
Binding and Kinetic Analysis of Human Protein Phosphatase PP2A Interactions with Caspase 9 Protein and the Interfering Peptide C9h
 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis and Sequence
2.2. Docking
2.3. Molecular Dynamics (MD) Simulation
2.4. Circular Dichroism Analysis
2.5. Characterization of PP2A and C9h Peptide Interaction by ELISA
2.6. Biolayer Interferometry and Kinetic Analysis
3. Results
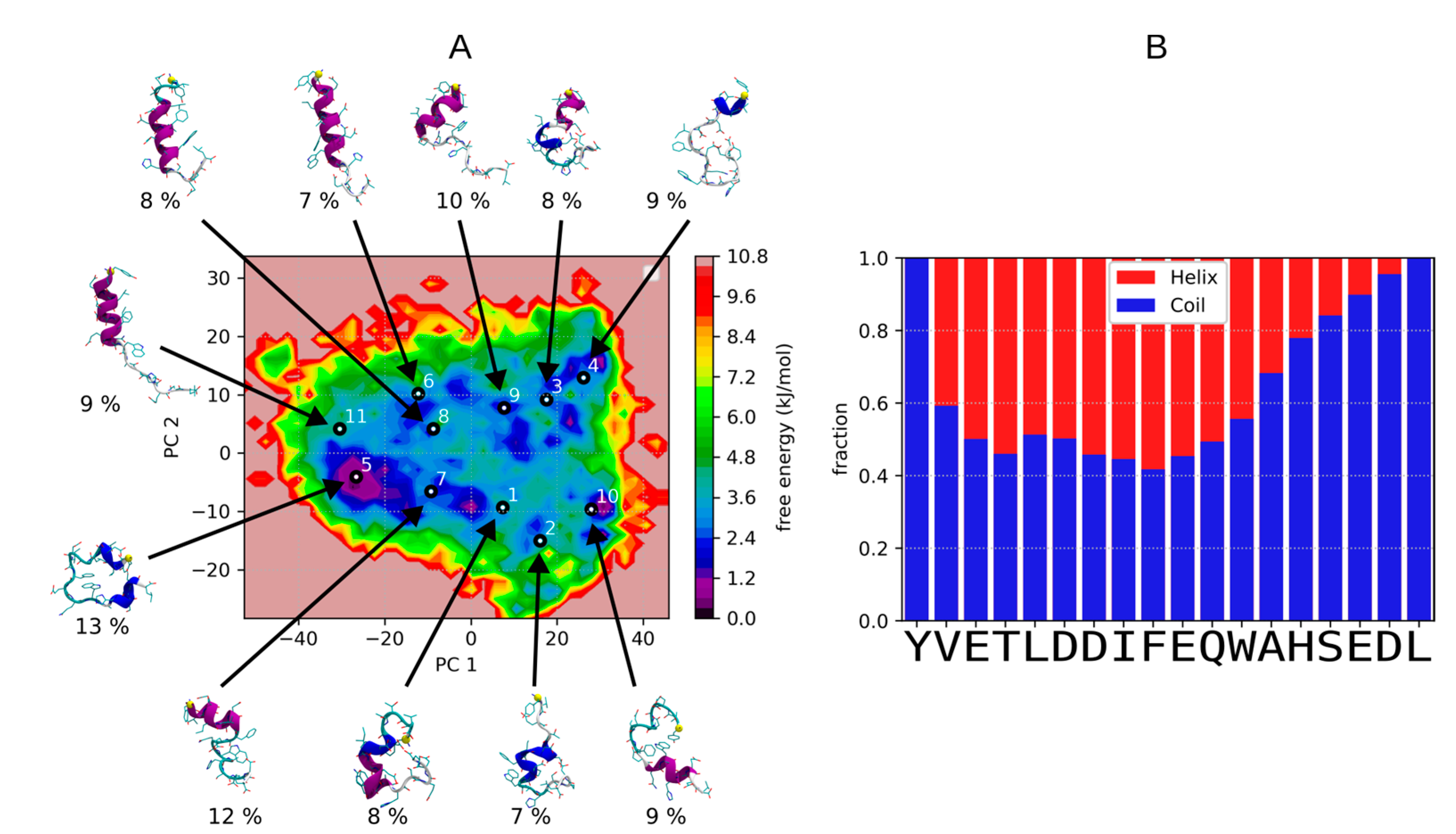
3.1. Dynamic Behavior of C9h Using ST Simulations
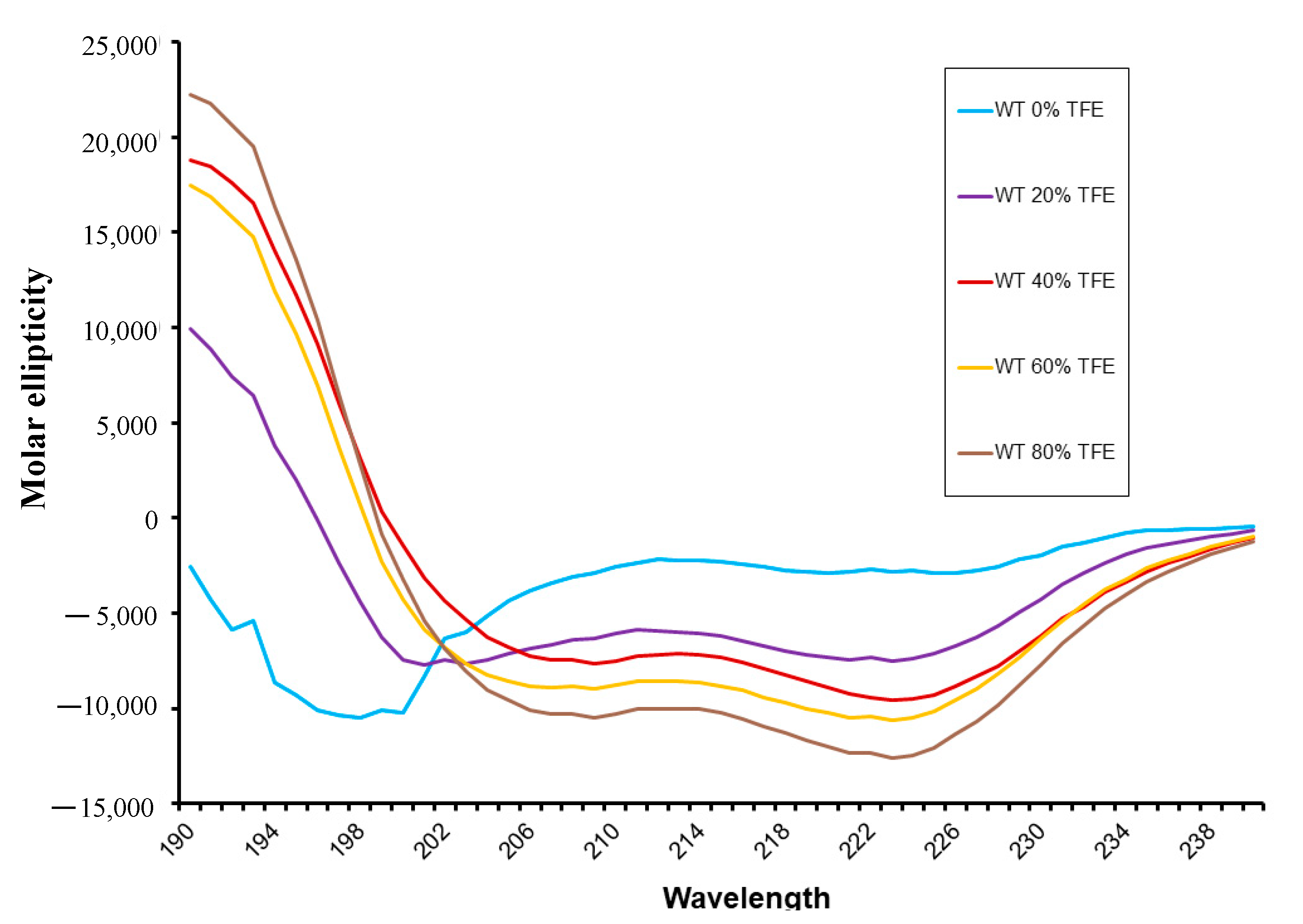
3.2. Peptide Secondary Structure Evaluation by Circular Dichroism (CD)
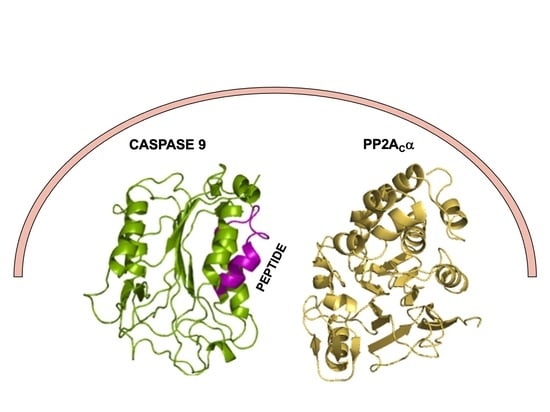
3.3. Molecular Modeling of Peptide Interaction with PP2A
3.4. Qualitative Interaction between PP2A and the Biotinylated C9h Peptide
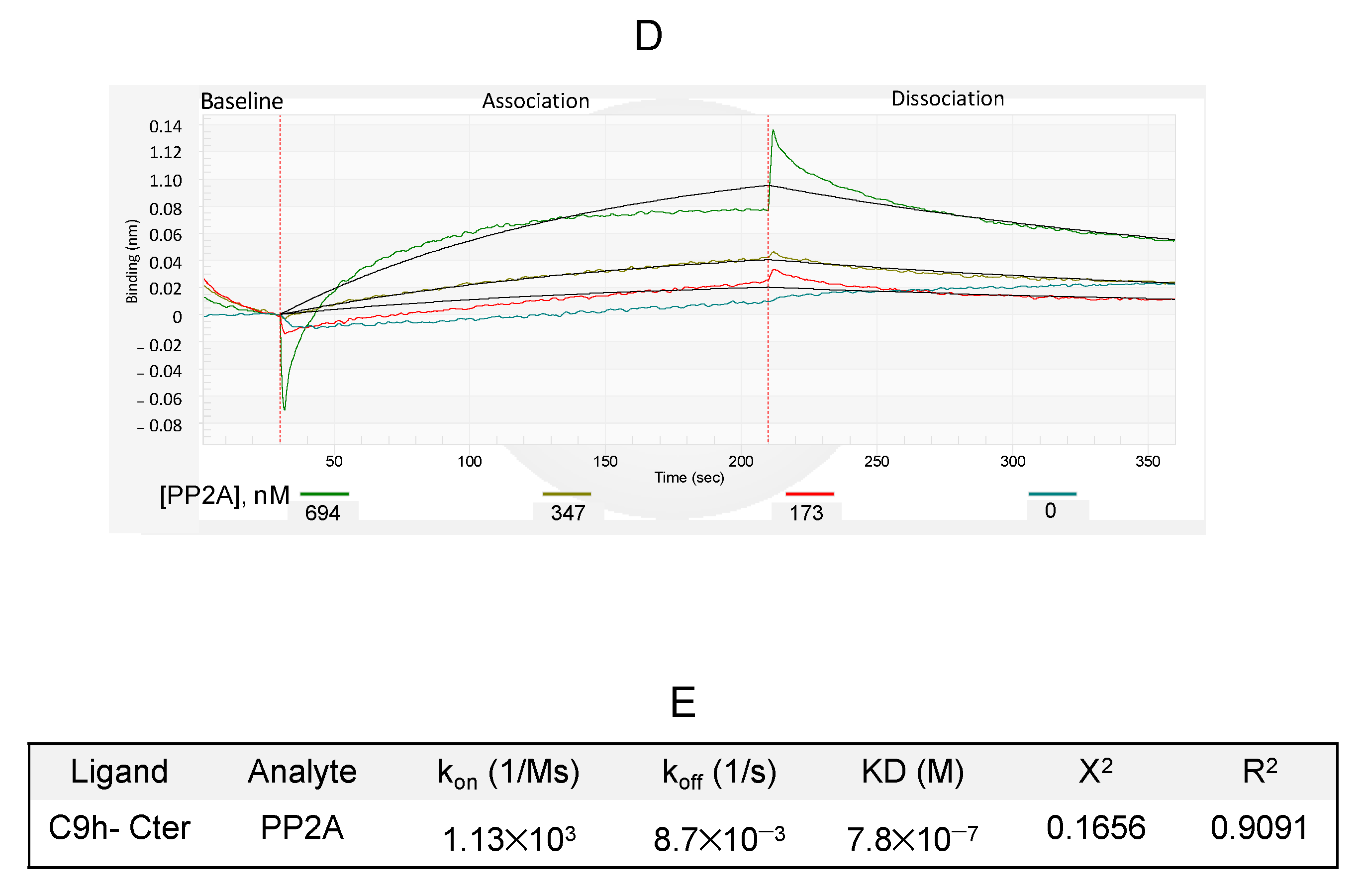
3.5. Affinity of PP2A Protein to Caspase 9 Protein and C9h Peptide
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein-protein interactions: The next generation of drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.Z.; Bruzzoni-Giovanelli, H.; Yu, Y.; Dorgham, K.; Parizot, C.; Zini, J.M.; Brossas, J.Y.; Tuffery, P.; Rebollo, A. Identification of peptides interfering with the LRRK2/PP1 interaction. PLoS ONE 2020, 15, e0237110. [Google Scholar] [CrossRef] [PubMed]
- Savier, E.; Simon-Gracia, L.; Charlotte, F.; Tuffery, P.; Teesalu, T.; Scatton, O.; Rebollo, A. Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma. Pharmaceutics 2021, 13, 1631. [Google Scholar] [CrossRef] [PubMed]
- Savier, E.; Tuffery, P.; Bruzzoni-Giovanelli, H.; Rebollo, A. Isolation of Primary Hepatocytes for Testing Tumor Penetrating Peptides. Methods Mol. Biol. 2022, 2383, 413–427. [Google Scholar] [CrossRef]
- Simon-Gracia, L.; Loisel, S.; Sidorenko, V.; Scodeller, P.; Parizot, C.; Savier, E.; Haute, T.; Teesalu, T.; Rebollo, A. Preclinical Validation of Tumor-Penetrating and Interfering Peptides against Chronic Lymphocytic Leukemia. Mol. Pharm. 2022, 19, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Rafferty, J.; Nagaraj, H.; McCloskey, A.P.; Huwaitat, R.; Porter, S.; Albadr, A.; Laverty, G. Peptide Therapeutics and the Pharmaceutical Industry: Barriers Encountered Translating from the Laboratory to Patients. Curr. Med. Chem. 2016, 23, 4231–4259. [Google Scholar] [CrossRef]
- Ayllon, V.; Martinez, A.C.; Garcia, A.; Cayla, X.; Rebollo, A. Protein phosphatase 1alpha is a Ras-activated Bad phosphatase that regulates interleukin-2 deprivation-induced apoptosis. EMBO J. 2000, 19, 2237–2246. [Google Scholar] [CrossRef]
- Kang, H.S.; Choi, I. Protein phosphatase 2A modulates the proliferation of human multiple myeloma cells via regulation of the production of reactive oxygen intermediates and anti-apoptotic factors. Cell Immunol. 2001, 213, 34–44. [Google Scholar] [CrossRef]
- Brautigan, D.L.; Farrington, C.; Narla, G. Targeting protein phosphatase PP2A for cancer therapy: Development of allosteric pharmaceutical agents. Clin. Sci. 2021, 135, 1545–1556. [Google Scholar] [CrossRef]
- Chen, W.; Possemato, R.; Campbell, K.T.; Plattner, C.A.; Pallas, D.C.; Hahn, W.C. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 2004, 5, 127–136. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J.; Van Hoof, C. PP2A: The expected tumor suppressor. Curr. Opin. Genet. Dev. 2005, 15, 34–41. [Google Scholar] [CrossRef]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef]
- Arrouss, I.; Nemati, F.; Roncal, F.; Wislez, M.; Dorgham, K.; Vallerand, D.; Rabbe, N.; Karboul, N.; Carlotti, F.; Bravo, J.; et al. Specific targeting of caspase-9/PP2A interaction as potential new anti-cancer therapy. PLoS ONE 2013, 8, e60816. [Google Scholar] [CrossRef]
- Avrutsky, M.I.; Troy, C.M. Caspase-9: A Multimodal Therapeutic Target with Diverse Cellular Expression in Human Disease. Front. Pharmacol. 2021, 12, 701301. [Google Scholar] [CrossRef]
- Binette, V.; Mousseau, N.; Tuffery, P. A Generalized Attraction-Repulsion Potential and Revisited Fragment Library Improves PEP-FOLD Peptide Structure Prediction. J. Chem. Theory Comput. 2022, 18, 2720–2736. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Rebollo, A.; Fliedel, L.; Tuffery, P. PEPscan: A Broad Spectrum Approach for the Characterization of Protein-Binder Interactions? Biomolecules 2022, 12, 178. [Google Scholar] [CrossRef]
- Rebollo, A.; Savier, E.; Tuffery, P. Pepscan Approach for the Identification of Protein-Protein Interfaces: Lessons from Experiment. Biomolecules 2021, 11, 772. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Marinari, E.; Parisi, G. Simulated tempering: A new Monte Carlo scheme. Europhys. Lett. 1992, 19, 451. [Google Scholar] [CrossRef]
- Park, S.; Pande, V.S. Choosing weights for simulated tempering. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007, 76, 016703. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Okamoto, Y.; Derreumaux, P. Communication: Simulated tempering with fast on-the-fly weight determination. J. Chem. Phys. 2013, 138, 061102. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Drew, E.D.; Janes, R.W. PDBMD2CD: Providing predicted protein circular dichroism spectra from multiple molecular dynamics-generated protein structures. Nucleic Acids Res. 2020, 48, W17–W24. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Kernya, L.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E3095–E3103. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat. Protoc. 2006, 1, 2527–2535. [Google Scholar] [CrossRef]
- Woollett, B.; Whitmore, L.; Janes, R.W.; Wallace, B.A. ValiDichro: A website for validating and quality control of protein circular dichroism spectra. Nucleic Acids Res. 2013, 41, W417–W421. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Sayed, A.M.M.; Tan, H.L.; Mok, Y.K.; Chen, E.S. Identifying Protein Interactions with Histone Peptides Using Bio-layer Interferometry. Bio. Protoc. 2018, 8, e3012. [Google Scholar] [CrossRef] [PubMed]
- Groner, M.; Ng, T.; Wang, W.; Udit, A.K. Bio-layer interferometry of a multivalent sulfated virus nanoparticle with heparin-like anticoagulant activity. Anal. Bioanal. Chem. 2015, 407, 5843–5847. [Google Scholar] [CrossRef]
- Sieker, F.; May, A.; Zacharias, M. Predicting affinity and specificity of antigenic peptide binding to major histocompatibility class I molecules. Curr. Protein Pept. Sci. 2009, 10, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Berggard, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Wierer, M.; Mann, M. Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Hum. Mol. Genet. 2016, 25, R106–R114. [Google Scholar] [CrossRef]
- Sultana, A.; Lee, J.E. Measuring protein-protein and protein-nucleic Acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19.25.1–19.25.26. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorgham, K.; Murail, S.; Tuffery, P.; Savier, E.; Bravo, J.; Rebollo, A. Binding and Kinetic Analysis of Human Protein Phosphatase PP2A Interactions with Caspase 9 Protein and the Interfering Peptide C9h. Pharmaceutics 2022, 14, 2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102055
Dorgham K, Murail S, Tuffery P, Savier E, Bravo J, Rebollo A. Binding and Kinetic Analysis of Human Protein Phosphatase PP2A Interactions with Caspase 9 Protein and the Interfering Peptide C9h. Pharmaceutics. 2022; 14(10):2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102055
Chicago/Turabian StyleDorgham, Karim, Samuel Murail, Pierre Tuffery, Eric Savier, Jeronimo Bravo, and Angelita Rebollo. 2022. "Binding and Kinetic Analysis of Human Protein Phosphatase PP2A Interactions with Caspase 9 Protein and the Interfering Peptide C9h" Pharmaceutics 14, no. 10: 2055. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102055