and Molecular Dynamics Simulations. Pharmaceutics. 2021; 13(12):2157. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122157
Chicago/Turabian StyleShafie, Alaa, Shama Khan, Zehra, Taj Mohammad, Farah Anjum, Gulam Mustafa Hasan, Dharmendra Kumar Yadav, and Md. Imtaiyaz Hassan. 2021. "Identification of Phytoconstituents as Potent Inhibitors of Casein Kinase-1 Alpha Using Virtual Screening and Molecular Dynamics Simulations" Pharmaceutics 13, no. 12: 2157. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13122157