
Application of Semi-Mechanistic Pharmacokinetic and Pharmacodynamic Model in Antimicrobial Resistance

 , ,
, ,
Abstract
:1. Introduction
2. Components of Semi-Mechanism PK/PD Model
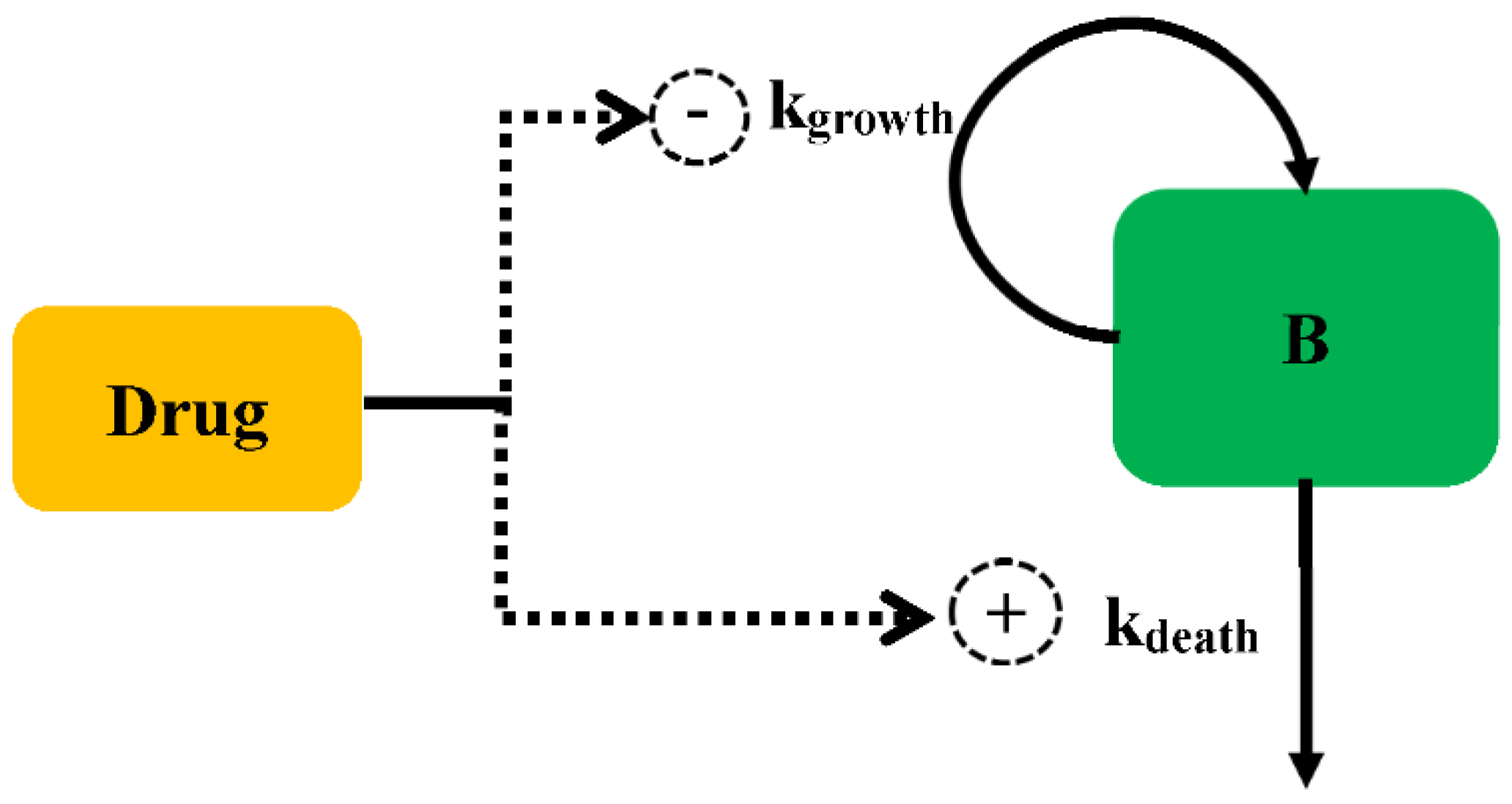
2.1. Bacterial Growth Model
2.2. Antibacterial Effect Model
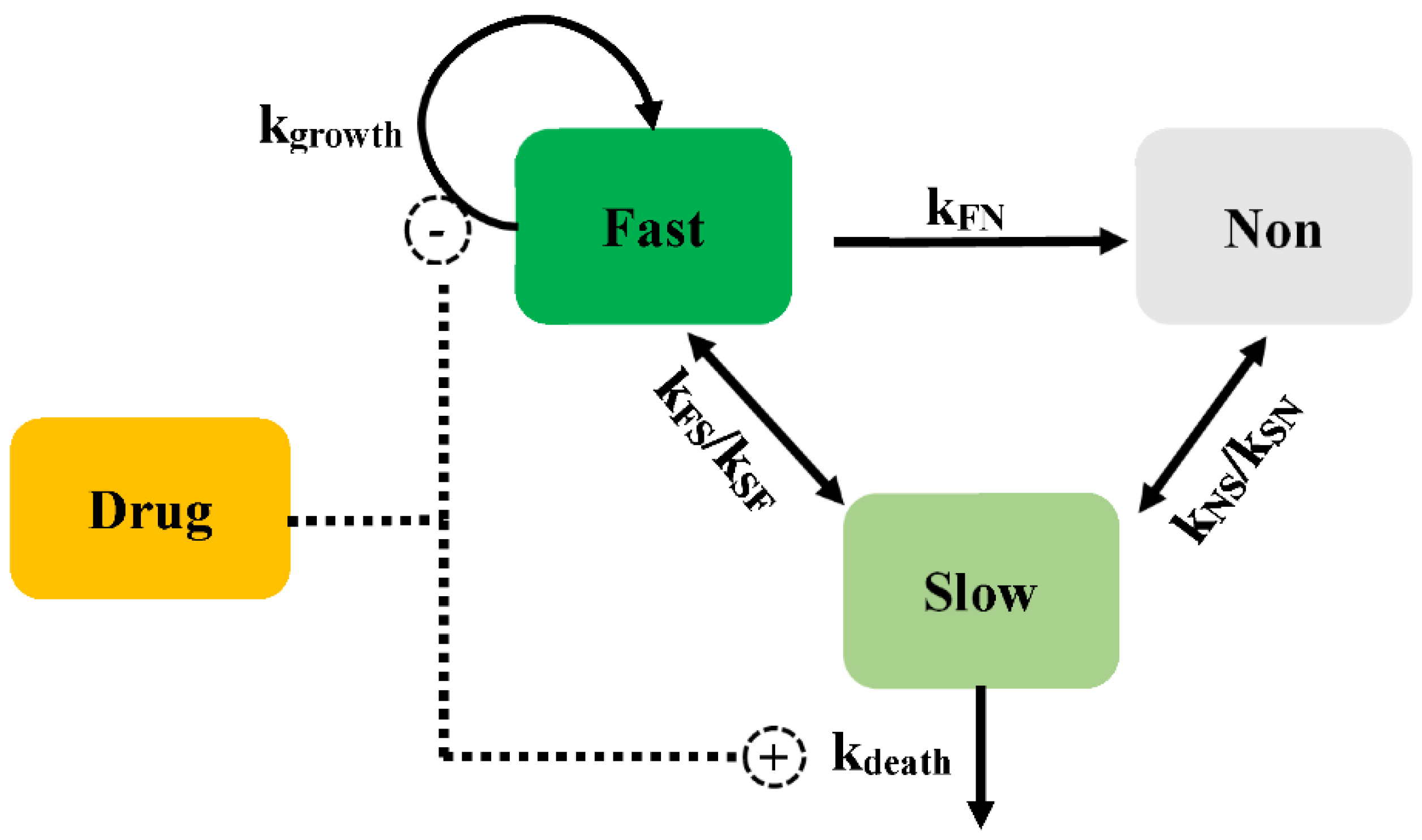
2.2.1. Persistent Resistance
2.2.2. Pre-Existing Resistance
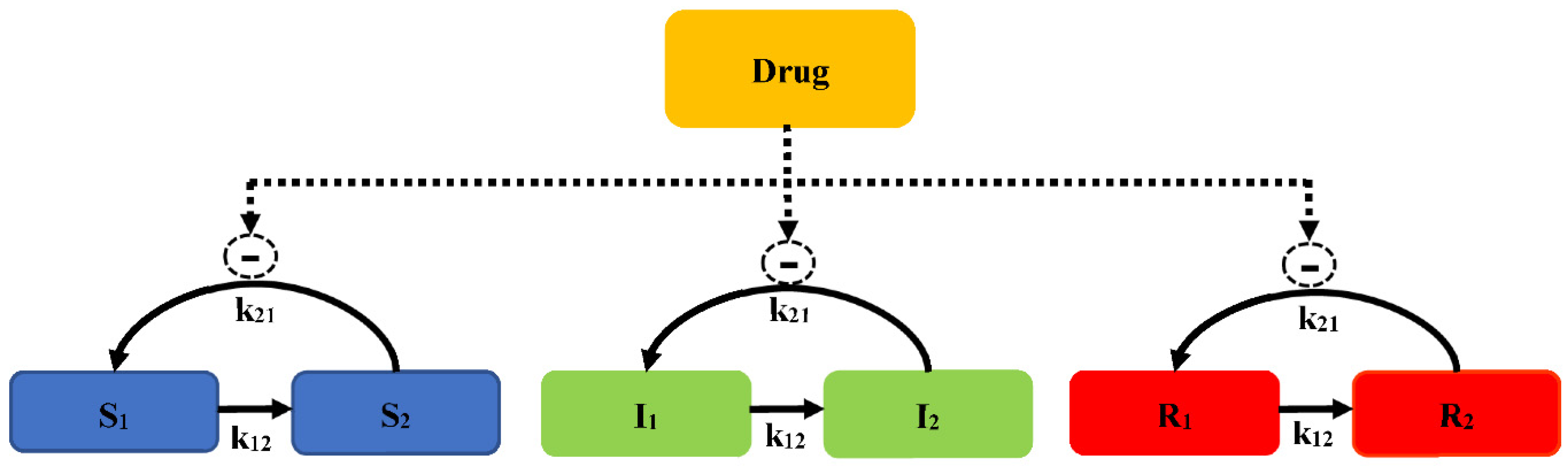
2.2.3. Adaptive Resistance
3. Methods for the Development of Semi-Mechanism PK/PD Model
4. The Factors Affecting Model Establishment
4.1. Inoculum Effect
4.2. Host Response
4.3. The Types of Pharmacodynamic Data for the Model
5. Application of Model in Dosing Regimen, Combination Therapy, and Determination of Breakpoint
5.1. Dosage Regimen
5.2. Combination Therapy
5.3. PK/PD Breakpoint and Cutoffs
5.4. Prediction the Kinetic of Bacterial in Guts
6. Overlook
- Regulation. The official regulations need to be published, which will play the role of encouragement and guidance.
- Education. It is very important to tell the modelers how to establish a model and judge the model. It is an efficient way to acquire the relevant knowledge from the tutorial. Rowland et al. summarized the inception, maturation, and future vision about Pharmacometrics and Systems Pharmacology. Twenty representative particles over the past 10 years were outlined [66]. Besides the tutorials, the software company and the public training courses also can offer some guidance. For example, many detailed courses can be found on the Metrum research group (www.metrumrg.com (accessed on 6 January 2022)).
- Share. It is critical and necessary to publish the model code for the subsequent model applications. This will help modelers to learn the programming languages. Of course, the excellent forms of programming languages are also important. Mathematical models are widely used in various fields that would require more competent modelers.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Innes, G.K.; Randad, P.R.; Korinek, A.; Davis, M.F.; Price, L.B.; So, A.D.; Heaney, C.D. External Societal Costs of Antimicrobial Resistance in Humans Attributable to Antimicrobial Use in Livestock. Annu. Rev. Public Health 2020, 41, 141–157. [Google Scholar] [CrossRef] [Green Version]
- CDC (Centers for Disease Control and Prevention); US DHHS (Department of Health and Human Services). 2013 Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA; US DHHS: Washington, DC, USA, 2013.
- Mouton, J.W.; Ambrose, P.G.; Canton, R.; Drusano, G.L.; Harbarth, S.; MacGowan, A.; Theuretzbacher, U.; Turnidge, J. Conserving antibiotics for the future: New ways to use old and new drugs from a pharmacokinetic and pharmacodynamic perspective. Drug Resist. Updates 2011, 14, 107–117. [Google Scholar] [CrossRef]
- Tängdén, T.; Ramos Martín, V.; Felton, T.W.; Nielsen, E.I.; Marchand, S.; Brüggemann, R.J.; Bulitta, J.B.; Bassetti, M.; Theuretzbacher, U.; Tsuji, B.T.; et al. The role of infection models and PK/PD modelling for optimising care of critically ill patients with severe infections. Intensive Care Med. 2017, 43, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.; Grégoire, N.; Couet, W.; Bulitta, J.B. Distinguishing Antimicrobial Models with Different Resistance Mechanisms via Population Pharmacodynamic Modeling. PLoS Comput. Biol. 2016, 12, e1004782. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.I.; Friberg, L.E. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol. Rev. 2013, 65, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Lees, P.; Pelligand, L.; Illambas, J.; Potter, T.; Lacroix, M.; Rycroft, A.; Toutain, P.L. Pharmacokinetic/pharmacodynamic integration and modelling of amoxicillin for the calf pathogens Mannheimia haemolytica and Pasteurella multocida. J. Vet. Pharmacol. Ther. 2015, 38, 457–470. [Google Scholar] [CrossRef]
- Nielsen, E.I.; Cars, O.; Friberg, L.E. Pharmacokinetic/pharmacodynamic (PK/PD) indices of antibiotics predicted by a semimechanistic PKPD model: A step toward model-based dose optimization. Antimicrob. Agents Chemother. 2011, 55, 4619–4630. [Google Scholar] [CrossRef] [Green Version]
- Rathi, C.; Lee, R.E.; Meibohm, B. Translational PK/PD of anti-infective therapeutics. Drug Discov. Today. Technol. 2016, 21–22, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhu, H.; Madabushi, R.; Liu, Q.; Huang, S.M.; Zineh, I. Model-Informed Drug Development: Current US Regulatory Practice and Future Considerations. Clin. Pharmacol. Ther. 2019, 105, 899–911. [Google Scholar] [CrossRef]
- Kristoffersson, A. Study Design and Dose Regimen Evaluation of Antibiotics Based on Pharmacokinetic and Pharmacodynamic Modelling. Doctoral Dissertation, Acta Universitatis Upsaliensis, Uppsala, Sweden, 2015. [Google Scholar]
- Fan, J.; de Lannoy, I.A. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-M.; Chan, M.; Chukwudebe, A.; Domoradzki, J.; Fisher, J.; Hack, C.E.; Hinderliter, P.; Hirasawa, K.; Leonard, J.; Lumen, A.; et al. PBPK model reporting template for chemical risk assessment applications. Regul. Toxicol. Pharmacol. RTP 2020, 115, 104691. [Google Scholar] [CrossRef] [PubMed]
- Kiang, T.K.; Sherwin, C.M.; Spigarelli, M.G.; Ensom, M.H. Fundamentals of population pharmacokinetic modelling: Modelling and software. Clin. Pharmacokinet. 2012, 51, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Jusko, W.J. Pharmacodynamics of chemotherapeutic effects: Dose-time-response relationships for phase-nonspecific agents. J. Pharm. Sci. 1971, 60, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Meagher, A.K.; Forrest, A.; Dalhoff, A.; Stass, H.; Schentag, J.J. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob. Agents Chemother. 2004, 48, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Campion, J.J.; McNamara, P.J.; Evans, M.E. Pharmacodynamic modeling of ciprofloxacin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.I.; Viberg, A.; Löwdin, E.; Cars, O.; Karlsson, M.O.; Sandström, M. Semimechanistic pharmacokinetic/pharmacodynamic model for assessment of activity of antibacterial agents from time-kill curve experiments. Antimicrob. Agents Chemother. 2007, 51, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Sykes, R. The 2009 Garrod lecture: The evolution of antimicrobial resistance: A Darwinian perspective. J. Antimicrob. Chemother. 2010, 65, 1842–1852. [Google Scholar] [CrossRef] [Green Version]
- Czock, D.; Keller, F. Mechanism-based pharmacokinetic-pharmacodynamic modeling of antimicrobial drug effects. J. Pharmacokinet. Pharmacodyn. 2007, 34, 727–751. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Reviews. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef]
- Khan, D.D.; Lagerbäck, P.; Cao, S.; Lustig, U.; Nielsen, E.I.; Cars, O.; Hughes, D.; Andersson, D.I.; Friberg, L.E. A mechanism-based pharmacokinetic/pharmacodynamic model allows prediction of antibiotic killing from MIC values for WT and mutants. J. Antimicrob. Chemother. 2015, 70, 3051–3060. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Reviews. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Sou, T.; Kukavica-Ibrulj, I.; Levesque, R.C.; Friberg, L.E.; Bergström, C.A.S. Model-Informed Drug Development in Pulmonary Delivery: Semimechanistic Pharmacokinetic-Pharmacodynamic Modeling for Evaluation of Treatments against Chronic Pseudomonas aeruginosa Lung Infections. Mol. Pharm. 2020, 17, 1458–1469. [Google Scholar] [CrossRef]
- Downing, K.J.; Mischenko, V.V.; Shleeva, M.O.; Young, D.I.; Young, M.; Kaprelyants, A.S.; Apt, A.S.; Mizrahi, V. Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 2005, 73, 3038–3043. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ortega, F.; Rullas, J.; Alameda, L.; Angulo-Barturen, I.; Ferrer, S.; Simonsson, U.S. The multistate tuberculosis pharmacometric model: A semi-mechanistic pharmacokinetic-pharmacodynamic model for studying drug effects in an acute tuberculosis mouse model. J. Pharmacokinet. Pharmacodyn. 2017, 44, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Jumbe, N.; Louie, A.; Leary, R.; Liu, W.; Deziel, M.R.; Tam, V.H.; Bachhawat, R.; Freeman, C.; Kahn, J.B.; Bush, K.; et al. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 2003, 112, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Erwin, S.; Foster, D.M.; Jacob, M.E.; Papich, M.G.; Lanzas, C. The effect of enrofloxacin on enteric Escherichia coli: Fitting a mathematical model to in vivo data. PLoS ONE 2020, 15, e0228138. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Breidenstein, E.B.; Hancock, R.E. Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Updates 2011, 14, 1–21. [Google Scholar] [CrossRef]
- Tam, V.H.; Schilling, A.N.; Nikolaou, M. Modelling time-kill studies to discern the pharmacodynamics of meropenem. J. Antimicrob. Chemother. 2005, 55, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.F.; Nielsen, E.I.; Cars, O.; Friberg, L.E. Pharmacokinetic-pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob. Agents Chemother. 2012, 56, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Mouksassi, M.S.; Holford, N.; Al-Huniti, N.; Freedman, I.; Hooker, A.C.; John, J.; Karlsson, M.O.; Mould, D.R.; Pérez Ruixo, J.J.; et al. Model Evaluation of Continuous Data Pharmacometric Models: Metrics and Graphics. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Bulitta, J.B.; Yang, J.C.; Yohonn, L.; Ly, N.S.; Brown, S.V.; D’Hondt, R.E.; Jusko, W.J.; Forrest, A.; Tsuji, B.T. Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob. Agents Chemother. 2010, 54, 2051–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsted, A.; Tano, E.; Kaivonen, K.; Sjölin, J.; Friberg, L.E.; Nielsen, E.I. Extension of Pharmacokinetic/Pharmacodynamic Time-Kill Studies To Include Lipopolysaccharide/Endotoxin Release from Escherichia coli Exposed to Cefuroxime. Antimicrob. Agents Chemother. 2020, 64, 4. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.M.; Alexander, E.; Salvatore, M. Translational research in infectious disease: Current paradigms and challenges ahead. Transl. Res. 2012, 159, 430–453. [Google Scholar] [CrossRef] [PubMed]
- Diep, J.K.; Russo, T.A.; Rao, G.G. Mechanism-Based Disease Progression Model Describing Host-Pathogen Interactions During the Pathogenesis of Acinetobacter baumannii Pneumonia. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Fors, J.; Strydom, N.; Fox, W.S.; Keizer, R.J.; Savic, R.M. Mathematical model and tool to explore shorter multi-drug therapy options for active pulmonary tuberculosis. PLoS Comput. Biol. 2020, 16, e1008107. [Google Scholar] [CrossRef]
- Thorsted, A.; Nielsen, E.I.; Friberg, L.E. Pharmacodynamics of immune response biomarkers of interest for evaluation of treatment effects in bacterial infections. Int. J. Antimicrob. Agents 2020, 56, 106059. [Google Scholar] [CrossRef]
- Sy, S.K.B.; Zhuang, L.; Xia, H.; Beaudoin, M.E.; Schuck, V.J.; Nichols, W.W.; Derendorf, H. A mathematical model-based analysis of the time-kill kinetics of ceftazidime/avibactam against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 1295–1304. [Google Scholar] [CrossRef]
- Nielsen, E.I.; Cars, O.; Friberg, L.E. Predicting in vitro antibacterial efficacy across experimental designs with a semimechanistic pharmacokinetic-pharmacodynamic model. Antimicrob. Agents Chemother. 2011, 55, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landersdorfer, C.B.; Rees, V.E.; Yadav, R.; Rogers, K.E.; Kim, T.H.; Bergen, P.J.; Cheah, S.E.; Boyce, J.D.; Peleg, A.Y.; Oliver, A.; et al. Optimization of a Meropenem-Tobramycin Combination Dosage Regimen against Hypermutable and Nonhypermutable Pseudomonas aeruginosa via Mechanism-Based Modeling and the Hollow-Fiber Infection Model. Antimicrob. Agents Chemother. 2018, 62, e02055-17. [Google Scholar] [CrossRef] [Green Version]
- Sadiq, M.W.; Nielsen, E.I.; Khachman, D.; Conil, J.M.; Georges, B.; Houin, G.; Laffont, C.M.; Karlsson, M.O.; Friberg, L.E. A whole-body physiologically based pharmacokinetic (WB-PBPK) model of ciprofloxacin: A step towards predicting bacterial killing at sites of infection. J. Pharmacokinet. Pharmacodyn. 2017, 44, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Sy, S.K.B.; Zhuang, L.; Xia, H.; Schuck, V.J.; Nichols, W.W.; Derendorf, H. A model-based analysis of pharmacokinetic-pharmacodynamic (PK/PD) indices of avibactam against Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2019, 25, 904.e9–904.e16. [Google Scholar] [CrossRef] [PubMed]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.; Zhou, Q.T.; Han, M.L.; Onufrak, N.J.; Chen, K.; Wang, J.; Forrest, A.; Chan, H.K.; Li, J. Mechanism-Based Pharmacokinetic/Pharmacodynamic Modeling of Aerosolized Colistin in a Mouse Lung Infection Model. Antimicrob. Agents Chemother. 2018, 62, e01965-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, V.H.; Kabbara, S.; Vo, G.; Schilling, A.N.; Coyle, E.A. Comparative pharmacodynamics of gentamicin against Staphylococcus aureus and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2626–2631. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.H.; Ledesma, K.R.; Vo, G.; Kabbara, S.; Lim, T.P.; Nikolaou, M. Pharmacodynamic modeling of aminoglycosides against Pseudomonas aeruginosa and Acinetobacter baumannii: Identifying dosing regimens to suppress resistance development. Antimicrob. Agents Chemother. 2008, 52, 3987–3993. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; He, Y.; Xia, H.; Liu, Y.; Sy, S.K.; Derendorf, H. Gentamicin dosing strategy in patients with end-stage renal disease receiving haemodialysis: Evaluation using a semi-mechanistic pharmacokinetic/pharmacodynamic model. J. Antimicrob. Chemother. 2016, 71, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Bulitta, J.B.; Ly, N.S.; Landersdorfer, C.B.; Wanigaratne, N.A.; Velkov, T.; Yadav, R.; Oliver, A.; Martin, L.; Shin, B.S.; Forrest, A.; et al. Two mechanisms of killing of Pseudomonas aeruginosa by tobramycin assessed at multiple inocula via mechanism-based modeling. Antimicrob. Agents Chemother. 2015, 59, 2315–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristoffersson, A.N.; David-Pierson, P.; Parrott, N.J.; Kuhlmann, O.; Lave, T.; Friberg, L.E.; Nielsen, E.I. Simulation-Based Evaluation of PK/PD Indices for Meropenem Across Patient Groups and Experimental Designs. Pharm. Res. 2016, 33, 1115–1125. [Google Scholar] [CrossRef]
- Mohamed, A.F.; Kristoffersson, A.N.; Karvanen, M.; Nielsen, E.I.; Cars, O.; Friberg, L.E. Dynamic interaction of colistin and meropenem on a WT and a resistant strain of Pseudomonas aeruginosa as quantified in a PK/PD model. J. Antimicrob. Chemother. 2016, 71, 1279–1290. [Google Scholar] [CrossRef]
- Ungphakorn, W.; Tängdén, T.; Sandegren, L.; Nielsen, E.I. A pharmacokinetic-pharmacodynamic model characterizing the emergence of resistant Escherichia coli subpopulations during ertapenem exposure. J. Antimicrob. Chemother. 2016, 71, 2521–2533. [Google Scholar] [CrossRef] [Green Version]
- Barbour, A.M.; Schmidt, S.; Zhuang, L.; Rand, K.; Derendorf, H. Application of pharmacokinetic/pharmacodynamic modelling and simulation for the prediction of target attainment of ceftobiprole against meticillin-resistant Staphylococcus aureus using minimum inhibitory concentration and time-kill curve based approaches. Int. J. Antimicrob. Agents 2014, 43, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Broeker, A.; Nowak, H.; Rahmel, T.; Nussbaumer-Pröll, A.; Österreicher, Z.; Zeitlinger, M.; Wicha, S.G. A pharmacometric approach to define target site-specific breakpoints for bacterial killing and resistance suppression integrating microdialysis, time-kill curves and heteroresistance data: A case study with moxifloxacin. Clin. Microbiol. Infect. 2020, 26, 1255.e1–1255.e8. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.; McNamara, P.J.; Campion, J.J.; Evans, M.E. Mechanism-based pharmacodynamic models of fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 2957–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grégoire, N.; Raherison, S.; Grignon, C.; Comets, E.; Marliat, M.; Ploy, M.C.; Couet, W. Semimechanistic pharmacokinetic-pharmacodynamic model with adaptation development for time-kill experiments of ciprofloxacin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2010, 54, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.F.; Cars, O.; Friberg, L.E. A pharmacokinetic/pharmacodynamic model developed for the effect of colistin on Pseudomonas aeruginosa in vitro with evaluation of population pharmacokinetic variability on simulated bacterial killing. J. Antimicrob. Chemother. 2014, 69, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Pelligand, L.; Lees, P.; Sidhu, P.K.; Toutain, P.L. Semi-Mechanistic Modeling of Florfenicol Time-Kill Curves and in silico Dose Fractionation for Calf Respiratory Pathogens. Front. Microbiol. 2019, 10, 1237. [Google Scholar] [CrossRef]
- Nguyen, K.; Bensman, T.J.; Wei, X.T.; Moore, J.N. Semimechanistic Modeling of Eravacycline Pharmacodynamics Using In Vitro Time-Kill Data with MIC Incorporated in an Adaptive Resistance Function. Antimicrob. Agents Chemother. 2020, 64, e01308-20. [Google Scholar] [CrossRef]
- Zhao, C.; Wistrand-Yuen, P.; Lagerbäck, P.; Tängdén, T.; Nielsen, E.I.; Friberg, L.E. Combination of polymyxin B and minocycline against multidrug-resistant Klebsiella pneumoniae: Interaction quantified by pharmacokinetic/pharmacodynamic modelling from in vitro data. Int. J. Antimicrob. Agents 2020, 55, 105941. [Google Scholar] [CrossRef]
- Aranzana-Climent, V.; Buyck, J.M.; Smani, Y.; Pachón-Diaz, J.; Marchand, S.; Couet, W.; Grégoire, N. Semi-mechanistic PK/PD modelling of combined polymyxin B and minocycline against a polymyxin-resistant strain of Acinetobacter baumannii. Clin. Microbiol. Infect. 2020, 26, 1254.e9–1254.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brill, M.J.E.; Kristoffersson, A.N.; Zhao, C.; Nielsen, E.I.; Friberg, L.E. Semi-mechanistic pharmacokinetic-pharmacodynamic modelling of antibiotic drug combinations. Clin. Microbiol. Infect. 2018, 24, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Drug Disease Model Resources (DDMoRe) Foundation. Available online: www.ddmore.foundation (accessed on 6 January 2022).
- Nguyen, T.T.; Guedj, J.; Chachaty, E.; de Gunzburg, J.; Andremont, A.; Mentré, F. Mathematical modeling of bacterial kinetics to predict the impact of antibiotic colonic exposure and treatment duration on the amount of resistant enterobacteria excreted. PLoS Comput. Biol. 2014, 10, e1003840. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Zachariasen, C.; Christiansen, L.E.; Græsbøll, K.; Toft, N.; Matthews, L.; Nielsen, S.S.; Olsen, J.E. Modeling the growth dynamics of multiple Escherichia coli strains in the pig intestine following intramuscular ampicillin treatment. BMC Microbiol. 2016, 16, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland Yeo, K.; Hennig, S.; Krishnaswami, S.; Strydom, N.; Ayyar, V.S.; French, J.; Sinha, V.; Sobie, E.; Zhao, P.; Friberg, L.E.; et al. CPT: Pharmacometrics & Systems Pharmacology-Inception, Maturation, and Future Vision. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 649–657. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Class | Drug | Bacteria | Resistant Species | References |
|---|---|---|---|---|
| Aminoglycosides | Gentamicin | Staphylococcus aureus | Adaptive resistance | [42] |
| Pseudomonas aeruginosa Acinetobacter baumannii | Adaptive resistance | [43] | ||
| Escherichia coli | Adaptive resistance Persistent resistance | [31] | ||
| Pseudomonas aeruginosa | Pre-existing resistance | [44] | ||
| Tobramycin | Pseudomonas aeruginosa | Persistent resistance | [24] | |
| Pre-existing resistance | [45] | |||
| Beta-lactams | Benzylpenicillin Cefuroxime | Streptococcus pyogenes | Persistent resistance | [18] |
| Meropenem | Pseudomonas aeruginosa | Adaptive resistance | [30] | |
| Pre-existing resistance Persistent resistance | [46] | |||
| Pre-existing resistance Persistent resistance Adaptive resistance | [47] | |||
| Ertapenem | Escherichia coli | Pre-existing resistance Persistent resistance | [48] | |
| Ceftobiprole | Staphylococcus aureus | Persistent resistance | [49] | |
| Cefuroxime | Escherichia coli | Persistent resistance | [34] | |
| Fluoroquinolones | Moxifloxacin | Streptococcus pyogenes | Persistent resistance | [18] |
| Staphylococcus aureus | Pre-existing resistance Adaptive resistance | [50] | ||
| Ciprofloxacin | Staphylococcus aureus | Pre-existing resistance | [51] | |
| Pseudomonas aerugeinosa | Adaptive resistance | [52] | ||
| Escherichia coli | Pre-existing resistance Persistent resistance | [22] | ||
| Enrofloxacin | Escherichia coli | Pre-existing resistance | [28] | |
| Macrolides | Erythromycin | Streptococcus pyogenes | Persistent resistance | [18] |
| Polymyxin | Colistin | Pseudomonas aeruginosa | Adaptive resistance Persistent resistance | [53] |
| Pre-existing resistance | [54] | |||
| Chloramphenicols | Florfenicol | Pasteurella multocida Mannheimia haemolytica | Persistent resistance | [55] |
| Tetracyclines | Eravacycline | Escherichia coli Acinetobacter baumannii | Adaptive resistance | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mi, K.; Zhou, K.; Sun, L.; Hou, Y.; Ma, W.; Xu, X.; Huo, M.; Liu, Z.; Huang, L. Application of Semi-Mechanistic Pharmacokinetic and Pharmacodynamic Model in Antimicrobial Resistance. Pharmaceutics 2022, 14, 246. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020246
Mi K, Zhou K, Sun L, Hou Y, Ma W, Xu X, Huo M, Liu Z, Huang L. Application of Semi-Mechanistic Pharmacokinetic and Pharmacodynamic Model in Antimicrobial Resistance. Pharmaceutics. 2022; 14(2):246. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020246
Chicago/Turabian StyleMi, Kun, Kaixiang Zhou, Lei Sun, Yixuan Hou, Wenjin Ma, Xiangyue Xu, Meixia Huo, Zhenli Liu, and Lingli Huang. 2022. "Application of Semi-Mechanistic Pharmacokinetic and Pharmacodynamic Model in Antimicrobial Resistance" Pharmaceutics 14, no. 2: 246. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14020246