
Processing Impact on In Vitro and In Vivo Performance of Solid Dispersions—A Comparison between Hot-Melt Extrusion and Spray Drying

Abstract
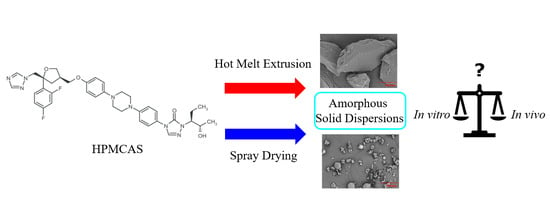
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of SDD Dispersion
2.2.2. Preparation of HME Dispersion
2.2.3. Physical Characterization of ASDs
2.2.4. In Vitro Dissolution
2.2.5. In Vivo Cynomolgus Monkey PK Study
3. Results and Discussion
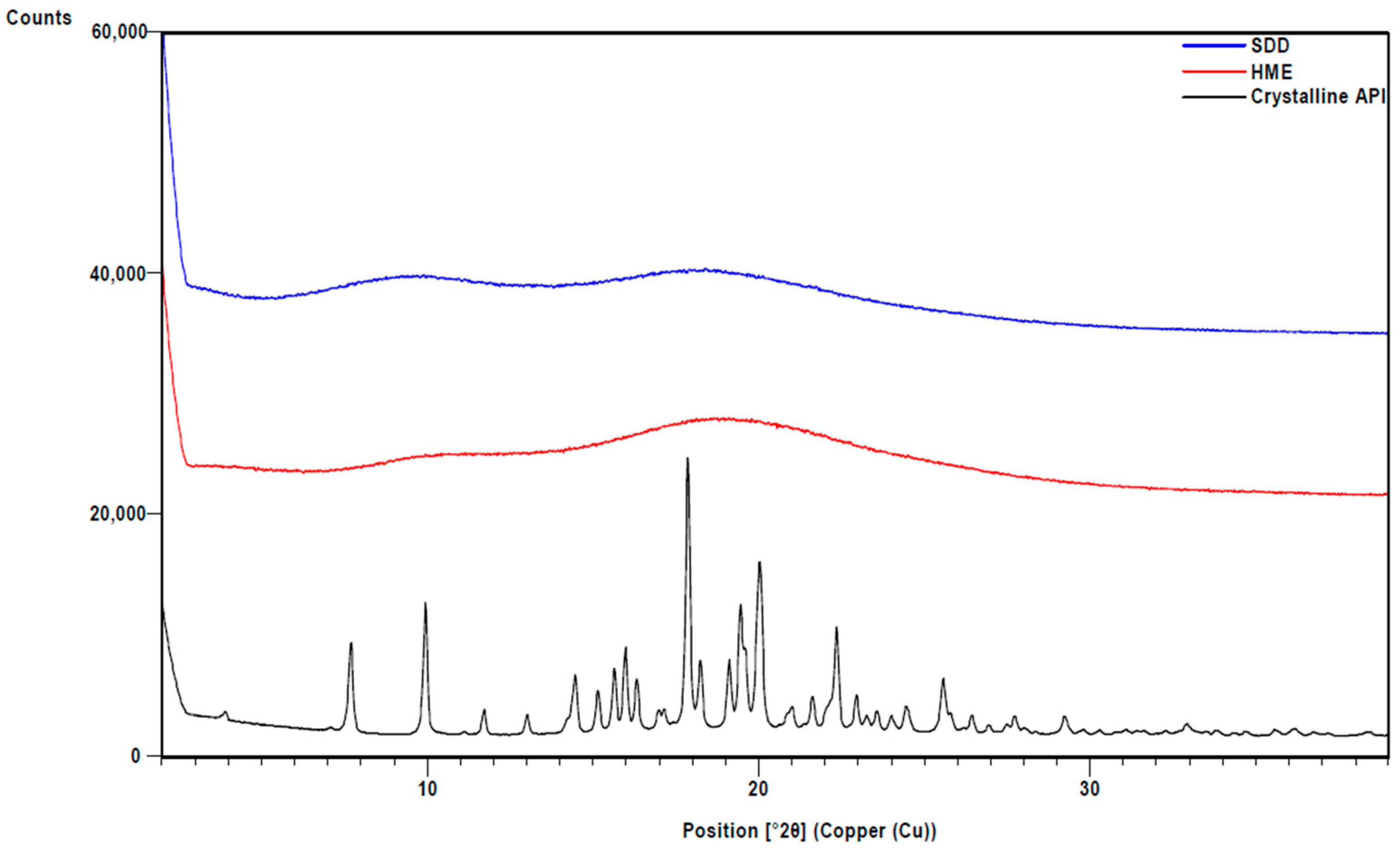
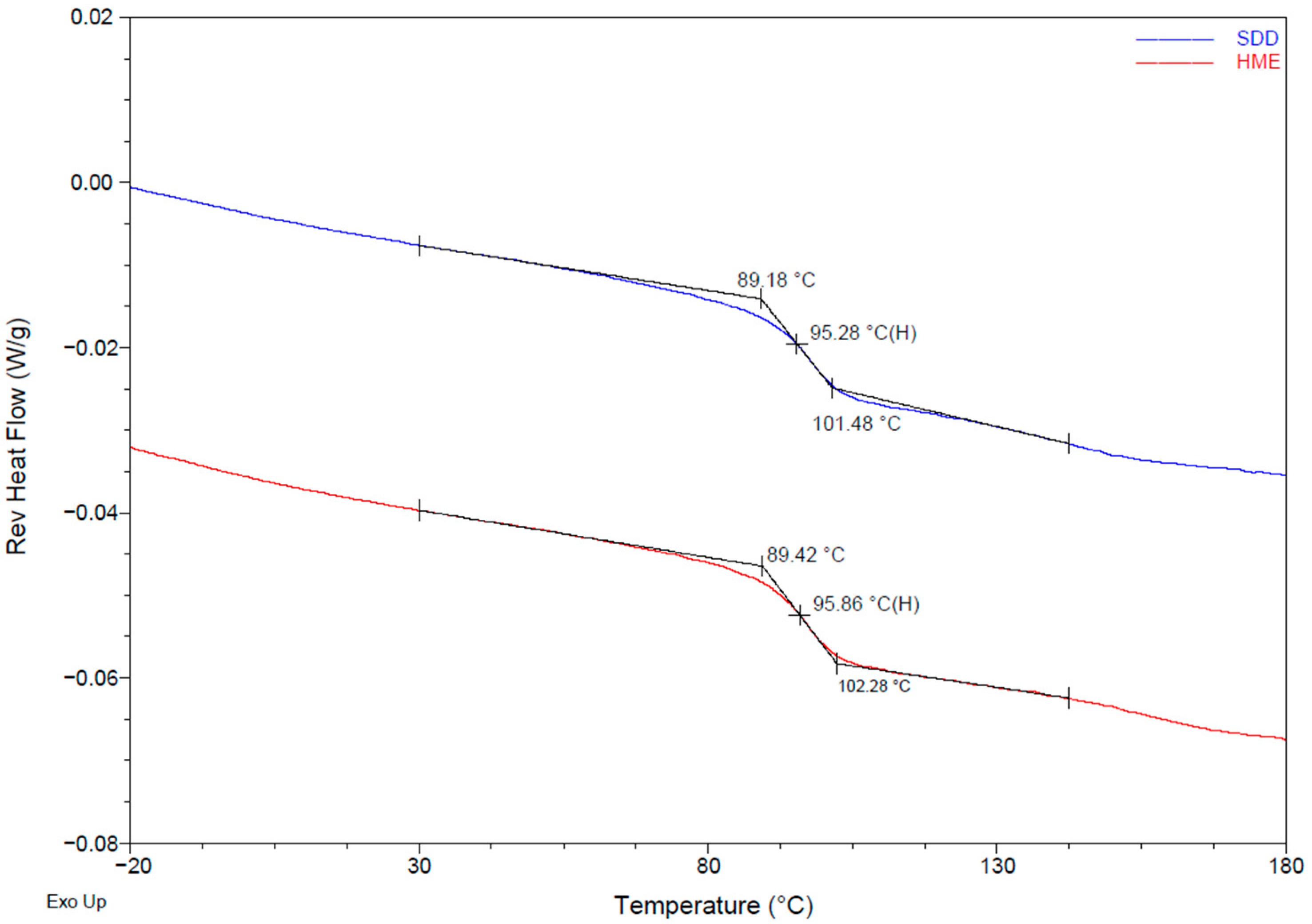
3.1. Physical Properties of SDD and HME
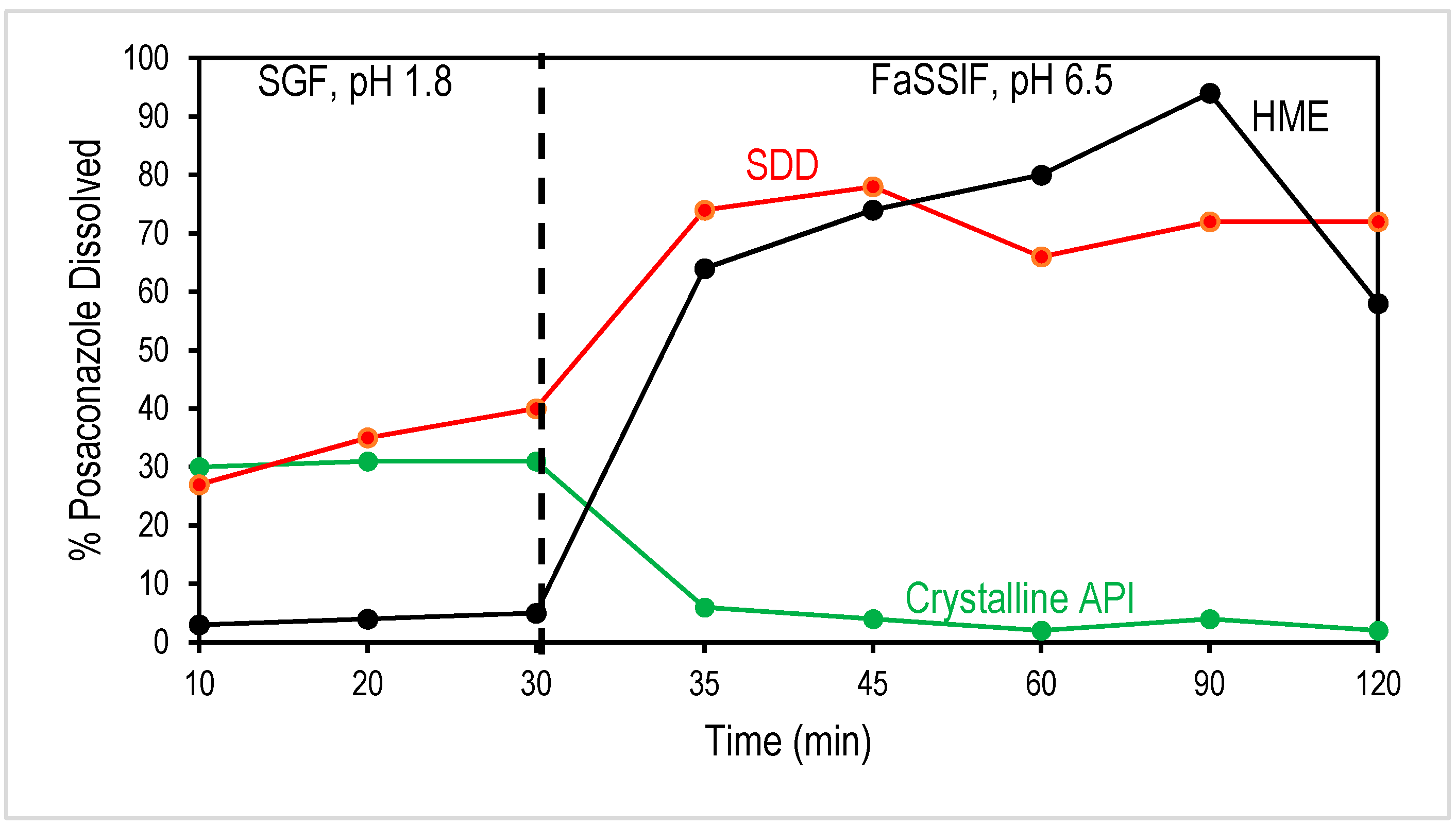
3.2. Comparison of In Vitro Dissolution of SDD and HME
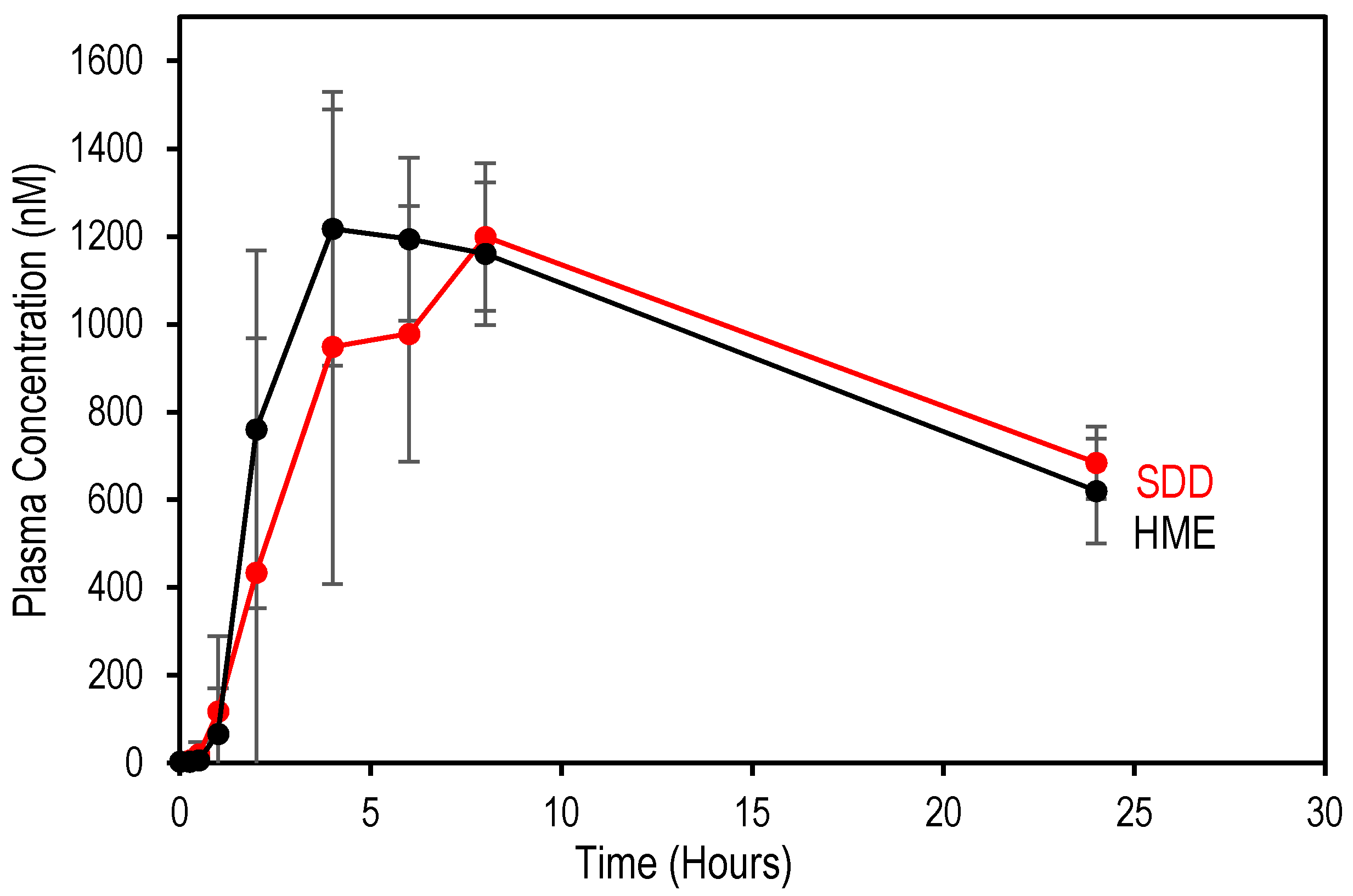
3.3. Comparison of In Vivo Performance of SDD and HME
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Patil, S.K.; Wagh, K.S.; Parik, V.B.; Akarte, A.M.; Baviskar, D.T. Strategies for solubility enhancement of poorly soluble drugs. Int. J. Pharm. Sci. Rev. Res. 2011, 8, 74–80. [Google Scholar]
- Fahr, A.; Liu, X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 2007, 4, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.W.; Podczeck, F.; Michael Newton, J.; Waddington, W.A.; Ell, P.J.; Lacey, L.F. The use of formulation technology to assess regional gastrointestinal drug absorption in humans. Eur. J. Pharm. Sci. 2004, 21, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Desai, J.; Alexander, K.; Riga, A. Characterization of polymeric dispersions of dimenhydrinate in ethyl cellulose for controlled release. Int. J. Pharm. 2006, 308, 115–123. [Google Scholar] [CrossRef]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility advantage of amorphous pharmaceuticals: I. A thermodynamic analysis. J. Pharm. Sci. 2010, 99, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Zhu, W.; Kesisoglou, F. Physiologically Based Absorption Modeling for Amorphous Solid Dispersion Formulations. Mol. Pharm. 2016, 13, 3206–3215. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Murdande, S.B.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility Advantage of Amorphous Pharmaceuticals: II. Application of Quantitative Thermodynamic Relationships for Prediction of Solubility Enhancement in Structurally Diverse Insoluble Pharmaceuticals. Pharm. Res. 2010, 27, 2704–2714. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Handa, T.; Alonzo, D.E.; Taylor, L.S. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur. J. Pharm. Biopharm. 2008, 70, 493–499. [Google Scholar] [CrossRef]
- Marsac, P.J.; Konno, H.; Taylor, L.S. A Comparison of the Physical Stability of Amorphous Felodipine and Nifedipine Systems. Pharm. Res. 2006, 23, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Knapik, J.; Wojnarowska, Z.; Grzybowska, K.; Tajber, L.; Mesallati, H.; Paluch, K.J.; Paluch, M. Molecular Dynamics and Physical Stability of Amorphous Nimesulide Drug and Its Binary Drug-Polymer Systems. Mol. Pharm. 2016, 13, 1937–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Mann, A.K.P.; Van Duong, T.; Ormes, J.D.; Okoh, G.A.; Hermans, A.; Taylor, L.S. Drug Release and Nanodroplet Formation from Amorphous Solid Dispersions: Insight into the Roles of Drug Physicochemical Properties and Polymer Selection. Mol. Pharm. 2021. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving Oral Bioavailability of Sorafenib by Optimizing the “Spring” and “Parachute” Based on Molecular Interaction Mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Newman, A. Rational design for amorphous solid dispersions. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Shi, N.-Q.; Wang, S.-R.; Zhang, Y.; Huo, J.-S.; Wang, L.-N.; Cai, J.-H.; Li, Z.-Q.; Xiang, B.; Qi, X.-R. Hot melt extrusion technology for improved dissolution, solubility and “spring-parachute” processes of amorphous self-micellizing solid dispersions containing BCS II drugs indomethacin and fenofibrate: Profiles and mechanisms. Eur. J. Pharm. Sci. 2019, 130, 78–90. [Google Scholar] [CrossRef]
- Mann, A.K.P.; Schenck, L.; Koynov, A.; Rumondor, A.C.F.; Jin, X.; Marota, M.; Dalton, C. Producing Amorphous Solid Dispersions via Co-Precipitation and Spray Drying: Impact to Physicochemical and Biopharmaceutical Properties. J. Pharm. Sci. 2018, 107, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Lee, Y.C.; Shabani, Z.; Frankenfeld Lamm, C.; Zhu, W.; Li, Y.; Templeton, A. Processing Impact on Performance of Solid Dispersions. Pharmaceutics 2018, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Duarte, I.; Corvo, M.L.; Serodio, P.; Vicente, J.; Pinto, J.F.; Temtem, M. Production of nano-solid dispersions using a novel solvent-controlled precipitation process—Benchmarking their in vivo performance with an amorphous micro-sized solid dispersion produced by spray drying. Eur. J. Pharm. Sci. 2016, 93, 203–214. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; Das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Dong, Z.; Chatterji, A.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Shah, N. Evaluation of solid state properties of solid dispersions prepared by hot-melt extrusion and solvent co-precipitation. Int. J. Pharm. 2008, 355, 141–149. [Google Scholar] [CrossRef]
- Lakshman, J.P.; Cao, Y.; Kowalski, J.; Serajuddin, A.T.M. Application of Melt Extrusion in the Development of a Physically and Chemically Stable High-Energy Amorphous Solid Dispersion of a Poorly Water-Soluble Drug. Mol. Pharm. 2008, 5, 994–1002. [Google Scholar] [CrossRef]
- Friesen, D.T.; Shanker, R.; Crew, M.; Smithey, D.T.; Curatolo, W.J.; Nightingale, J.A.S. Hydroxypropyl Methylcellulose Acetate Succinate-Based Spray-Dried Dispersions: An Overview. Mol. Pharm. 2008, 5, 1003–1019. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Wen, M.; Li, W.; He, M.; Yang, X.; Li, S. Preparation and characterization of baicalin-poly-vinylpyrrolidone coprecipitate. Int. J. Pharm. 2011, 408, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Broadhead, J.; Edmond Rouan, S.K.; Rhodes, C.T. The spray drying of pharmaceuticals. Drug Dev. Ind. Pharm. 1992, 18, 1169–1206. [Google Scholar] [CrossRef]
- Ré, M.-I. Formulating Drug Delivery Systems by Spray Drying. Dry. Technol. 2006, 24, 433–446. [Google Scholar] [CrossRef]
- Weuts, I.; Van Dycke, F.; Voorspoels, J.; De Cort, S.; Stokbroekx, S.; Leemans, R.; Brewster, M.E.; Xu, D.; Segmuller, B.; Turner, Y.T.A.; et al. Physicochemical properties of the amorphous drug, cast films, and spray dried powders to predict formulation probability of success for solid dispersions: Etravirine. J. Pharm. Sci. 2011, 100, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Worku, Z.A.; Meeus, J.; Guns, S.; Van den Mooter, G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: Formulation and process considerations. Int. J. Pharm. 2013, 453, 253–284. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.M.; Zhang, F.; Repka, M.A.; Thumma, S.; Upadhye, S.B.; Kumar Battu, S.; McGinity, J.W.; Martin, C. Pharmaceutical Applications of Hot-Melt Extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Schenck, L.; Troup, G.M.; Lowinger, M.; Li, L.; McKelvey, C. Achieving a Hot Melt Extrusion Design Space for the Production of Solid Solutions. In Chemical Engineering in the Pharmaceutical Industry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 819–836. [Google Scholar]
- Shah, N.; Iyer, R.M.; Mair, H.-J.; Choi, D.S.; Tian, H.; Diodone, R.; Fähnrich, K.; Pabst-Ravot, A.; Tang, K.; Scheubel, E.; et al. Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J. Pharm. Sci. 2013, 102, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.R.C.; Loebenberg, R.; Almukainzi, M. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolut. Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Vertzoni, M.; Fotaki, N.; Nicolaides, E.; Reppas, C.; Kostewicz, E.; Stippler, E.; Leuner, C.; Dressman, J. Dissolution media simulating the intralumenal composition of the small intestine: Physiological issues and practical aspects. J. Pharm. Pharmacol. 2004, 56, 453–462. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Structure |  |
| Melting Point (°C) | 167.9 |
| pKa | 3.6 (piperazine N) and 4.6 (triazole N) |
| Log P | 5 |
| BCS Classification | II |
| Solubility (mg/mL) | pH 1: 0.79 pH 3: 0.003 pH 7: 0.001 Fasted state simulated intestinal fluid (FaSSIF): 0.002 |
| Sample | ~API Loading on Surface Based on F and N |
|---|---|
| SDD | 18% |
| HME | 21% |
| Posaconazole standard | 100% |
| HPMCAS standard | 0% |
| Sample | Cmax (nmol/L) | AUC (h·nmol/L) | Tmax (h) | Cmax Ratio * | AUC Ratio * |
|---|---|---|---|---|---|
| SDD | 1267 ± 217 | 18,147 ± 5889 | 6 | 0.98 | 0.85 |
| HME | 1293 ± 100 | 21,423 ± 1285 | 4 | 1.00 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Mann, A.K.P.; Zhang, D.; Yang, Z. Processing Impact on In Vitro and In Vivo Performance of Solid Dispersions—A Comparison between Hot-Melt Extrusion and Spray Drying. Pharmaceutics 2021, 13, 1307. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13081307
Li Y, Mann AKP, Zhang D, Yang Z. Processing Impact on In Vitro and In Vivo Performance of Solid Dispersions—A Comparison between Hot-Melt Extrusion and Spray Drying. Pharmaceutics. 2021; 13(8):1307. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13081307
Chicago/Turabian StyleLi, Yongjun, Amanda K. P. Mann, Dan Zhang, and Zhen Yang. 2021. "Processing Impact on In Vitro and In Vivo Performance of Solid Dispersions—A Comparison between Hot-Melt Extrusion and Spray Drying" Pharmaceutics 13, no. 8: 1307. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13081307