
Dendrimer-Conjugated nSMase2 Inhibitor Reduces Tau Propagation in Mice
 ,
,  and
and 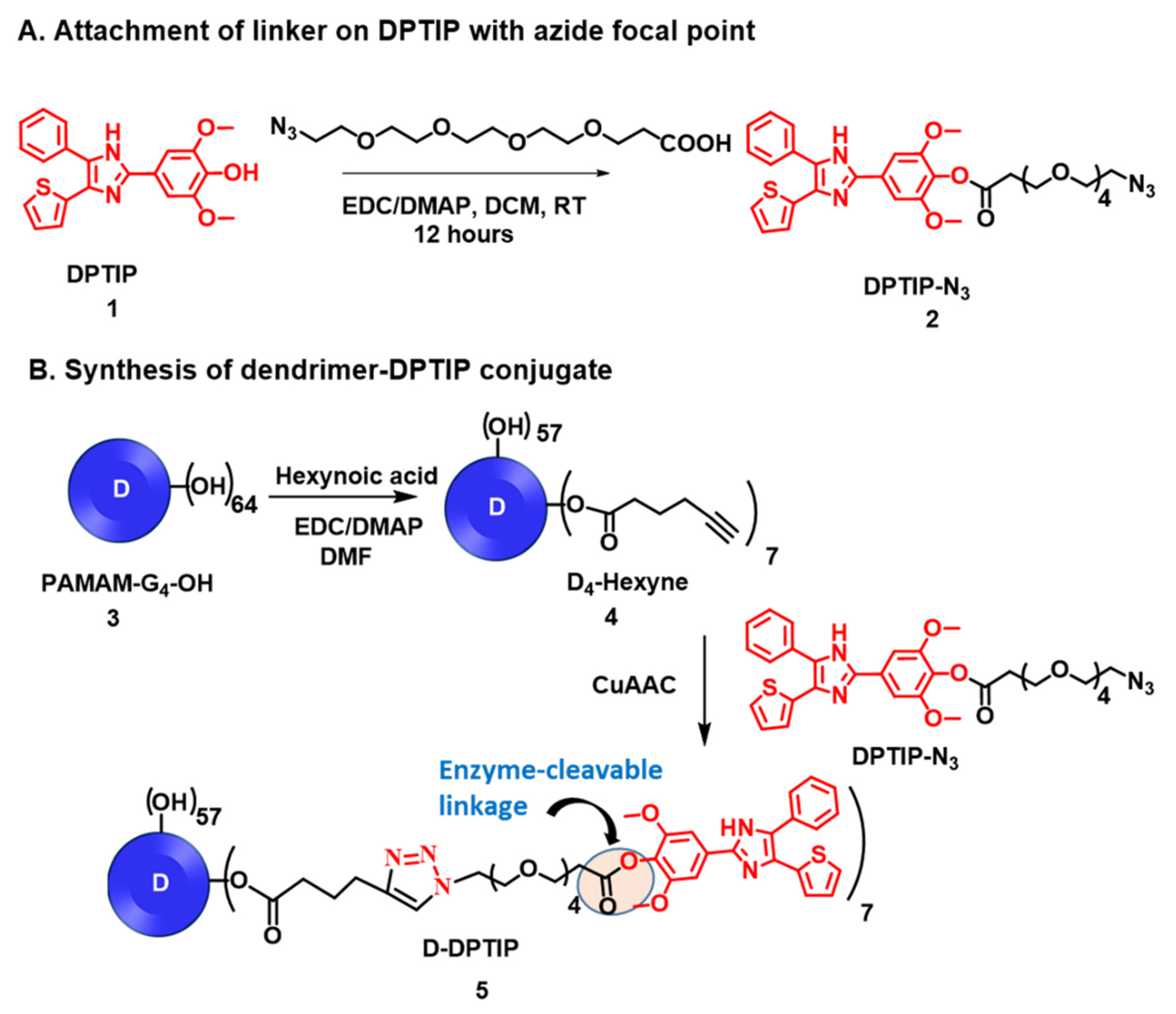{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of D-DPTIP Conjugates
2.2. High Performance Liquid Chromatography (HPLC)
2.3. Mass Spectroscopy
2.4. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.5. Dynamic Light Scattering (DLS) and Zeta Potential (ζ)
2.6. In Vitro Drug Release
2.7. Animal Studies
2.8. DPTIP and D-DPTIP Preparations for In Vivo Studies
2.9. Brain Levels of DPTIP Following Oral D-DPTIP in AD Mice
2.10. DPTIP Bioanalysis
2.11. IL-1β Induced EV Secretion in an Acute Brain Injury Model
2.12. Quantitation of Plasma EVs in IL-1β Induced Acute Brain Injury Model
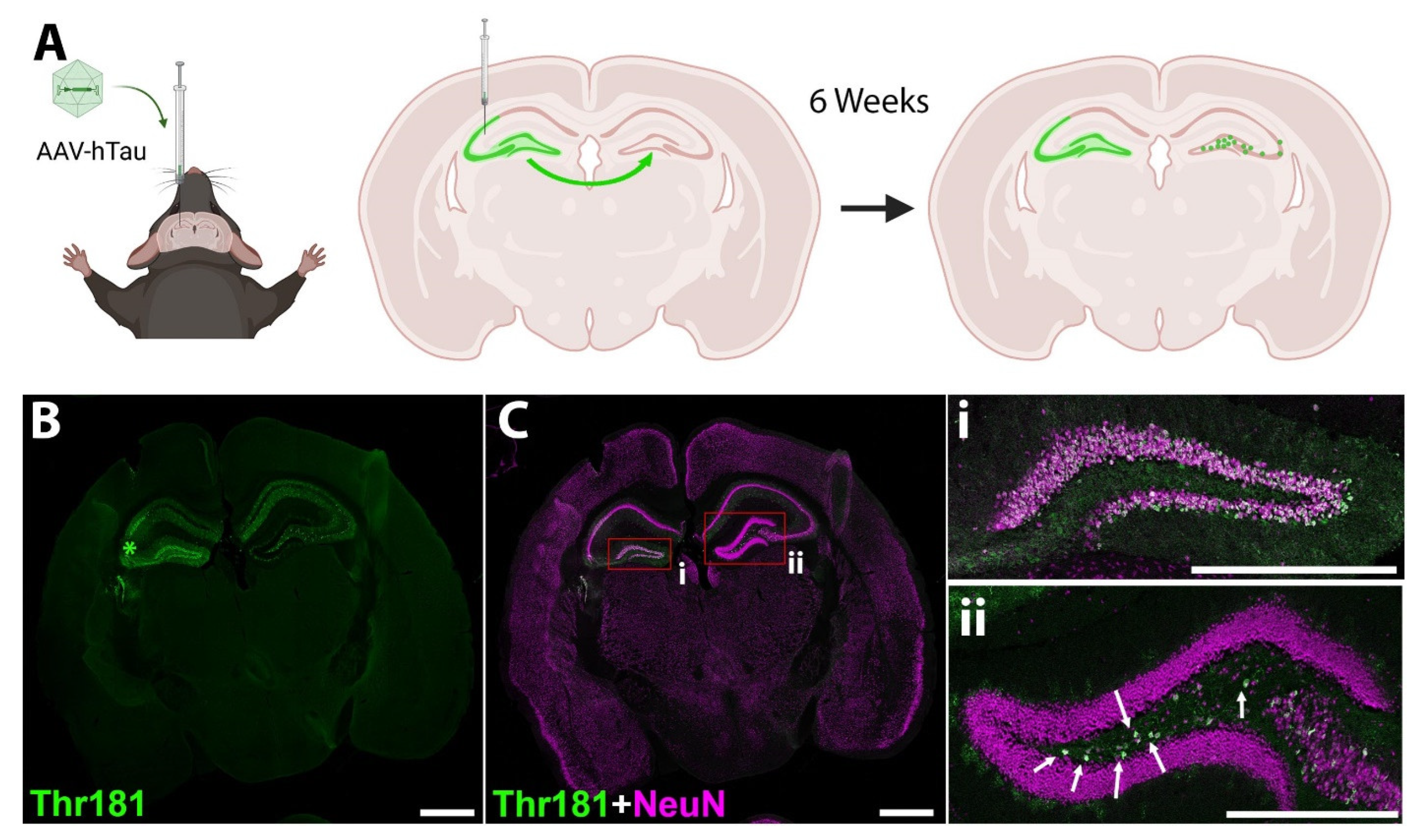
2.13. Stereotaxic Injection of AAV-hTau Vector
2.14. Immunofluorescence Imaging in AAV-hTau Model
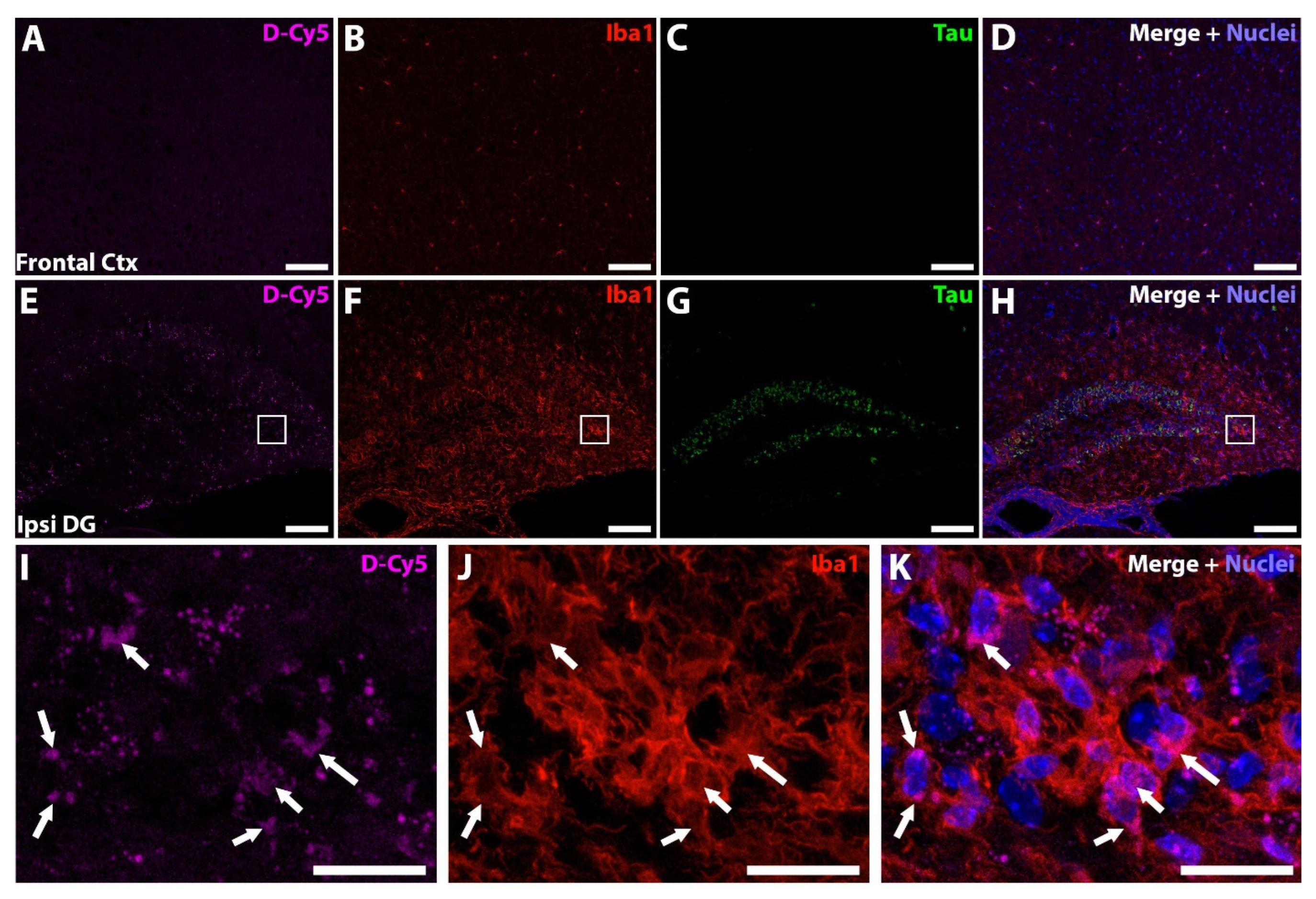
2.15. Dendrimer-Cy5 Localization Studies
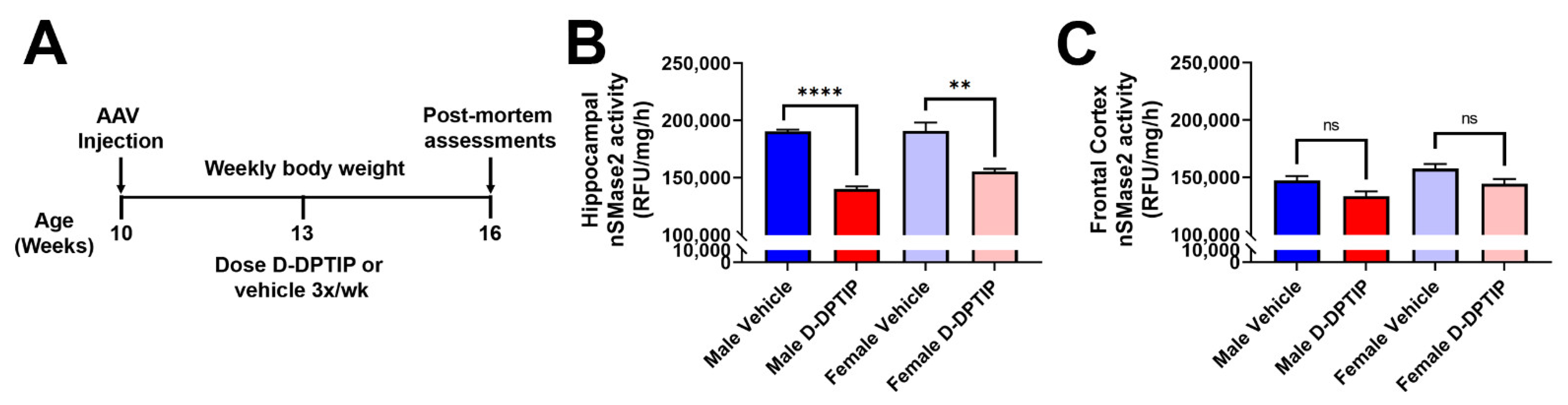
2.16. AAV-hTau Mice Dosing Regimen
2.17. nSMase2 Activity Assessments in AAV-hTau Mice
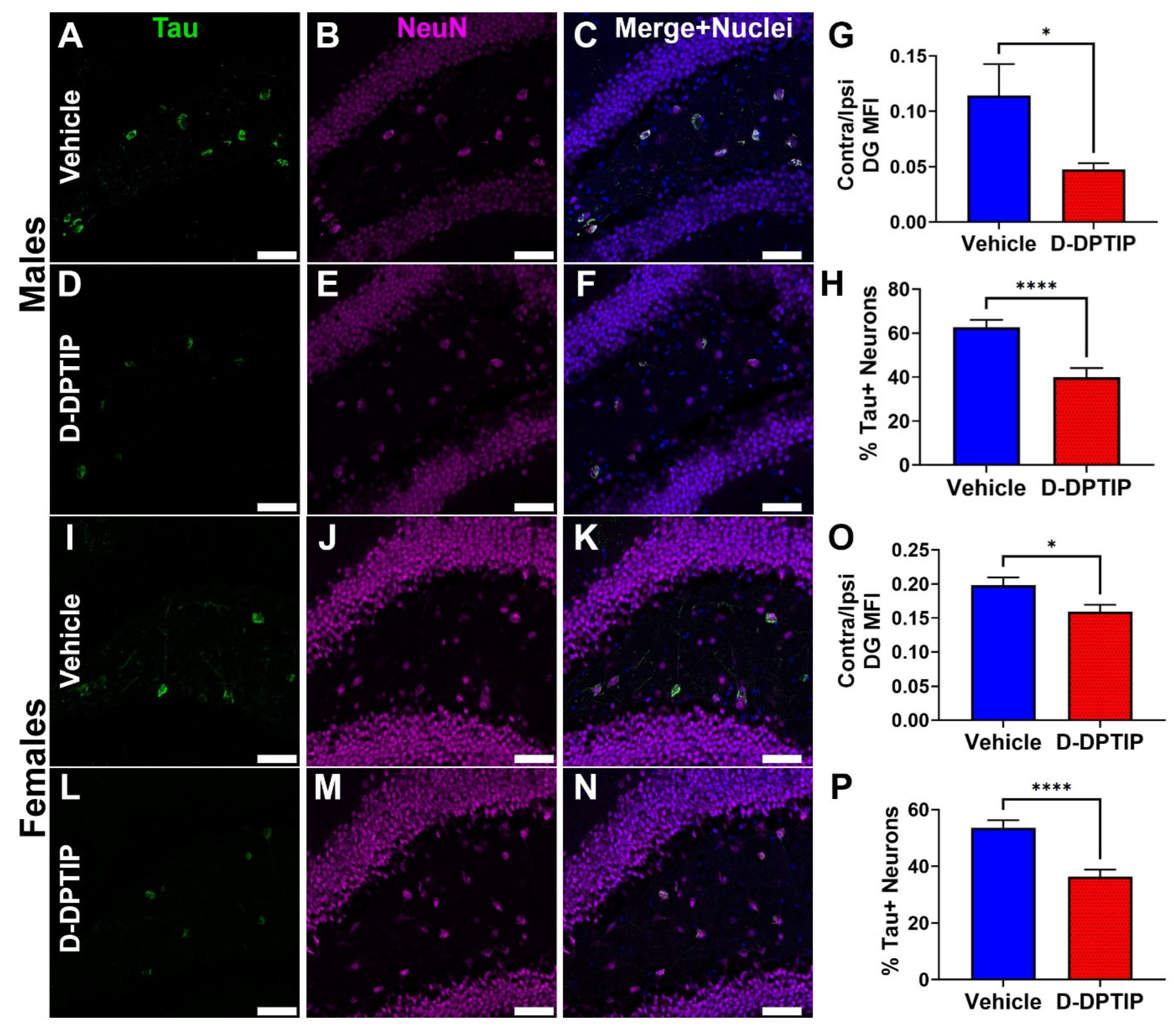
2.18. Efficacy Analysis in AAV-hTau Mice
2.19. Clinical Chemistries
2.20. Statistical Analysis
3. Results
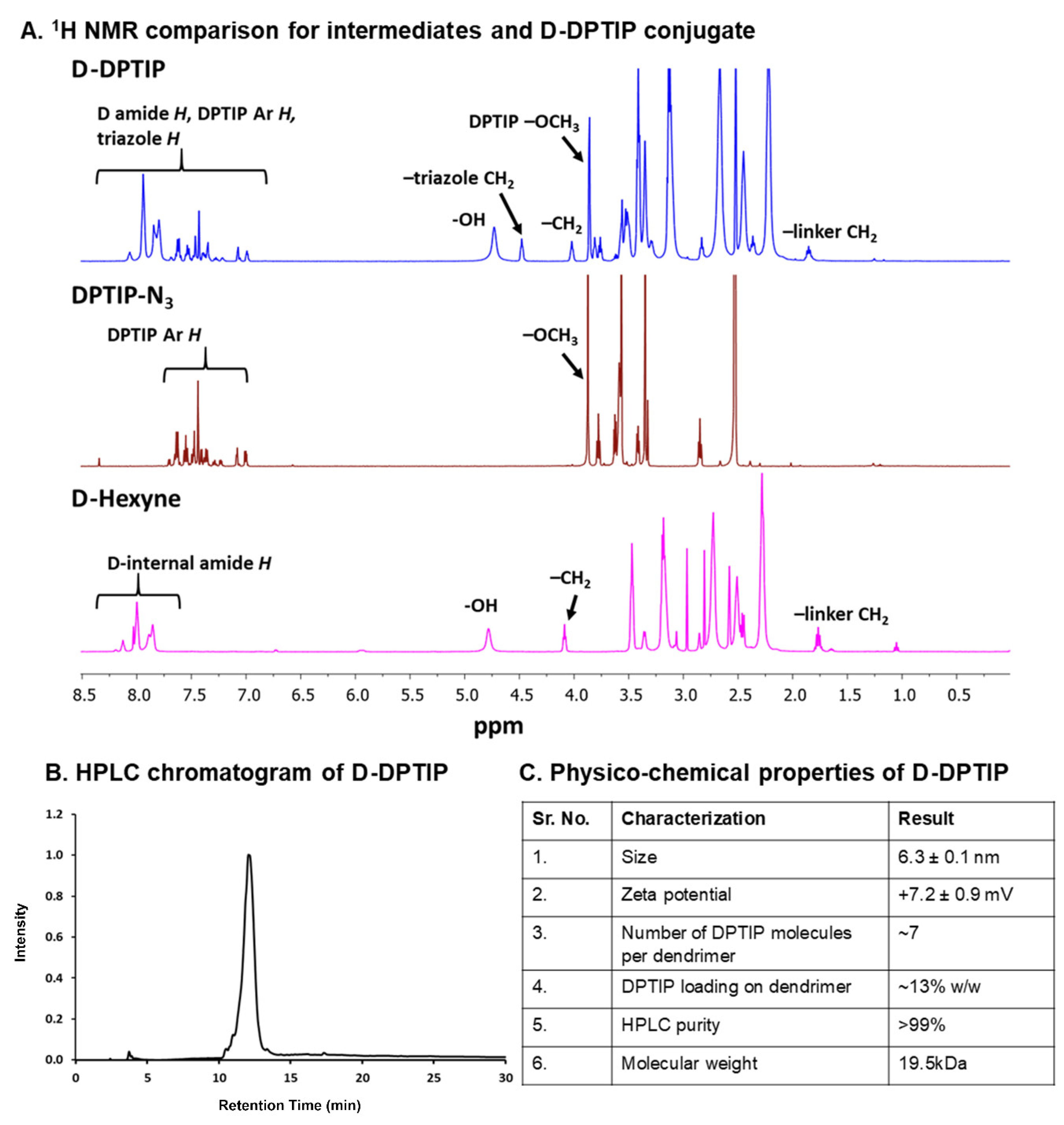
3.1. Synthesis and Characterization of D-DPTIP
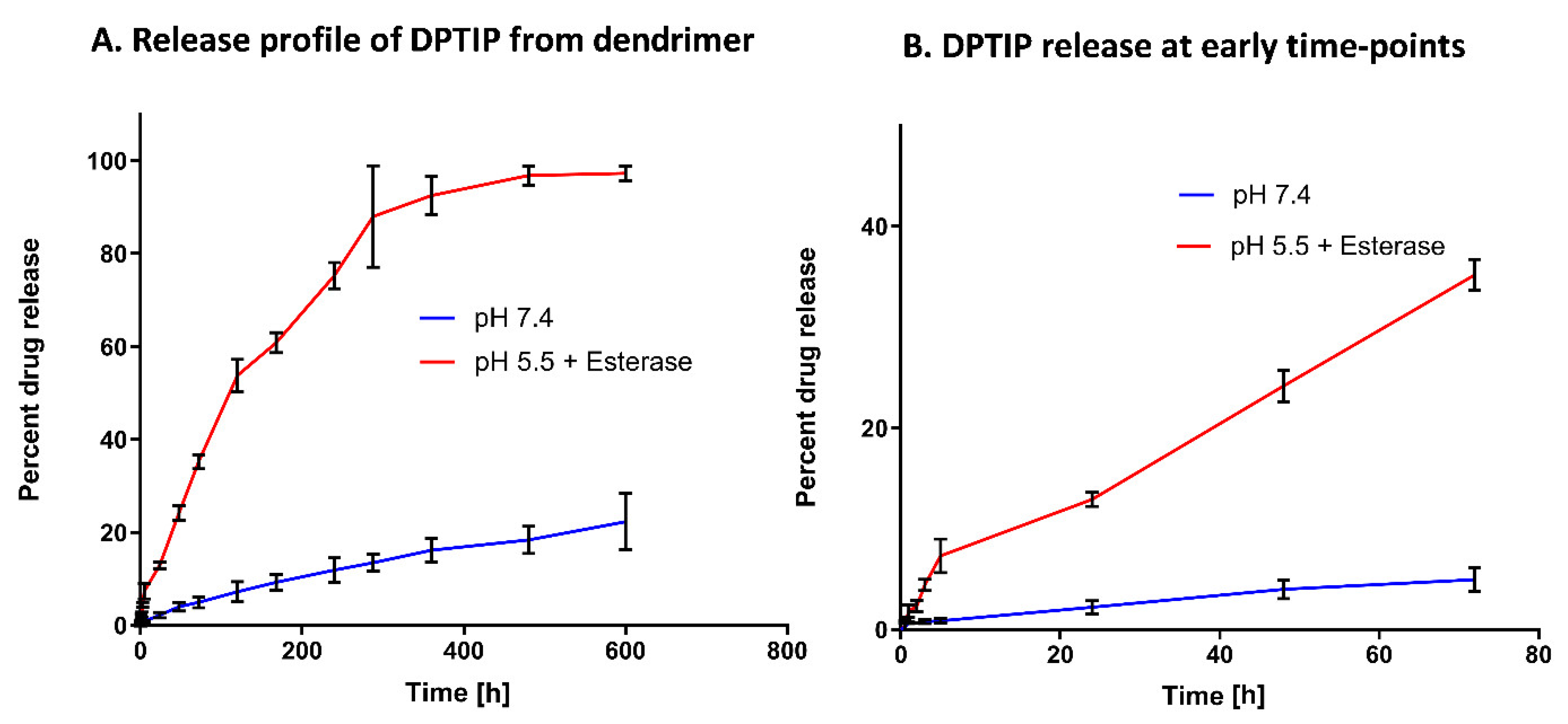
3.2. In Vitro Release of DPTIP from D-DPTIP
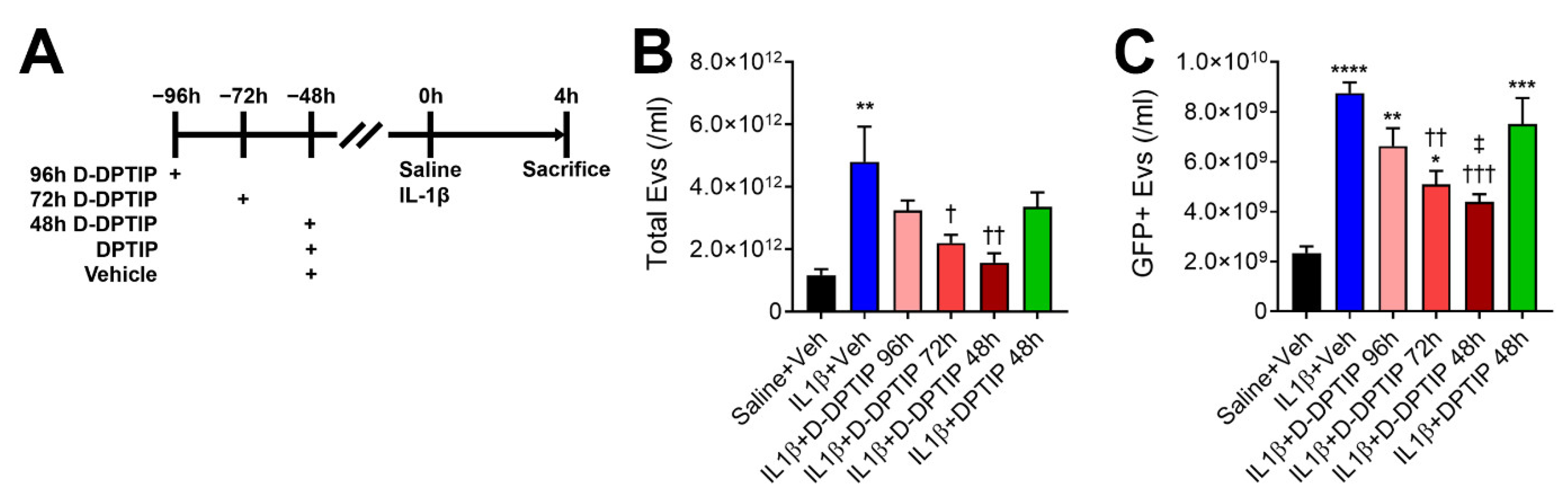
3.3. Oral D-DPTIP Inhibits EV Release into Plasma Following Acute Brain Injury in Mice
3.4. Oral D-DPTIP Is Localized to Areas of Active Neuroinflammation
3.5. Oral D-DPTIP Inhibits nSMase2 Activity in the Hippocampus and Is Well Tolerated
3.6. D-DPTIP Significantly Inhibits Tau Propagation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in brain function occur years before the onset of cognitive impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Joe, E.; Ringman, J.M. Cognitive symptoms of Alzheimer’s disease: Clinical management and prevention. BMJ 2019, 367, l6217. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W.J.; Landau, S.M.; Alzheimer’s Disease Neuroimaging Initiative. Temporal Dynamics of beta-Amyloid Accumulation in Aging and Alzheimer Disease. Neurology 2021, 96, e1347–e1357. [Google Scholar] [CrossRef]
- Lowe, V.J.; Wiste, H.J.; Senjem, M.L.; Weigand, S.D.; Therneau, T.M.; Boeve, B.F.; Josephs, K.A.; Fang, P.; Pandey, M.K.; Murray, M.E.; et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 2018, 141, 271–287. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Schonhaut, D.R.; Schöll, M.; Lockhart, S.N.; Ayakta, N.; Baker, S.L.; O’Neil, J.P.; Janabi, M.; Lazaris, A.; Cantwell, A.; et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016, 139, 1551–1567. [Google Scholar] [CrossRef]
- Tallon, C.; Hollinger, K.R.; Pal, A.; Bell, B.J.; Rais, R.; Tsukamoto, T.; Witwer, K.W.; Haughey, N.J.; Slusher, B.S. Nipping disease in the bud: nSMase2 inhibitors as therapeutics in extracellular vesicle-mediated diseases. Drug Discov. Today 2021, 26, 1656–1668. [Google Scholar] [CrossRef]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrügger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 2019, 8, 1684862. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Wu, B.X.; Clarke, C.J.; Hannun, Y.A. Mammalian neutral sphingomyelinases: Regulation and roles in cell signaling responses. Neuromolecular. Med. 2010, 12, 320–330. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef]
- Longobardi, A.; Nicsanu, R.; Bellini, S.; Squitti, R.; Catania, M.; Tiraboschi, P.; Saraceno, C.; Ferrari, C.; Zanardini, R.; Binetti, G.; et al. Cerebrospinal Fluid EV Concentration and Size Are Altered in Alzheimer’s Disease and Dementia with Lewy Bodies. Cells 2022, 11, 462. [Google Scholar] [CrossRef]
- Ruan, Z. Extracellular vesicles drive tau spreading in Alzheimer’s disease. Neural. Regen. Res. 2022, 17, 328–329. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef]
- Vogel, J.W.; Iturria-Medina, Y.; Strandberg, O.T.; Smith, R.; Levitis, E.; Evans, A.C.; Hansson, O.; Weiner, M.; Aisen, P.; Petersen, R.; et al. Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 2020, 11, 2612. [Google Scholar] [CrossRef]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef]
- Clavaguera, F.; Hench, J.; Goedert, M.; Tolnay, M. Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 2015, 41, 47–58. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Bilousova, T.; Elias, C.; Miyoshi, E.; Alam, M.P.; Zhu, C.; Campagna, J.; Vadivel, K.; Jagodzinska, B.; Gylys, K.H.; John, V. Suppression of tau propagation using an inhibitor that targets the DK-switch of nSMase2. Biochem. Biophys. Res. Commun. 2018, 499, 751–757. [Google Scholar] [CrossRef]
- Luberto, C.; Hassler, D.F.; Signorelli, P.; Okamoto, Y.; Sawai, H.; Boros, E.; Hazen-Martin, D.J.; Obeid, L.M.; Hannun, Y.A.; Smith, G.K. Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 2002, 277, 41128–41139. [Google Scholar] [CrossRef]
- Figuera-Losada, M.; Stathis, M.; Dorskind, J.M.; Thomas, A.G.; Bandaru, V.V.; Yoo, S.W.; Westwood, N.J.; Rogers, G.W.; McArthur, J.C.; Haughey, N.J.; et al. Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS ONE 2015, 10, e0124481. [Google Scholar] [CrossRef]
- Taguchi, M.; Goda, K.; Sugimoto, K.; Akama, T.; Yamamoto, K.; Suzuki, T.; Tomishima, Y.; Nishiguchi, M.; Arai, K.; Takahashi, K.; et al. Biological evaluation of sphingomyelin analogues as inhibitors of sphingomyelinase. Bioorg. Med. Chem. Lett. 2003, 13, 3681–3684. [Google Scholar] [CrossRef]
- Nara, F.; Tanaka, M.; Hosoya, T.; Suzuki-Konagai, K.; Ogita, T. Scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima: Taxonomy of the producing organism, fermentation, isolation, and physico-chemical properties. J. Antibiot. (Tokyo) 1999, 52, 525–530. [Google Scholar] [CrossRef]
- Rojas, C.; Sala, M.; Thomas, A.G.; Datta Chaudhuri, A.; Yoo, S.W.; Li, Z.; Dash, R.P.; Rais, R.; Haughey, N.J.; Nencka, R.; et al. A novel and potent brain penetrant inhibitor of extracellular vesicle release. Br. J. Pharm. 2019, 176, 3857–3870. [Google Scholar] [CrossRef]
- Sala, M.; Hollinger, K.R.; Thomas, A.G.; Dash, R.P.; Tallon, C.; Veeravalli, V.; Lovell, L.; Kögler, M.; Hrebabecky, H.; Prochazkova, E.; et al. Novel human neutral sphingomyelinase 2 inhibitors as potential therapeutics for Alzheimer disease. J. Med. Chem. 2020, 63, 6028–6056. [Google Scholar] [CrossRef]
- Rojas, C.; Barnaeva, E.; Thomas, A.G.; Hu, X.; Southall, N.; Marugan, J.; Chaudhuri, A.D.; Yoo, S.-W.; Hin, N.; Stepanek, O.; et al. DPTIP, a newly identified potent brain penetrant neutral sphingomyelinase 2 inhibitor, regulates astrocyte-peripheral immune communication following brain inflammation. Sci. Rep. 2018, 8, 17715. [Google Scholar] [CrossRef]
- Pal, A.; Gori, S.; Yoo, S.-w.; Thomas, A.G.; Wu, Y.; Friedman, J.; Tenora, L.; Bhasin, H.; Alt, J.; Haughey, N.; et al. Discovery of Orally Bioavailable and Brain-Penetrable Prodrugs of the Potent nSMase2 Inhibitor DPTIP. J. Med. Chem. 2022, 65, 11111–11125. [Google Scholar] [CrossRef]
- Stepanek, O.; Hin, N.; Thomas, A.G.; Dash, R.P.; Alt, J.; Rais, R.; Rojas, C.; Slusher, B.S.; Tsukamoto, T. Neutral sphingomyelinase 2 inhibitors based on the 4-(1H-imidazol-2-yl)-2,6-dialkoxyphenol scaffold. Eur. J. Med. Chem. 2019, 170, 276–289. [Google Scholar] [CrossRef]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.J.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Menjoge, A.R.; Kannan, R.M.; Tomalia, D.A. Dendrimer-based drug and imaging conjugates: Design considerations for nanomedical applications. Drug Discov. Today 2010, 15, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Hayder, M.; Poupot, M.; Baron, M.; Nigon, D.; Turrin, C.-O.; Caminade, A.-M.; Majoral, J.-P.; Eisenberg, R.A.; Fournié, J.-J.; Cantagrel, A.; et al. A Phosphorus-Based Dendrimer Targets Inflammation and Osteoclastogenesis in Experimental Arthritis. Sci. Transl. Med. 2011, 3, ra35–ra81. [Google Scholar] [CrossRef] [PubMed]
- Shaunak, S. Dendrimer drugs prevent scar tissue formation. Discov. Med. 2004, 4, 464–469. [Google Scholar] [PubMed]
- Nigavekar, S.S.; Sung, L.Y.; Llanes, M.; El-Jawahri, A.; Lawrence, T.S.; Becker, C.W.; Balogh, L.; Khan, M.K. 3H dendrimer nanoparticle organ/tumor distribution. Pharm. Res. 2004, 21, 476–483. [Google Scholar] [CrossRef]
- Sarin, H.; Kanevsky, A.S.; Wu, H.; Brimacombe, K.R.; Fung, S.H.; Sousa, A.A.; Auh, S.; Wilson, C.M.; Sharma, K.; Aronova, M.A.; et al. Effective transvascular delivery of nanoparticles across the blood-brain tumor barrier into malignant glioma cells. J. Transl. Med. 2008, 6, 80. [Google Scholar] [CrossRef]
- Kukowska-Latallo, J.F.; Candido, K.A.; Cao, Z.; Nigavekar, S.S.; Majoros, I.J.; Thomas, T.P.; Balogh, L.P.; Khan, M.K.; Baker, J.R., Jr. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005, 65, 5317–5324. [Google Scholar] [CrossRef]
- Kannan, R.M.; Nance, E.; Kannan, S.; Tomalia, D.A. Emerging concepts in dendrimer-based nanomedicine: From design principles to clinical applications. J. Intern. Med. 2014, 276, 579–617. [Google Scholar] [CrossRef]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Niño, D.F.; Zhou, Q.; Yamaguchi, Y.; Martin, L.Y.; Wang, S.; Fulton, W.B.; Jia, H.; Lu, P.; Prindle, T.; Zhang, F.; et al. Cognitive impairments induced by necrotizing enterocolitis can be prevented by inhibiting microglial activation in mouse brain. Sci. Transl. Med. 2018, 10, eaan0237. [Google Scholar] [CrossRef]
- Qi, R.; Li, Y.-z.; Chen, C.; Cao, Y.-n.; Yu, M.-m.; Xu, L.; He, B.; Jie, X.; Shen, W.-w.; Wang, Y.-n.; et al. G5-PEG PAMAM dendrimer incorporating nanostructured lipid carriers enhance oral bioavailability and plasma lipid-lowering effect of probucol. J. Control. Release 2015, 210, 160–168. [Google Scholar] [CrossRef]
- Qi, R.; Zhang, H.; Xu, L.; Shen, W.; Chen, C.; Wang, C.; Cao, Y.; Wang, Y.; van Dongen, M.A.; He, B.; et al. G5 PAMAM dendrimer versus liposome: A comparison study on the in vitro transepithelial transport and in vivo oral absorption of simvastatin. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1141–1151. [Google Scholar] [CrossRef]
- Hollinger, K.R.; Sharma, A.; Tallon, C.; Lovell, L.; Thomas, A.G.; Zhu, X.; Wiseman, R.; Wu, Y.; Kambhampati, S.P.; Liaw, K.; et al. Dendrimer-2PMPA selectively blocks upregulated microglial GCPII activity and improves cognition in a mouse model of multiple sclerosis. Nanotheranostics 2022, 6, 126–142. [Google Scholar] [CrossRef]
- Tallon, C.; Sharma, A.; Zhang, Z.; Thomas, A.G.; Ng, J.; Zhu, X.; Donoghue, A.; Schulte, M.; Joe, T.R.; Kambhampati, S.P.; et al. Dendrimer-2PMPA Delays Muscle Function Loss and Denervation in a Murine Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2022, 19, 274–288. [Google Scholar] [CrossRef]
- Nance, E.; Kambhampati, S.P.; Smith, E.S.; Zhang, Z.; Zhang, F.; Singh, S.; Johnston, M.V.; Kannan, R.M.; Blue, M.E.; Kannan, S. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J. Neuroinflammation 2017, 14, 252. [Google Scholar] [CrossRef]
- Singh, A.K.; Gothwal, A.; Rani, S.; Rana, M.; Sharma, A.K.; Yadav, A.K.; Gupta, U. Dendrimer Donepezil Conjugates for Improved Brain Delivery and Better in Vivo Pharmacokinetics. ACS Omega 2019, 4, 4519–4529. [Google Scholar] [CrossRef]
- Kannan, S.; Dai, H.; Navath, R.S.; Balakrishnan, B.; Jyoti, A.; Janisse, J.; Romero, R.; Kannan, R.M. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 2012, 4, 130ra46. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, Y.A.; Kim, S.Y.; Su, L.; Liu, J.; Kannan, R.M.; Kannan, S. Systemic dendrimer-drug nanomedicines for long-term treatment of mild-moderate cerebral palsy in a rabbit model. J. Neuroinflammation 2020, 17, 319. [Google Scholar] [CrossRef]
- Mishra, M.K.; Beaty, C.A.; Lesniak, W.G.; Kambhampati, S.P.; Zhang, F.; Wilson, M.A.; Blue, M.E.; Troncoso, J.C.; Kannan, S.; Johnston, M.V.; et al. Dendrimer brain uptake and targeted therapy for brain injury in a large animal model of hypothermic circulatory arrest. ACS Nano 2014, 8, 2134–2147. [Google Scholar] [CrossRef]
- Inapagolla, R.; Guru, B.R.; Kurtoglu, Y.E.; Gao, X.; Lieh-Lai, M.; Bassett, D.J.P.; Kannan, R.M. In vivo efficacy of dendrimer–methylprednisolone conjugate formulation for the treatment of lung inflammation. Int. J. Pharm. 2010, 399, 140–147. [Google Scholar] [CrossRef]
- Kambhampati, S.P.; Kannan, R.M. Dendrimer nanoparticles for ocular drug delivery. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2013, 29, 151–165. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Faraday, N.; Aita, J.S.; Kumar, S.; Mehta, I.; Choi, H.A.; Cleland, J.L.; Robinson, K.; McCullough, L.D.; Ng, D.K.; et al. Dendrimer nanotherapy for severe COVID-19 attenuates inflammation and neurological injury markers and improves outcomes in a phase2a clinical trial. Sci. Transl. Med. 2022, 14, eabo2652. [Google Scholar] [CrossRef]
- Sharma, R.; Kambhampati, S.P.; Zhang, Z.; Sharma, A.; Chen, S.; Duh, E.I.; Kannan, S.; Tso, M.O.M.; Kannan, R.M. Dendrimer mediated targeted delivery of sinomenine for the treatment of acute neuroinflammation in traumatic brain injury. J. Control. Release 2020, 323, 361–375. [Google Scholar] [CrossRef]
- Sharma, A.; Liaw, K.; Sharma, R.; Spriggs, T.; Appiani La Rosa, S.; Kannan, S.; Kannan, R.M. Dendrimer-Mediated Targeted Delivery of Rapamycin to Tumor-Associated Macrophages Improves Systemic Treatment of Glioblastoma. Biomacromolecules 2020, 21, 5148–5161. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Wang, R.; Tang, H.; Dong, Y.; Chan, A.; Sareddy, G.R.; Vadlamudi, R.K.; Brann, D.W. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol. Cell. Endocrinol. 2014, 389, 84–91. [Google Scholar] [CrossRef]
- Dickens, A.M.; Tovar-y-Romo, L.B.; Yoo, S.-W.; Trout, A.L.; Bae, M.; Kanmogne, M.; Megra, B.; Williams, D.W.; Witwer, K.W.; Gacias, M.; et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 2017, 10, eaai7696. [Google Scholar] [CrossRef]
- Tallon, C.; Picciolini, S.; Yoo, S.-W.; Thomas, A.G.; Pal, A.; Alt, J.; Carlomagno, C.; Gualerzi, A.; Rais, R.; Haughey, N.J.; et al. Inhibition of neutral sphingomyelinase 2 reduces extracellular vesicle release from neurons, oligodendrocytes, and activated microglial cells following acute brain injury. Biochem. Pharmacol. 2021, 194, 114796. [Google Scholar] [CrossRef]
- Koller, E.J.; Gonzalez De La Cruz, E.; Machula, T.; Ibanez, K.R.; Lin, W.-L.; Williams, T.; Riffe, C.J.; Ryu, D.; Strang, K.H.; Liu, X.; et al. Combining P301L and S320F tau variants produces a novel accelerated model of tauopathy. Hum. Mol. Genet. 2019, 28, 3255–3269. [Google Scholar] [CrossRef]
- Geng, Z.; Shin, J.J.; Xi, Y.; Hawker, C.J. Click chemistry strategies for the accelerated synthesis of functional macromolecules. J. Polym. Sci. 2021, 59, 963–1042. [Google Scholar] [CrossRef]
- Sharma, A.; Liaw, K.; Sharma, R.; Thomas, A.G.; Slusher, B.S.; Kannan, S.; Kannan, R.M. Targeting Mitochondria in Tumor-Associated Macrophages using a Dendrimer-Conjugated TSPO Ligand that Stimulates Antitumor Signaling in Glioblastoma. Biomacromolecules 2020, 21, 3909–3922. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Porterfield, J.E.; Smith, E.; Sharma, R.; Kannan, S.; Kannan, R.M. Effect of mannose targeting of hydroxyl PAMAM dendrimers on cellular and organ biodistribution in a neonatal brain injury model. J. Control Release 2018, 283, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, R.; Zhang, Z.; Liaw, K.; Kambhampati, S.P.; Porterfield, J.E.; Lin, K.C.; DeRidder, L.B.; Kannan, S.; Kannan, R.M. Dense hydroxyl polyethylene glycol dendrimer targets activated glia in multiple CNS disorders. Sci. Adv. 2020, 6, eaay8514. [Google Scholar] [CrossRef] [PubMed]
- Khoury, E.S.; Sharma, A.; Ramireddy, R.R.; Thomas, A.G.; Alt, J.; Fowler, A.; Rais, R.; Tsukamoto, T.; Blue, M.E.; Slusher, B.; et al. Dendrimer-conjugated glutaminase inhibitor selectively targets microglial glutaminase in a mouse model of Rett syndrome. Theranostics 2020, 10, 5736–5748. [Google Scholar] [CrossRef]
- Sharma, R.; Kim, S.-Y.; Sharma, A.; Zhang, Z.; Kambhampati, S.P.; Kannan, S.; Kannan, R.M. Activated Microglia Targeting Dendrimer–Minocycline Conjugate as Therapeutics for Neuroinflammation. Bioconjug. Chem. 2017, 28, 2874–2886. [Google Scholar] [CrossRef]
- Perumal, O.P.; Inapagolla, R.; Kannan, S.; Kannan, R.M. The effect of surface functionality on cellular trafficking of dendrimers. Biomaterials 2008, 29, 3469–3476. [Google Scholar] [CrossRef]
- Bell, B.J.; Malvankar, M.M.; Tallon, C.; Slusher, B.S. Sowing the Seeds of Discovery: Tau-Propagation Models of Alzheimer’s Disease. ACS Chem. Neurosci. 2020, 11, 3499–3509. [Google Scholar] [CrossRef]
- Oh, S.W.; Harris, J.A.; Ng, L.; Winslow, B.; Cain, N.; Mihalas, S.; Wang, Q.; Lau, C.; Kuan, L.; Henry, A.M.; et al. A mesoscale connectome of the mouse brain. Nature 2014, 508, 207–214. [Google Scholar] [CrossRef]
- Harris, J.A.; Mihalas, S.; Hirokawa, K.E.; Whitesell, J.D.; Choi, H.; Bernard, A.; Bohn, P.; Caldejon, S.; Casal, L.; Cho, A.; et al. Hierarchical organization of cortical and thalamic connectivity. Nature 2019, 575, 195–202. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Barnes, L.L.; Wilson, R.S.; Bienias, J.L.; Schneider, J.A.; Evans, D.A.; Bennett, D.A. Sex Differences in the Clinical Manifestations of Alzheimer Disease Pathology. Arch. Gen. Psychiatry 2005, 62, 685–691. [Google Scholar] [CrossRef]
- Oveisgharan, S.; Arvanitakis, Z.; Yu, L.; Farfel, J.; Schneider, J.A.; Bennett, D.A. Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol. 2018, 136, 887–900. [Google Scholar] [CrossRef]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Otto, G.P.; Rathkolb, B.; Oestereicher, M.A.; Lengger, C.J.; Moerth, C.; Micklich, K.; Fuchs, H.; Gailus-Durner, V.; Wolf, E.; Hrabě de Angelis, M. Clinical Chemistry Reference Intervals for C57BL/6J, C57BL/6N, and C3HeB/FeJ Mice (Mus musculus). J. Am. Assoc. Lab. Anim. Sci. JAALAS 2016, 55, 375–386. [Google Scholar]
- The Jackson Laboratory. Physiological Data Summary—C57BL/6J (000664). Available online: https://jackson.jax.org/rs/444-BUH-304/images/physiological_data_000664.pdf (accessed on 2 August 2022).
- Charles River Larboratories International, Inc. C57BL/6 Mice Datasheet. Available online: https://www.criver.com/sites/default/files/resources/C57BL6MouseModelInformationSheet.pdf (accessed on 2 August 2022).
- Taconic Biosciences, Inc. Automated Clinical Chemistry Analysis (ACCA). Available online: https://www.taconic.com/phenotypic-data/automated-clinical-chemistry-analysis/ (accessed on 2 August 2022).
- Kumar, A.; Henry, R.J.; Stoica, B.A.; Loane, D.J.; Abulwerdi, G.; Bhat, S.A.; Faden, A.I. Neutral Sphingomyelinase Inhibition Alleviates LPS-Induced Microglia Activation and Neuroinflammation after Experimental Traumatic Brain Injury. J. Pharm. Exp. 2019, 368, 338–352. [Google Scholar] [CrossRef]
- Mielke, M.M. Sex and Gender Differences in Alzheimer’s Disease Dementia. Psychiatr. Times 2018, 35, 14–17. [Google Scholar]
- Mielke, M.M.; Bandaru, V.V.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum ceramides increase the risk of Alzheimer disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Han, D.; An, Y.; Bandaru, V.V.R.; Lyketsos, C.G.; Ferrucci, L.; Resnick, S.M. The Association Between Plasma Ceramides and Sphingomyelins and Risk of Alzheimer’s Disease Differs by Sex and APOE in the Baltimore Longitudinal Study of Aging. J. Alzheimers Dis. 2017, 60, 819–828. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallon, C.; Bell, B.J.; Sharma, A.; Pal, A.; Malvankar, M.M.; Thomas, A.G.; Yoo, S.-W.; Hollinger, K.R.; Coleman, K.; Wilkinson, E.L.; et al. Dendrimer-Conjugated nSMase2 Inhibitor Reduces Tau Propagation in Mice. Pharmaceutics 2022, 14, 2066. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102066
Tallon C, Bell BJ, Sharma A, Pal A, Malvankar MM, Thomas AG, Yoo S-W, Hollinger KR, Coleman K, Wilkinson EL, et al. Dendrimer-Conjugated nSMase2 Inhibitor Reduces Tau Propagation in Mice. Pharmaceutics. 2022; 14(10):2066. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102066
Chicago/Turabian StyleTallon, Carolyn, Benjamin J. Bell, Anjali Sharma, Arindom Pal, Medhinee M. Malvankar, Ajit G. Thomas, Seung-Wan Yoo, Kristen R. Hollinger, Kaleem Coleman, Elizabeth L. Wilkinson, and et al. 2022. "Dendrimer-Conjugated nSMase2 Inhibitor Reduces Tau Propagation in Mice" Pharmaceutics 14, no. 10: 2066. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14102066