
Predicting Drug Release Rate of Implantable Matrices and Better Understanding of the Underlying Mechanisms through Experimental Design and Artificial Neural Network-Based Modelling

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Tablet Preparation
2.2.2. Physical Properties
2.2.3. Physicochemical Characterization
2.2.4. Dissolution Tests
2.2.5. Design of Experiments and Artificial Neural Networks
3. Results and Discussion
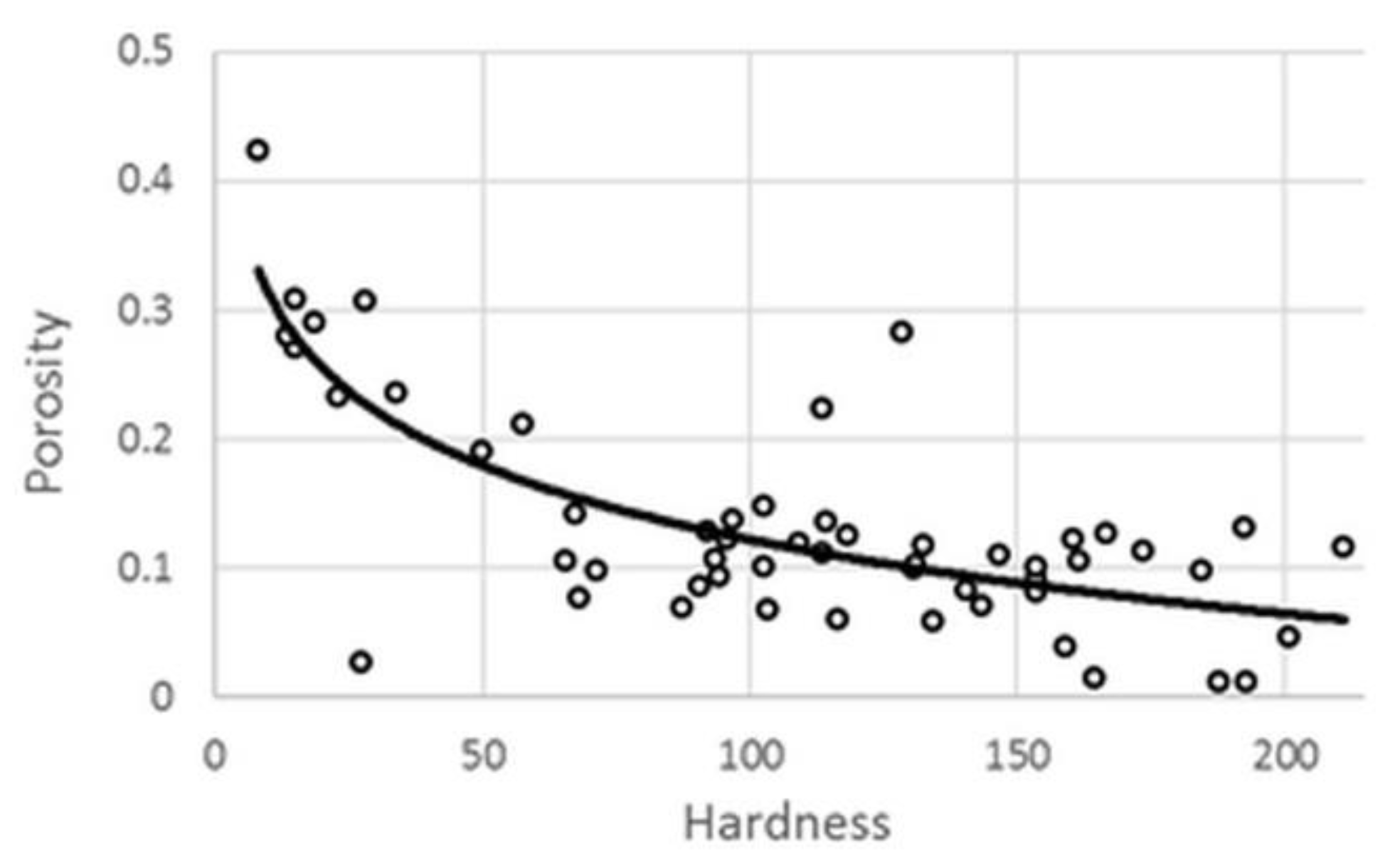
3.1. Physical Parameters
R2 = 0.9743 adj. R2 = 0.9599 MS Res = 123.02
R2 = 0.7627 adj R2 = 0.6775 MS Res = 0.0023
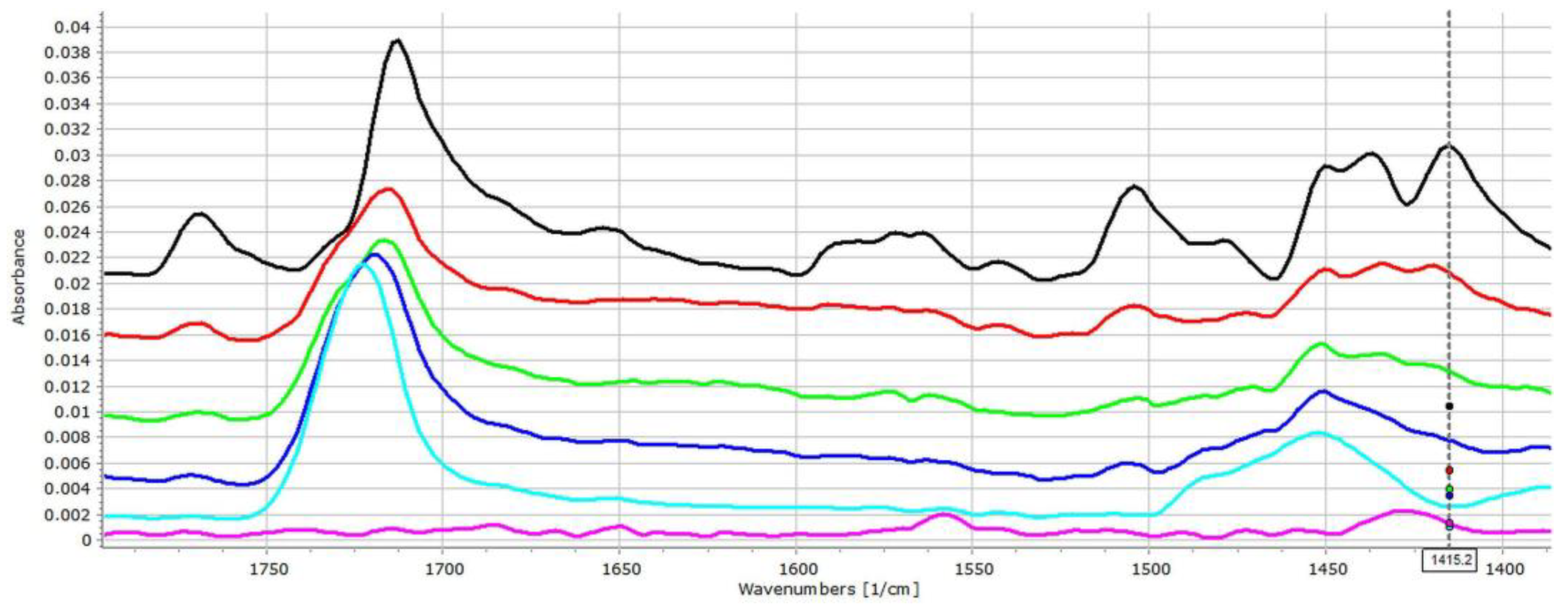
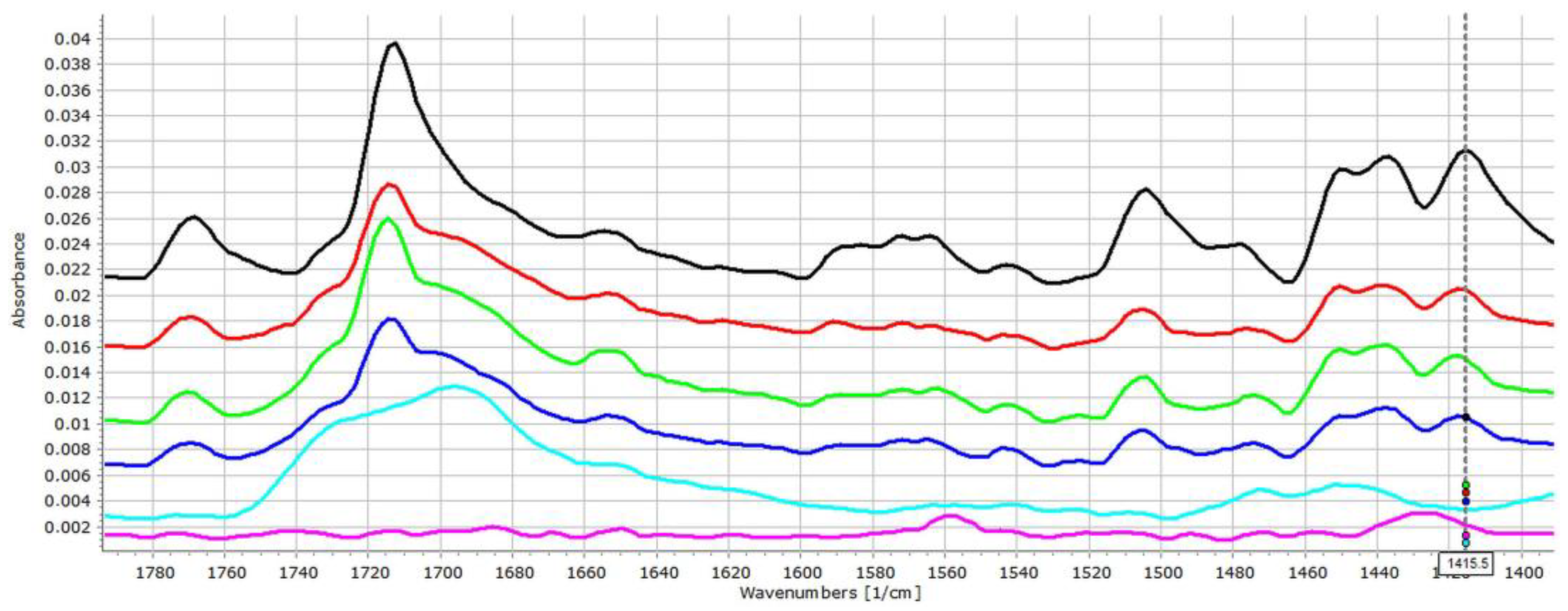
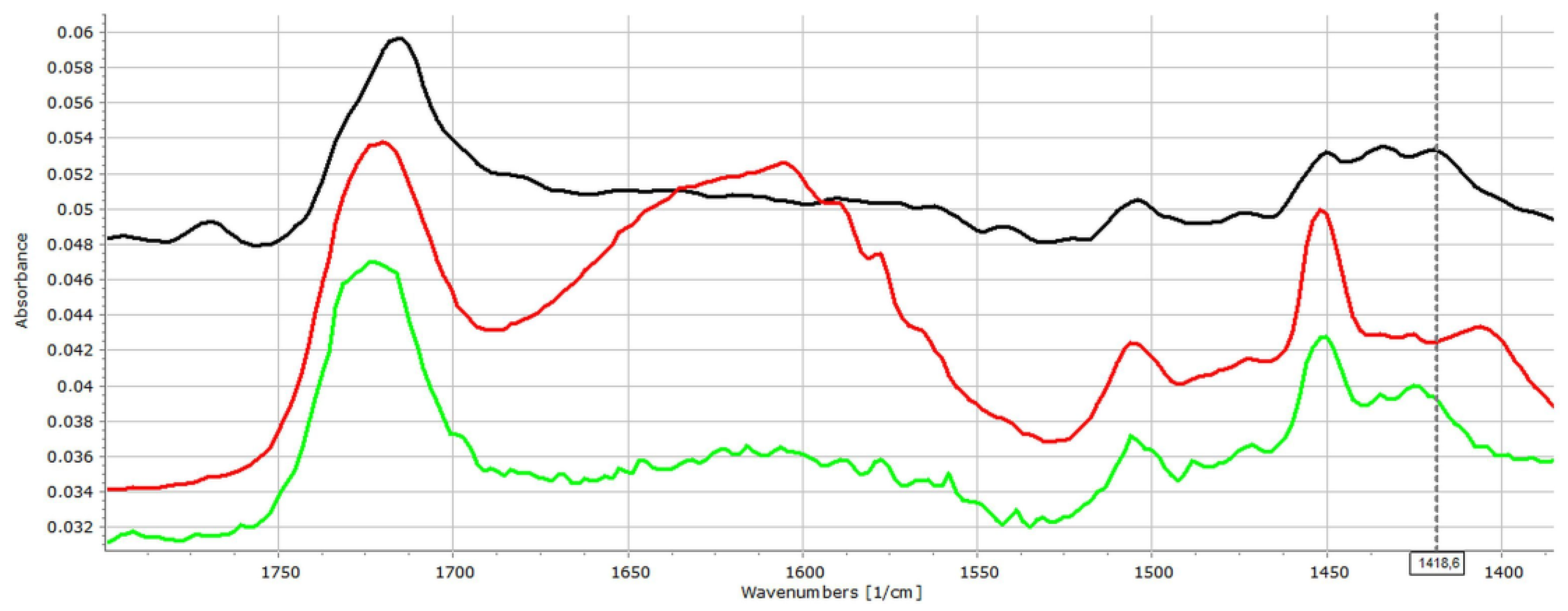
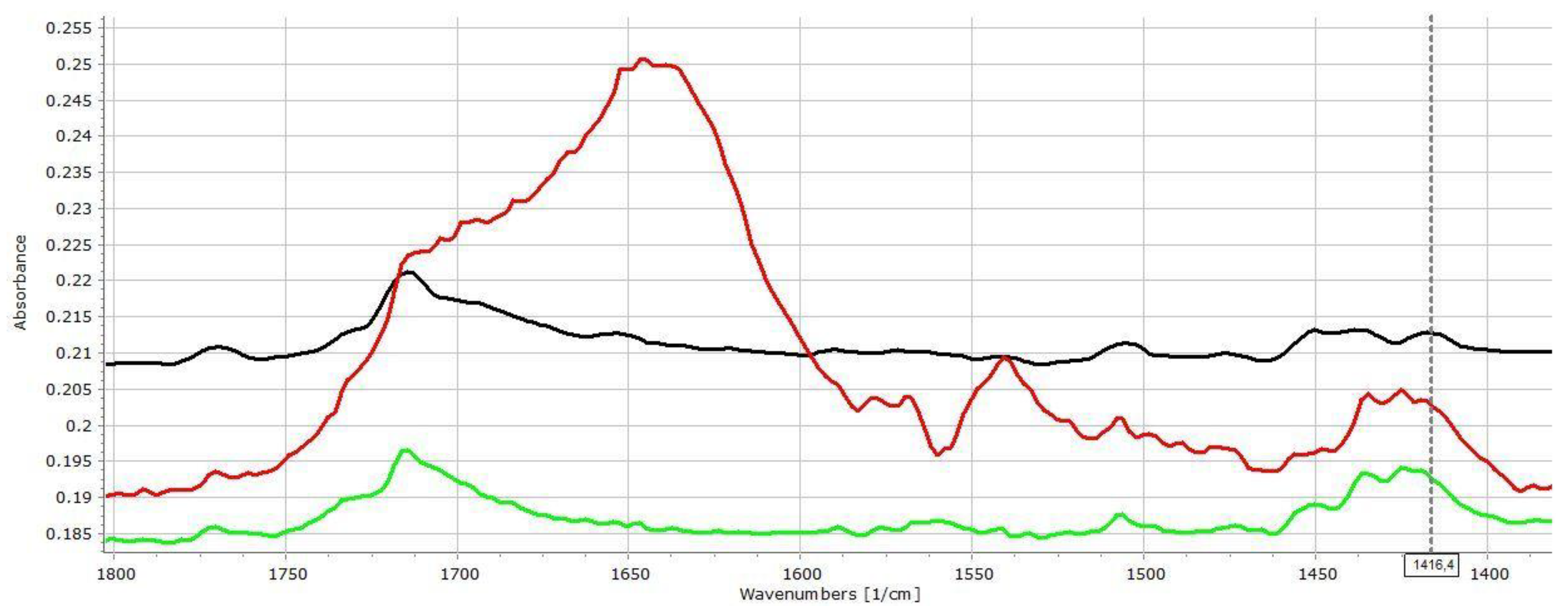
3.2. Investigation of Drug–Carrier Interactions
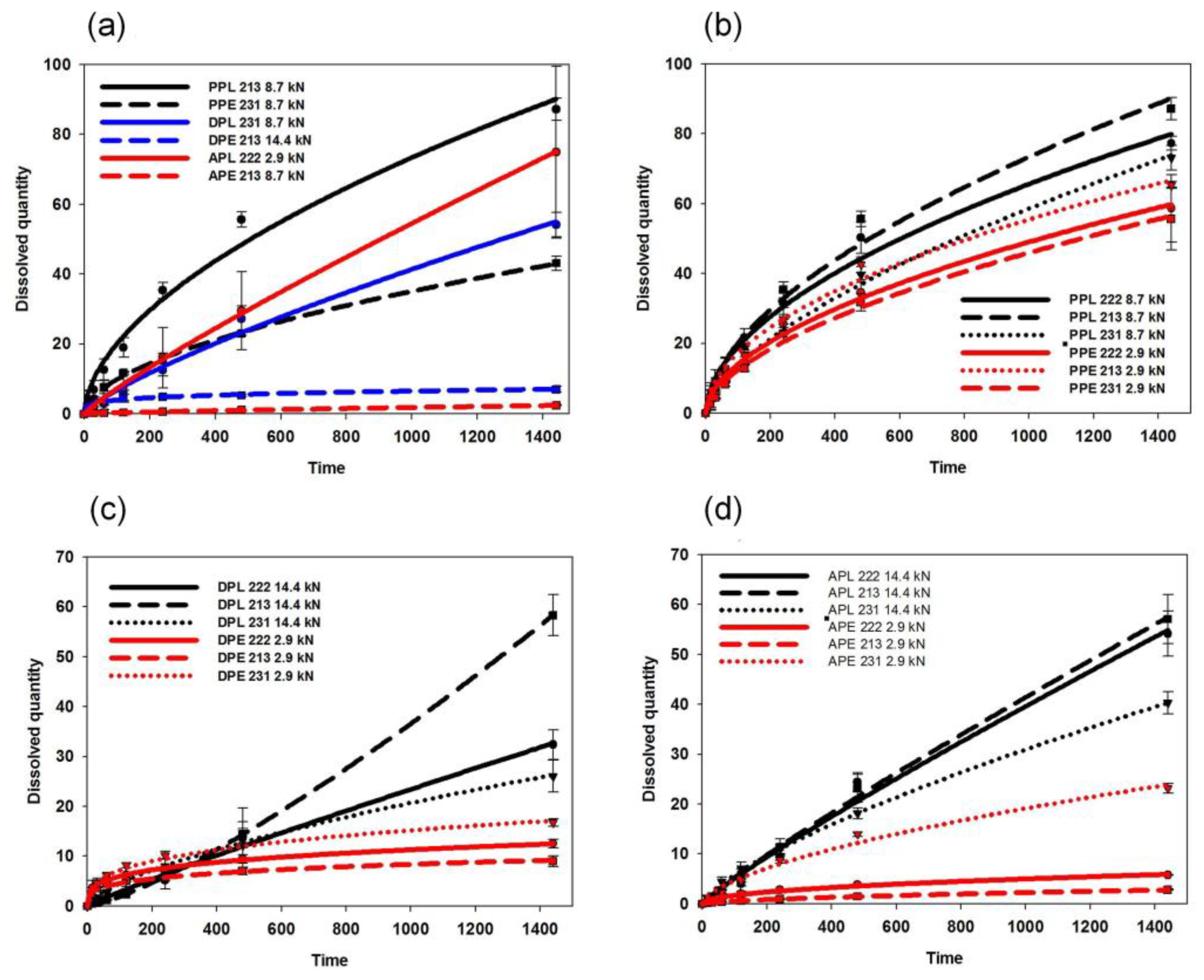
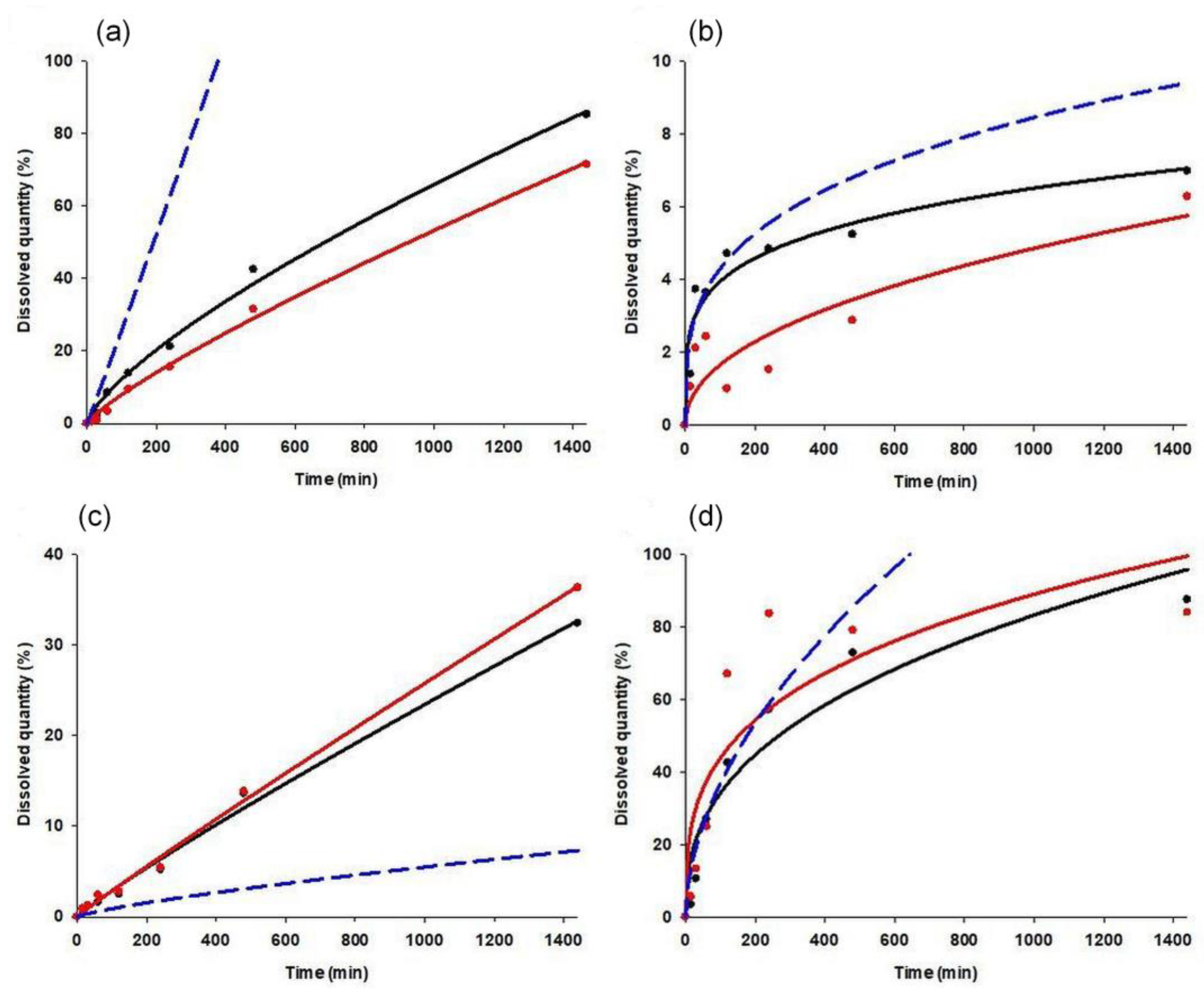
3.3. Dissolution Tests and Kinetic Study
R2 = 0.7669 adj R2 = 0.6661 MS Res = 0.5176
3.4. ANN Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crowley, M.M.; Schroeder, B.; Fredersdorf, A.; Obara, S.; Talarico, M.; Kucera, S.; McGinity, J.W. Physicochemical properties and mechanism of drug release from ethyl cellulose matrix tablets prepared by direct compression and hot-melt extrusion. Int. J. Pharm. 2004, 269, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Lee, P.I.; Peppas, N.A. Prediction of polymer dissolution in swellable controlled-release systems. J. Control. Release 1987, 6, 207–215. [Google Scholar] [CrossRef]
- Campos-Aldrete, M.E.; Villafuerte-Robles, L. Influence of the viscosity grade and the particle size of HPMC on metronidazole release from matrix tablets. Eur. J. Pharm. Biopharm. 1997, 43, 173–178. [Google Scholar] [CrossRef]
- Huanbutta, K.; Cheewatanakornkool, K.; Terada, K.; Nunthanid, J.; Sriamornsak, P. Impact of salt form and molecular weight of chitosan on swelling and drug release from chitosan matrix tablets. Carbohydr. Polym. 2013, 97, 26–33. [Google Scholar] [CrossRef]
- Thomas, N.; Windle, A. A theory of case II diffusion. Polymers 1982, 23, 529–542. [Google Scholar] [CrossRef]
- Kaunisto, E.; Alami, S.A.; Borgquist, P.; Larsson, A.; Nilsson, B.; Axelsson, A. A mechanistic modelling approach to polymer dissolution using magnetic resonance microimaging. J. Control. Release 2010, 147, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Dhiman, A.; Kumar, A. Slow release of ciprofloxacin from β-cyclodextrin containing drug delivery system through network formation and supramolecular interactions. Int. J. Biol. Macromol. 2016, 92, 390–400. [Google Scholar] [CrossRef]
- Deaconu, M.; Brezoiu, A.-M.; Mitran, R.-A.; Nicu, I.; Manolescu, B.; Matei, C.; Berger, D. Exploiting the zwitterionic properties of lomefloxacin to tailor its delivery from functionalized MCM-41 silica. Microporous Mesoporous Mater. 2020, 305, 110323. [Google Scholar] [CrossRef]
- Huang, J.; Jacobsen, J.; Larsen, S.; Genina, N.; van de Weert, M.; Müllertz, A.; Nielsen, H.; Mu, H. Graphene oxide as a functional excipient in buccal films for delivery of clotrimazole: Effect of molecular interactions on drug release and antifungal activity in vitro. Int. J. Pharm. 2020, 589, 119811. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, S.; Bonferoni, M.C.; Elviri, L.; Sandri, G.; Caramella, C.; Bettini, R. Advances in oral controlled drug delivery: The role of drug–polymer and interpolymer non-covalent interactions. Expert Opin. Drug Deliv. 2014, 12, 441–453. [Google Scholar] [CrossRef]
- Sovány, T.; Csüllög, A.; Benkő, E.; Regdon, G.; Pintye-Hódi, K. Comparison of the properties of implantable matrices prepared from degradable and non-degradable polymers for bisphosphonate delivery. Int. J. Pharm. 2017, 533, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Brocca-Cofano, E.; Castaldello, A.; De Michele, R.; Altavilla, G.; Marchisio, M.; Gavioli, R.; Rolen, U.; Chiarantini, L.; Cerasi, A.; et al. Novel biocompatible anionic polymeric microspheres for the delivery of the HIV-1 Tat protein for vaccine application. Vaccine 2004, 22, 2910–2924. [Google Scholar] [CrossRef] [PubMed]
- Schlocker, W.; Gschließer, S.; Bernkop-Schnürch, A. Evaluation of the potential of air jet milling of solid protein-poly(acrylate) complexes for microparticle preparation. Eur. J. Pharm. Biopharm. 2006, 62, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.E.; Lamprecht, A. Polyethylene glycol as an alternative polymer solvent for nanoparticle preparation. Int. J. Pharm. 2013, 456, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Hari, B.V.; Narayanan, N.; Dhevendaran, K.; Ramyadevi, D. Engineered nanoparticles of Efavirenz using methacrylate co-polymer (Eudragit-E100) and its biological effects in-vivo. Mater. Sci. Eng. C 2016, 67, 522–532. [Google Scholar] [CrossRef]
- Thomas, T.T.; Kohane, D.S.; Wang, A.; Langer, R. Microparticulate Formulations for the Controlled Release of Interleukin. J. Pharm. Sci. 2004, 93, 1100–1109. [Google Scholar] [CrossRef]
- Basarkar, A.; Singh, J. Poly (lactide-co-glycolide)-Polymethacrylate Nanoparticles for Intramuscular Delivery of Plasmid Encoding Interleukin-10 to Prevent Autoimmune Diabetes in Mice. Pharm. Res. 2009, 26, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Balaguer-Fernández, C.; Femenía-Font, A.; del Rio-Sancho, S.; Merino, V.; López-Castellano, A. Sumatriptan Succinate Transdermal Delivery Systems for The Treatment of Migraine. J. Pharm. Sci. 2008, 97, 2102–2109. [Google Scholar] [CrossRef] [PubMed]
- Sovány, T.; Kása, P.; Pintye-Hódi, K. Modeling of subdivision of scored tablets with the application of artificial neural networks. J. Pharm. Sci. 2010, 99, 905–915. [Google Scholar] [CrossRef]
- Sovány, T.; Papós, K.; Kása, P., Jr.; Ilič, I.; Srčič, S.; Pintye-Hódi, K. Application of physicochemical properties and process parameters in the development of a neural network model for prediction of tablet characteristics. AAPS PharmSciTech 2013, 14, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Galata, D.L.; Farkas, A.; Könyves, Z.; Mészáros, L.A.; Szabó, E.; Csontos, I.; Pálos, A.; Marosi, G.; Nagy, Z.K.; Nagy, B. Fast, Spectroscopy-Based Prediction of In Vitro Dissolution Profile of Extended Release Tablets Using Artificial Neural Networks. Pharmaceutics 2019, 11, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jangir, D.K.; Charak, S.; Mehrotra, R.; Kundu, S. FTIR and circular dichroism spectroscopic study of interaction of 5-fluorouracil with DNA. J. Photochem. Photobiol. B Biol. 2011, 105, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mihaylov, M.Y.; Ivanova, E.; Vayssilov, G.N.; Hadjiivanov, K.I. Revisiting ceria-NOx interaction: FTIR studies. Catal. Today 2020, 357, 613–620. [Google Scholar] [CrossRef]
- Liu, X.; Ma, X.; Kun, E.; Guo, X.; Yu, Z.; Zhang, F. Influence of lidocaine forms (salt vs. freebase) on properties of drug–eudragit® L100-55 extrudates prepared by reactive melt extrusion. Int. J. Pharm. 2018, 547, 291–302. [Google Scholar] [CrossRef]
- Obeidat, W.M.; Nokhodchi, A.; Alkhatib, H. Evaluation of Matrix Tablets Based on Eudragit®E100/Carbopol®971P Combinations for Controlled Release and Improved Compaction Properties of Water Soluble Model Drug Paracetamol. AAPS PharmSciTech 2015, 16, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Pavli, M.; Baumgartner, S.; Kos, P.; Kogej, K. Doxazosin–carrageenan interactions: A novel approach for studying drug–polymer interactions and relation to controlled drug release. Int. J. Pharm. 2011, 421, 110–119. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Moustafine, R.I.; Zaharov, I.M.; Kemenova, V.A. Physicochemical characterization and drug release properties of Eudragit® E PO/Eudragit® L 100-55 interpolyelectrolyte complexes. Eur. J. Pharm. Biopharm. 2006, 63, 26–36. [Google Scholar] [CrossRef]
- Priemel, P.A.; Laitinen, R.; Grohganz, H.; Rades, T.; Strachan, C. In situ amorphisation of indomethacin with Eudragit® E during dissolution. Eur. J. Pharm. Biopharm. 2013, 85, 1259–1265. [Google Scholar] [CrossRef]
- Moustafine, R.I.; Sitenkov, A.Y.; Bukhovets, A.V.; Nasibullin, S.F.; Appeltans, B.; Kabanova, T.V.; Khutoryanskiy, V.; Mooter, G.V.D. Indomethacin-containing interpolyelectrolyte complexes based on Eudragit ® E PO/S 100 copolymers as a novel drug delivery system. Int. J. Pharm. 2017, 524, 121–133. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paracetamol | Diclofenac Sodium | Aceclofenac | EUDR-E | EUDR-L | PVC | |
|---|---|---|---|---|---|---|
| Solubility (pH 7.4) | 18–21 mg/mL | 5.15 mg/mL | 4.0–12.8 mg/mL | <10 µg/mL (>pH 5) | <0.03 mg/mL (>pH 6) | <10 µg/mL |
| logP | 0.34 | 3.10 | 4.16 | - | - | - |
| pKa | 9.46 | 4.00 | 3.44 | 10 | 6 | - |
| H+ acceptor | 2 | 2 | 4 | 3/n* | 3/n * | - |
| H+ donor | 2 | 2 | 2 | 0/n* | 1/n * | - |
| Rotable bonds | 3 | 3 | 5 | - | - | - |
| Compression Pressure (MPa) (x1) | Eudragit/PVC Ratio (%/%) (x2) | API (x3) | Eudragit Type (x4) | ||||
|---|---|---|---|---|---|---|---|
| Level | Value | Level | Value | Level | Value | Level | Value |
| −1 | 75 | −1 | 25/75 | −1 | Paracetamol | −1 | Eudragit E |
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| 0 | 50/50 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | 75/25 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| 0 | 225 | −1 | 25/75 | −1 | Paracetamol | −1 | Eudragit E |
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| 0 | 50/50 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | 75/25 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | 375 | −1 | 25/75 | −1 | Paracetamol | −1 | Eudragit E |
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| 0 | 50/50 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | 75/25 | −1 | Paracetamol | −1 | Eudragit E | ||
| +1 | Eudragit L | ||||||
| 0 | Diclofenac Sodium | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| +1 | Aceclofenac | −1 | Eudragit E | ||||
| +1 | Eudragit L | ||||||
| Modelling Type | Approach 1 | Approach 2 | Approach 3 | Approach 4 | Approach 5 | Approach 6 |
|---|---|---|---|---|---|---|
| Kinetic Based | Kinetic Based | Kinetic Based | Point-to-Point | Point-to-Point | Point-to-Point | |
| Input variable | ||||||
| Drug | x | x | ||||
| Drug solubility (mg/mL) | x | x | x | x | ||
| Drug pKa | x | x | x | x | ||
| Excipient | x | x | ||||
| Excipient solubility (mg/mL) | x | x | x | x | ||
| Excipient pKa | x | x | x | x | ||
| Excipient amount (%) | x | x | x | x | x | x |
| Compression pressure (MPa) | x | x | x | x | x | x |
| Hardness | x | x | ||||
| Porosity | x | x | ||||
| Peak Shift | x | x | x | x | x | x |
| Eudragit/PVC Ratio (%/%) | Compression Pressure (MPa) | Composition | Mass (mg) | Hardness (N) | Poro-Sity | Composition | Mass (mg) | Hardness (N) | Porosity | Composition | Mass (mg) | Hardness (N) | Porosity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25/75 | 75 | PAR-PVC-EL | 147.0 | 15.0 | 0.307 | DIS-PVC-EL | 152.1 | 8.0 | 0.423 | ACE-PVC-EL | 144.5 | 13.5 | 0.279 |
| 225 | 147.9 | 114.3 | 0.136 | 135.2 | 118.5 | 0.125 | 131.4 | 68.2 | 0.077 | ||||
| 375 | 141.6 | 109.1 | 0.118 | 202.6 | 140.7 | 0.082 | 131.2 | 103.6 | 0.067 | ||||
| 50/50 | 75 | 149.0 | 18.7 | 0.290 | 154.2 | 34.0 | 0.236 | 152.7 | 22.9 | 0.232 | |||
| 225 | 135.9 | 95.8 | 0.122 | 138.7 | 92.0 | 0.128 | 137.8 | 65.8 | 0.104 | ||||
| 375 | 142.2 | 133.6 | 0.111 | 146.9 | 134.4 | 0.058 | 142.4 | 71.3 | 0.097 | ||||
| 75/25 | 75 | 147.0 | 28.2 | 0.306 | 153.8 | 27.3 | 0.026 | 147.7 | 15.2 | 0.027 | |||
| 225 | 151.7 | 132.6 | 0.118 | 136.9 | 97.0 | 0.137 | 134.9 | 90.7 | 0.086 | ||||
| 375 | 149.7 | 153.8 | 0.091 | 121.5 | 153.8 | 0.100 | 137.1 | 116.7 | 0.060 | ||||
| 25/75 | 75 | PAR-PVC-EE | 154.8 | 113.6 | 0.223 | DIS-PVC-EE | 150.4 | 94.4 | 0.092 | ACE-PVC-EE | 147.3 | 93.4 | 0.106 |
| 225 | 142.5 | 166.6 | 0.126 | 129.0 | 130.7 | 0.098 | 151.2 | 159.1 | 0.038 | ||||
| 375 | 136.1 | 184.5 | 0.098 | 130.7 | 173.7 | 0.113 | 152.7 | 164.7 | 0.014 | ||||
| 50/50 | 75 | 149.0 | 50.1 | 0.191 | 151.7 | 57.4 | 0.210 | 151.1 | 67.6 | 0.141 | |||
| 225 | 152.2 | 146.7 | 0.109 | 138.8 | 102.6 | 0.101 | 137.6 | 87.5 | 0.068 | ||||
| 375 | 145.2 | 161.8 | 0.105 | 138.7 | 153.8 | 0.080 | 139.0 | 143.6 | 0.071 | ||||
| 75/25 | 75 | 149.5 | 128.6 | 0.282 | 137.4 | 102.9 | 0.147 | 146.3 | 131.2 | 0.102 | |||
| 225 | 137.0 | 192.4 | 0.131 | 135.8 | 160.6 | 0.121 | 145.9 | 187.9 | 0.011 | ||||
| 375 | 148.6 | 211.2 | 0.115 | 151.7 | 201.0 | 0.046 | 141.7 | 192.8 | 0.011 |
| Eudragit/PVC Ratio (%/%) | Compression Pressure (MPa) | Composition | R2 | k | n | Composition | R2 | k | n | Composition | R2 | k | n |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25/75 | 75 | PAR-PVC-EL | 0.9230 | 5.7937 | 0.3858 | DIS-PVC-EL | 0.9966 | 0.0419 | 0.9833 | ACE-PVC-EL | 0.9945 | 0.4184 | 0.7325 |
| 225 | 0.9840 | 1.4970 | 0.5635 | 0.9997 | 0.0092 | 1.1609 | 0.9979 | 0.0540 | 0.9340 | ||||
| 375 | 0.9870 | 1.7224 | 0.5345 | 0.9999 | 0.0054 | 1.2765 | 0.9976 | 0.0850 | 0.8940 | ||||
| 50/50 | 75 | 0.9410 | 6.8732 | 0.3534 | 0.9867 | 1.4394 | 0.4317 | 0.9994 | 0.1289 | 0.8756 | |||
| 225 | 0.9845 | 1.6004 | 0.5377 | 0.9979 | 0.0265 | 0.9560 | 0.9691 | 0.1877 | 0.8271 | ||||
| 375 | 0.9712 | 1.5522 | 0.5594 | 0.9942 | 0.0435 | 0.9105 | 0.9899 | 0.0829 | 0.8931 | ||||
| 75/25 | 75 | 0.9920 | 1.7155 | 0.5058 | 0.9789 | 1.5287 | 0.5567 | 0.9906 | 0.2466 | 0.7311 | |||
| 225 | 0.9974 | 0.7570 | 0.6298 | 0.9902 | 0.1828 | 0.7850 | 0.9990 | 0.2002 | 0.7389 | ||||
| 375 | 0.9928 | 1.5108 | 0.5239 | 0.9976 | 0.2207 | 0.6573 | 0.9991 | 0.2055 | 0.7260 | ||||
| 25/75 | 75 | PAR-PVC-EE | 0.9936 | 0.9010 | 0.5693 | DIS-PVC-EE | 0.9660 | 1.3715 | 0.2623 | ACE-PVC-EE | 0.9998 | 0.0376 | 0.5936 |
| 225 | 0.9977 | 0.6775 | 0.5774 | 0.9609 | 1.4599 | 0.2306 | 0.9933 | 0.0097 | 0.7596 | ||||
| 375 | 0.9755 | 0.8078 | 0.5483 | 0.9286 | 1.4502 | 0.2176 | 0.9820 | 0.0106 | 0.7241 | ||||
| 50/50 | 75 | 0.9915 | 1.1441 | 0.5441 | 0.9974 | 1.7063 | 0.2739 | 0.9877 | 0.2063 | 0.4619 | |||
| 225 | 0.9973 | 0.4156 | 0.6751 | 0.9846 | 0.9046 | 0.3542 | 0.9972 | 0.0704 | 0.5474 | ||||
| 375 | 0.9978 | 0.3755 | 0.6785 | 0.9823 | 0.9284 | 0.3404 | 0.9969 | 0.0805 | 0.4910 | ||||
| 75/25 | 75 | 0.9920 | 1.6898 | 0.5056 | 0.9867 | 1.5953 | 0.3265 | 0.9839 | 0.2829 | 0.6100 | |||
| 225 | 0.9983 | 0.7675 | 0.5535 | 0.9659 | 1.0545 | 0.3185 | 0.9933 | 0.1695 | 0.5134 | ||||
| 375 | 0.9918 | 0.8430 | 0.5239 | 0.9641 | 1.5575 | 0.2903 | 0.9670 | 0.2069 | 0.4661 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benkő, E.; Ilič, I.G.; Kristó, K.; Regdon, G., Jr.; Csóka, I.; Pintye-Hódi, K.; Srčič, S.; Sovány, T. Predicting Drug Release Rate of Implantable Matrices and Better Understanding of the Underlying Mechanisms through Experimental Design and Artificial Neural Network-Based Modelling. Pharmaceutics 2022, 14, 228. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020228
Benkő E, Ilič IG, Kristó K, Regdon G Jr., Csóka I, Pintye-Hódi K, Srčič S, Sovány T. Predicting Drug Release Rate of Implantable Matrices and Better Understanding of the Underlying Mechanisms through Experimental Design and Artificial Neural Network-Based Modelling. Pharmaceutics. 2022; 14(2):228. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020228
Chicago/Turabian StyleBenkő, Ernő, Ilija German Ilič, Katalin Kristó, Géza Regdon, Jr., Ildikó Csóka, Klára Pintye-Hódi, Stane Srčič, and Tamás Sovány. 2022. "Predicting Drug Release Rate of Implantable Matrices and Better Understanding of the Underlying Mechanisms through Experimental Design and Artificial Neural Network-Based Modelling" Pharmaceutics 14, no. 2: 228. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020228