
Prodrug Therapies for Infectious and Neurodegenerative Diseases

 , , and
, , and
Abstract
:1. Introduction
2. Prodrug Strategies for Neurodegenerative Diseases
2.1. Current and Prospective Therapies for Neurodegenerative Diseases
2.2. Prodrugs for Alzheimer’s Disease
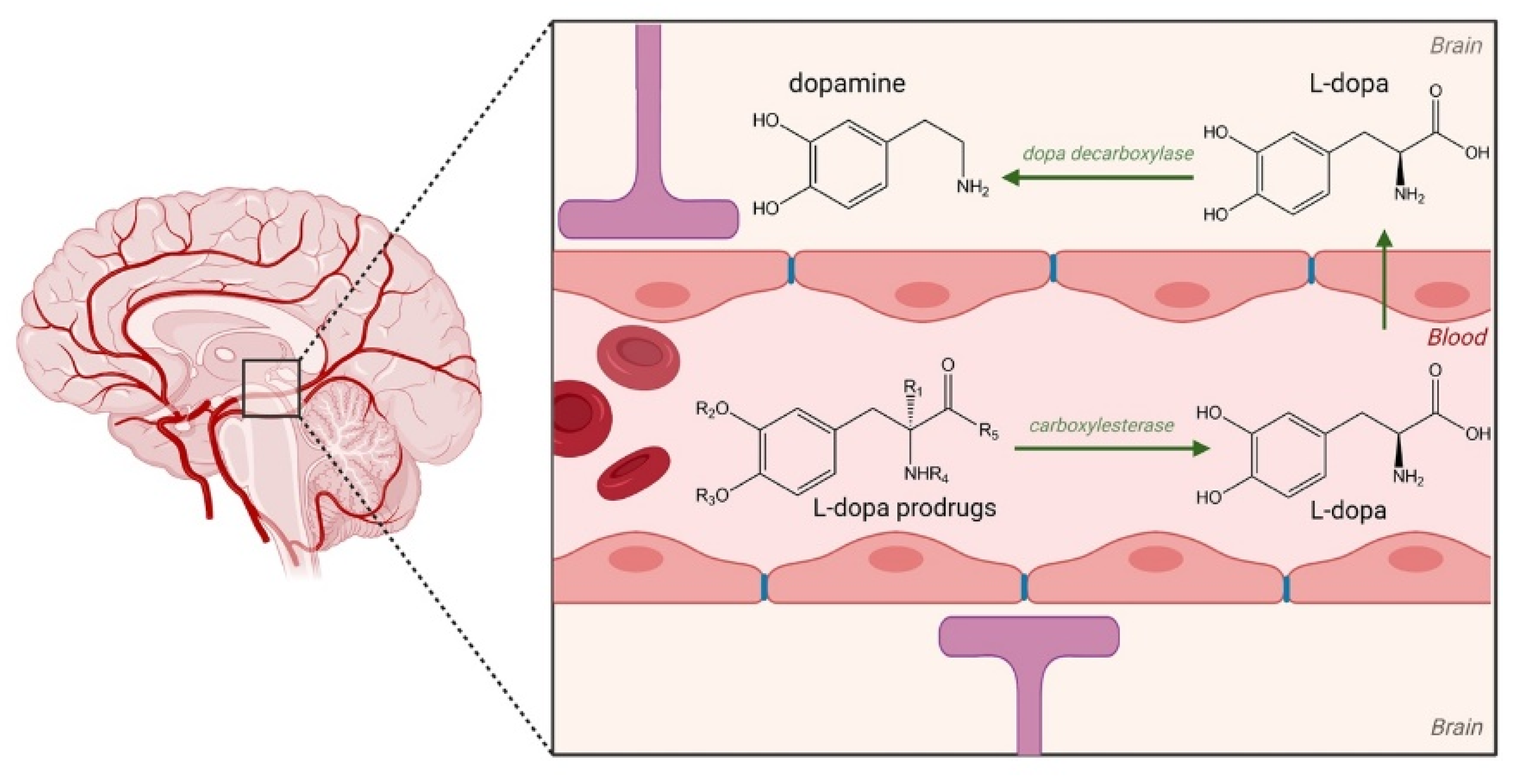
2.3. Prodrugs for Parkinson’s Disease (PD)
3. The Use of Prodrugs for Infectious Diseases
3.1. Prodrug Therapies for Herpesviridae Infection: Herpes Simplex Virus (HSV) and Varicella Zoster Virus (VZV)
3.2. Prodrug Therapies for Human Immunodeficiency Virus (HIV)
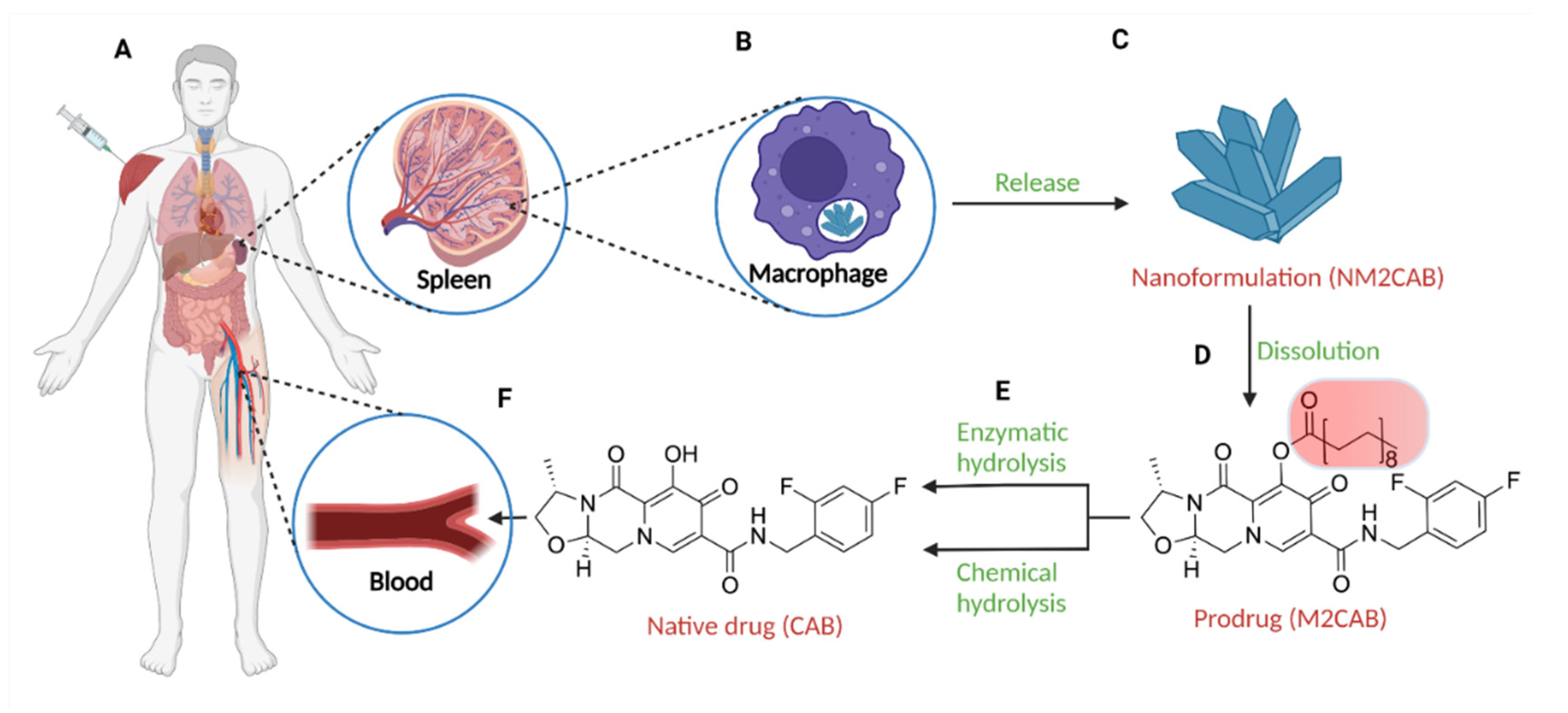
3.2.1. Long-Acting Slow Effective Release Antiretroviral Therapy (LASER ART)
3.2.2. Prodrug Therapies for Human Immunodeficiency Virus (HIV)-Associated Neurocognitive Disorders (HAND)
3.3. Prodrugs Therapies for Hepatitis B and C Infection
3.4. Prodrug Therapies for COVID-19
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stella, V.J. Prodrugs as therapeutics. Expert Opin. Ther. Pat. 2004, 14, 277–280. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Prodrugs for Improved Drug Delivery: Lessons Learned from Recently Developed and Marketed Products. Pharmaceutics 2020, 12, 1031. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipidic prodrug approach for improved oral drug delivery and therapy. Med. Res. Rev. 2019, 39, 579–607. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef]
- Mullin, R. Chemical & Engineering News. Cost to Develop New Pharmaceutical Drug Now Exceeds $2.5B. In: Scientific American. Available online: https://www.scientificamerican.com/article/cost-to-develop-new-pharmaceutical-drug-now-exceeds-2-5b/ (accessed on 1 November 2021).
- Stella, V.J. A Case for prodrugs. In Prodrugs: Challenges and Rewards—Part 1; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 3–33. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.; dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef] [Green Version]
- Markovic, M.; Ben-Shabat, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Lipids and Lipid-Processing Pathways in Drug Delivery and Therapeutics. Int. J. Mol. Sci. 2020, 21, 3248. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Epstein, S.; Cohen, N.; Zimmermann, E.M.; Aponick, A.; Ben-Shabat, S. Phospholipid-drug conjugates as a novel oral drug targeting approach for the treatment of inflammatory bowel disease. Eur. J. Pharm. Sci. 2017, 108, 78–85. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs design based on inter- and intramolecular chemical processes. Chem. Biol. Drug Des. 2013, 82, 643–668. [Google Scholar] [CrossRef]
- Manda, J.N.; Markovic, M.; Zimmermann, E.M.; Ben-Shabat, S.; Dahan, A.; Aponick, A. Phospholipid Cyclosporine Prodrugs Targeted at Inflammatory Bowel Disease (IBD) Treatment: Design, Synthesis, and in Vitro Validation. ChemMedChem 2020, 15, 1639–1644. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs. Int. J. Mol. Sci. 2019, 20, 2210. [Google Scholar] [CrossRef] [Green Version]
- Najjar, A.; Karaman, R. The prodrug approach in the era of drug design. Expert Opin. Drug Deliv. 2019, 16, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cobb, D.A.; Smith, N.A.; Edagwa, B.J.; McMillan, J.M. Long-acting approaches for delivery of antiretroviral drugs for prevention and treatment of HIV: A review of recent research. Expert Opin. Drug Deliv. 2020, 17, 1227–1238. [Google Scholar] [CrossRef]
- Palombo, M.S.; Singh, Y.; Sinko, P.J. Prodrug and conjugate drug delivery strategies for improving HIV/AIDS therapy. J. Drug Deliv. Sci. Technol. 2009, 19, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Giardiello, M.; Liptrott, N.J.; McDonald, T.O.; Moss, D.; Siccardi, M.; Martin, P.; Smith, D.; Gurjar, R.; Rannard, S.P.; Owen, A. Accelerated oral nanomedicine discovery from miniaturized screening to clinical production exemplified by paediatric HIV nanotherapies. Nat. Commun. 2016, 7, 13184. [Google Scholar] [CrossRef] [Green Version]
- Gendelman, H.E.; McMillan, J.; Bade, A.N.; Edagwa, B.; Kevadiya, B.D. The Promise of Long-Acting Antiretroviral Therapies: From Need to Manufacture. Trends Microbiol. 2019, 27, 593–606. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Wimo, A.A.G.-C.; Guerchet, M.; Prince, M.; Prina, M.; Wu, Y.-T. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. Available online: https://www.alzint.org/u/WorldAlzheimerReport2015.pdf (accessed on 20 December 2021).
- 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 2020, 12, 459–509. [CrossRef]
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 2021, 17, 327–406. [CrossRef]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Straif-Bourgeois, S.; Ratard, R.; Kretzschmar, M. Infectious disease epidemiology. In Handbook of Epidemiology; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- World Health Organisation. COVID-19 Dashboard. Available online: https://covid19.who.int (accessed on 2 November 2021).
- Andrews, J.A.; Gordon, P.H. Neurodegenerative diseases. In Neuroimmune Pharmacology; Gendelman, H.E., Ikezu, T., Eds.; Springer: Boston, MA, USA, 2008; pp. 565–588. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M.Y. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Schwab, A.D.; Thurston, M.J.; Machhi, J.; Olson, K.E.; Namminga, K.L.; Gendelman, H.E.; Mosley, R.L. Immunotherapy for Parkinson’s disease. Neurobiol. Dis. 2020, 137, 104760. [Google Scholar] [CrossRef]
- Vlieghe, P.; Khrestchatisky, M. Medicinal chemistry based approaches and nanotechnology-based systems to improve CNS drug targeting and delivery. Med. Res. Rev. 2013, 33, 457–516. [Google Scholar] [CrossRef]
- Machhi, J.; Sinha, A.; Patel, P.; Kanhed, A.M.; Upadhyay, P.; Tripathi, A.; Parikh, Z.S.; Chruvattil, R.; Pillai, P.P.; Gupta, S.; et al. Neuroprotective Potential of Novel Multi-Targeted Isoalloxazine Derivatives in Rodent Models of Alzheimer’s Disease Through Activation of Canonical Wnt/β-Catenin Signalling Pathway. Neurotox. Res. 2016, 29, 495–513. [Google Scholar] [CrossRef]
- Sinha, A.; Tamboli, R.S.; Seth, B.; Kanhed, A.M.; Tiwari, S.K.; Agarwal, S.; Nair, S.; Giridhar, R.; Chaturvedi, R.K.; Yadav, M.R. Neuroprotective Role of Novel Triazine Derivatives by Activating Wnt/β Catenin Signaling Pathway in Rodent Models of Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 638–652. [Google Scholar] [CrossRef]
- Alexander, G.C.; Karlawish, J. The Problem of Aducanumab for the Treatment of Alzheimer Disease. Ann. Intern. Med. 2021, 174, 1303–1304. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 22 February 2021).
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current Pharmaceutical Treatments and Alternative Therapies of Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef]
- Machhi, J.; Kevadiya, B.D.; Muhammad, I.K.; Herskovitz, J.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. Harnessing regulatory T cell neuroprotective activities for treatment of neurodegenerative disorders. Mol. Neurodegen. 2020, 15, 32. [Google Scholar] [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 25, CD005593. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Marotta, G.; Basagni, F.; Rosini, M.; Minarini, A. Memantine Derivatives as Multitarget Agents in Alzheimer’s Disease. Molecules 2020, 25, 4005. [Google Scholar] [CrossRef] [PubMed]
- Sestito, S.; Daniele, S.; Pietrobono, D.; Citi, V.; Bellusci, L.; Chiellini, G.; Calderone, V.; Martini, C.; Rapposelli, S. Memantine prodrug as a new agent for Alzheimer’s Disease. Sci. Rep. 2019, 9, 4612. [Google Scholar] [CrossRef] [Green Version]
- Maelicke, A.; Hoeffle-Maas, A.; Ludwig, J.; Maus, A.; Samochocki, M.; Jordis, U.; Koepke, A.K. Memogain is a galantamine pro-drug having dramatically reduced adverse effects and enhanced efficacy. J. Mol. Neurosci. 2010, 40, 135–137. [Google Scholar] [CrossRef]
- Bakker, C.; van der Aart, J.; Hart, E.P.; Klaassen, E.S.; Bergmann, K.R.; van Esdonk, M.J.; Kay, D.G.; Groeneveld, G.J. Safety, pharmacokinetics, and pharmacodynamics of Gln-1062, a prodrug of galantamine. Alzheimers Dement. 2020, 6, e12093. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, J.; Noakes, P.G.; Bellingham, M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2019, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Ohno, M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Lee, M.; Nam, G.; Kim, M.; Kang, J.; Choi, B.J.; Jeong, M.S.; Park, K.H.; Han, W.H.; Tak, E.; et al. N,N’-Diacetyl-p-phenylenediamine restores microglial phagocytosis and improves cognitive defects in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2019, 116, 23426–23436. [Google Scholar] [CrossRef]
- Kim, M.; Park, M.H.; Nam, G.; Lee, M.; Kang, J.; Song, I.S.; Choi, M.K.; Jin, H.K.; Bae, J.S.; Lim, M.H. A Glycosylated Prodrug to Attenuate Neuroinflammation and Improve Cognitive Deficits in Alzheimer’s Disease Transgenic Mice. Mol. Pharm. 2021, 18, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Manzano, S.; Agüera, L.; Aguilar, M.; Olazarán, J. A Review on Tramiprosate (Homotaurine) in Alzheimer’s Disease and Other Neurocognitive Disorders. Front. Neurol. 2020, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—A randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]
- Aisen, P.S.; Saumier, D.; Briand, R.; Laurin, J.; Gervais, F.; Tremblay, P.; Garceau, D. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 2006, 67, 1757–1763. [Google Scholar] [CrossRef]
- Hey, J.A.; Yu, J.Y.; Versavel, M.; Abushakra, S.; Kocis, P.; Power, A.; Kaplan, P.L.; Amedio, J.; Tolar, M. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin. Pharmacokinet. 2018, 57, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Tambasco, N.; Romoli, M.; Calabresi, P. Levodopa in Parkinson’s Disease: Current Status and Future Developments. Curr. Neuropharmacol. 2018, 16, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Cacciatore, I.; Ciulla, M.; Marinelli, L.; Eusepi, P.; Di Stefano, A. Advances in prodrug design for Parkinson’s disease. Expert Opin. Drug Discov. 2018, 13, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Sozio, P.; Cerasa, L.S.; Iannitelli, A. L-Dopa prodrugs: An overview of trends for improving Parkinson’s disease treatment. Curr. Pharm. Des. 2011, 17, 3482–3493. [Google Scholar] [CrossRef]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and Levodopa Prodrugs for the Treatment of Parkinson’s Disease. Molecules 2017, 23, 40. [Google Scholar] [CrossRef] [Green Version]
- LeWitt, P.A.; Huff, F.J.; Hauser, R.A.; Chen, D.; Lissin, D.; Zomorodi, K.; Cundy, K.C. Double-blind study of the actively transported levodopa prodrug XP21279 in Parkinson’s disease. Mov. Disord. 2014, 29, 75–82. [Google Scholar] [CrossRef]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Rautio, J.; Laine, K. Design, Synthesis and Brain Uptake of LAT1-Targeted Amino Acid Prodrugs of Dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Calignano, A.; La Rana, G.; De Caprariis, P. Glycosyl derivatives of dopamine and L-dopa as anti-Parkinson prodrugs: Synthesis, pharmacological activity and in vitro stability studies. J. Drug Target. 2003, 11, 25–36. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, K.H.; Yoon, I.K.; Lee, K.E.; Chun, I.K.; Rhie, J.Y.; Gwak, H.S. Pharmacokinetic evaluation of formulated levodopa methyl ester nasal delivery systems. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; McFarthing, K.; Simuni, T. Clinical Trial Highlights—Infusion Therapies. J. Parkinsons Dis. 2020, 10, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosebraugh, M.; Liu, W.; Neenan, M.; Facheris, M.F. Foslevodopa/Foscarbidopa Is Well Tolerated and Maintains Stable Levodopa and Carbidopa Exposure Following Subcutaneous Infusion. J. Parkinsons Dis. 2021, 11, 1695–1702. [Google Scholar] [CrossRef]
- Rosebraugh, M.; Kym, P.; Liu, W.; Facheris, M.; Benesh, J. A Novel Levodopa/Carbidopa Prodrug (ABBV-951) 24-Hour Continuous Subcutaneous Infusion Treatment for Parkinson’s Disease (P3.8-037). Neurology 2019, 92, P3.8-037. [Google Scholar]
- Facheris, M.; Benesh, J.; Streit, J.; Robieson, W.; Zadikoff, C.; Standaert, D. Safety and Tolerability in Parkinson’s Disease Patients Treated with a Continuous Subcutaneous Infusion of ABBV-951: Design of a 52-Week Phase 3 Study [abstract]. Available online: https://www.mdsabstracts.org/abstract/safety-and-tolerability-in-parkinsons-disease-patients-treated-with-a-continuous-subcutaneous-infusion-of-abbv-951-design-of-a-52-week-phase-3-study/ (accessed on 2 November 2021).
- Nyholm, D.; Adnan, M.; Senek, M. Real-Life Use of Levodopa/Carbidopa Intestinal Gel in Parkinson’s Disease According to Analysis of Pump Data. J. Parkinsons Dis. 2020, 10, 1529–1534. [Google Scholar] [CrossRef]
- de Oliveira Junior, E.R.; Truzzi, E.; Ferraro, L.; Fogagnolo, M.; Pavan, B.; Beggiato, S.; Rustichelli, C.; Maretti, E.; Lima, E.M.; Leo, E.; et al. Nasal administration of nanoencapsulated geraniol/ursodeoxycholic acid conjugate: Towards a new approach for the management of Parkinson’s disease. J. Control. Release 2020, 321, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Rekha, K.R.; Selvakumar, G.P.; Sethupathy, S.; Santha, K.; Sivakamasundari, R.I. Geraniol ameliorates the motor behavior and neurotrophic factors inadequacy in MPTP-induced mice model of Parkinson’s disease. J. Mol. Neurosci. 2013, 51, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.S.; Low, W.C. Ursodeoxycholic acid suppresses mitochondria-dependent programmed cell death induced by sodium nitroprusside in SH-SY5Y cells. Toxicology 2012, 292, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.G.; Marosi, K.; Halpern, J.; Mattson, M.P. DNP, mitochondrial uncoupling, and neuroprotection: A little dab’ll do ya. Alzheimer’s Dement. 2017, 13, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, W.B.; Harwood, C.L.; Geisler, J.G.; Vekaria, H.J.; Sullivan, P.G. Mitochondrial uncoupling prodrug improves tissue sparing, cognitive outcome, and mitochondrial bioenergetics after traumatic brain injury in male mice. J. Neurosci. Res. 2018, 96, 1677–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, Y.; Johnson, J.; Fang, W.; Halpern, J.; Marosi, K.; Liu, D.; Geisler, J.G.; Mattson, M.P. A mitochondrial uncoupler prodrug protects dopaminergic neurons and improves functional outcome in a mouse model of Parkinson’s disease. Neurobiol. Aging 2020, 85, 123–130. [Google Scholar] [CrossRef]
- Kaufmann, H.; Freeman, R.; Biaggioni, I.; Low, P.; Pedder, S.; Hewitt, L.A.; Mauney, J.; Feirtag, M.; Mathias, C.J.; Investigators, N.O.H. Droxidopa for neurogenic orthostatic hypotension: A randomized, placebo-controlled, phase 3 trial. Neurology 2014, 83, 328–335. [Google Scholar] [CrossRef]
- Cermelli, C.; Vinceti, M.; Beretti, F.; Pietrini, V.; Nacci, G.; Pietrosemoli, P.; Bartoletti, A.; Guidetti, D.; Sola, P.; Bergomi, M.; et al. Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesviruses and echovirus-7. Eur. J. Epidemiol. 2003, 18, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.C.; Tsan, Y.T.; Tsai, S.L.; Chang, C.J.; Wang, J.D.; Chen, P.C. Hepatitis C viral infection and the risk of dementia. Eur. J. Neurol. 2014, 21, 1068-e1059. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Herrero, M.; Soto-Rojas, L.O.; Harrington, C.R.; Flores-Martinez, Y.M.; Villegas-Rojas, M.M.; León-Aguilar, A.M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Corniel-Taveras, C.N.; et al. Elucidating the Neuropathologic Mechanisms of SARS-CoV-2 Infection. Front. Neurol. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Dahan, A.; Amidon, G.L. Enhancing the intestinal absorption of molecules containing the polar guanidino functionality: A double-targeted prodrug approach. J. Med. Chem. 2010, 53, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Dahan, A.; Walls, Z.F.; Lai, L.; Lee, K.D.; Amidon, G.L. Specificity of a prodrug-activating enzyme hVACVase: The leaving group effect. Mol. Pharm. 2010, 7, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Miller, J.M.; Beig, A.; Rozen, L.; Amidon, G.L.; Dahan, A. Mechanistic enhancement of the intestinal absorption of drugs containing the polar guanidino functionality. Expert Opin. Drug Metab. Toxicol. 2011, 7, 313–323. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Fellay, J. HIV-1 and human genetic variation. Nat. Rev. Genet. 2021, 22, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, K.; Kalichman, S.C. Barriers to HIV Medication Adherence as a Function of Regimen Simplification. Ann. Behav. Med. 2017, 51, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baril, J.G.; Angel, J.B.; Gill, M.J.; Gathe, J.; Cahn, P.; van Wyk, J.; Walmsley, S. Dual Therapy Treatment Strategies for the Management of Patients Infected with HIV: A Systematic Review of Current Evidence in ARV-Naive or ARV-Experienced, Virologically Suppressed Patients. PLoS ONE 2016, 11, e0148231. [Google Scholar] [CrossRef] [PubMed]
- Mofenson, L.M.; Brady, M.T.; Danner, S.P.; Dominguez, K.L.; Hazra, R.; Handelsman, E.; Havens, P.; Nesheim, S.; Read, J.S.; Serchuck, L.; et al. Guidelines for the Prevention and Treatment of Opportunistic Infections among HIV-exposed and HIV-infected children: Recommendations from CDC, the National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics. MMWR Recomm. Rep. 2009, 58, 1–166. [Google Scholar]
- Havlir, D.; Gandhi, M. Implementation challenges for long-acting antivirals as treatment. Curr. Opin. HIV AIDS 2015, 10, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, M.D.; Zack, J.A. HIV/AIDS eradication. Bioorg. Med. Chem. Lett. 2013, 23, 4003–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef]
- Gallois-Montbrun, S.; Veron, M.; Deville-Bonne, D. Antiviral nucleoside analogs phosphorylation by nucleoside diphosphate kinase. Mini Rev. Med. Chem. 2004, 4, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Hillaireau, H.; Dereuddre-Bosquet, N.; Skanji, R.; Bekkara-Aounallah, F.; Caron, J.; Lepêtre, S.; Argote, S.; Bauduin, L.; Yousfi, R.; Rogez-Kreuz, C.; et al. Anti-HIV efficacy and biodistribution of nucleoside reverse transcriptase inhibitors delivered as squalenoylated prodrug nanoassemblies. Biomaterials 2013, 34, 4831–4838. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, S. Is the expression of deoxynucleoside kinases and 5’-nucleotidases in animal tissues related to the biological effects of nucleoside analogs? Curr. Med. Chem. 2013, 20, 4241–4248. [Google Scholar] [CrossRef]
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Steuart, C.D.; Burke, P.J. Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nat. New Biol. 1971, 233, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Calogeropoulou, T.; Detsi, A.; Lekkas, E.; Koufaki, M. Strategies in the design of prodrugs of anti-HIV agents. Curr. Top. Med. Chem. 2003, 3, 1467–1495. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Nickson, C.; Petrik, J.; Karpas, A. Phosphate derivatives of AZT display enhanced selectivity of action against HIV 1 by comparison to the parent nucleoside. FEBS Lett. 1992, 310, 171–174. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, C.; Pathirana, R.N.; Mahmood, N.; Devine, K.G.; Hay, A.J. Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antivir. Res. 1992, 17, 311–321. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Serpi, M.; Pertusati, F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 2018, 26, 2040206618775243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [Green Version]
- Perry, C.M.; Noble, S. Didanosine: An updated review of its use in HIV infection. Drugs 1999, 58, 1099–1135. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Sun, J.; Chang, Y.; Liu, Y.; Fu, Q.; Xu, Y.; Sun, Y.; Pu, X.; Zhang, Y.; Jing, Y.; et al. Bifunctional peptidomimetic prodrugs of didanosine for improved intestinal permeability and enhanced acidic stability: Synthesis, transepithelial transport, chemical stability and pharmacokinetics. Mol. Pharm. 2011, 8, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Sinko, P.J. Involvement of multiple transporters in the oral absorption of nucleoside analogues. Adv. Drug Deliv. Rev. 1999, 39, 183–209. [Google Scholar] [CrossRef]
- Sinko, P.J.; Hu, P.; Waclawski, A.P.; Patel, N.R. Oral absorption of anti-AIDS nucleoside analogues. 1. Intestinal transport of didanosine in rat and rabbit preparations. J. Pharm. Sci. 1995, 84, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; Staszewski, S.; Pozniak, A.L.; DeJesus, E.; Suleiman, J.M.; Miller, M.D.; Coakley, D.F.; Lu, B.; Toole, J.J.; Cheng, A.K. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: A 3-year randomized trial. JAMA 2004, 292, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Skanji, R.; Andrieux, K.; Lalanne, M.; Caron, J.; Bourgaux, C.; Degrouard, J.; Brisset, F.; Gueutin, C.; Chacun, H.; Dereuddre-Bosquet, N.; et al. A new nanomedicine based on didanosine glycerolipidic prodrug enhances the long term accumulation of drug in a HIV sanctuary. Int. J. Pharm. 2011, 414, 285–297. [Google Scholar] [CrossRef]
- Jin, Y.; Xin, R.; Tong, L.; Du, L.; Li, M. Combination anti-HIV therapy with the self-assemblies of an asymmetric bolaamphiphilic zidovudine/didanosine prodrug. Mol. Pharm. 2011, 8, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.M.; Adiseshaiah, P.; Dasa, S.S.K.; Potter, T.M.; Skoczen, S.L.; Snapp, K.S.; Cedrone, E.; Patel, N.; Busman-Sahay, K.; Rosen, E.P.; et al. Application of a Scavenger Receptor A1-Targeted Polymeric Prodrug Platform for Lymphatic Drug Delivery in HIV. Mol. Pharm. 2020, 17, 3794–3812. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Fogagnolo, M.; Ferraro, L.; Capuzzo, A.; Pavan, B.; Rassu, G.; Salis, A.; Giunchedi, P.; Gavini, E. Nasal chitosan microparticles target a zidovudine prodrug to brain HIV sanctuaries. Antivir. Res. 2015, 123, 146–157. [Google Scholar] [CrossRef]
- Toti, K.S.; Derudas, M.; Pertusati, F.; Sinnaeve, D.; Van den Broeck, F.; Margamuljana, L.; Martins, J.C.; Herdewijn, P.; Balzarini, J.; McGuigan, C.; et al. Synthesis of an apionucleoside family and discovery of a prodrug with anti-HIV activity. J. Org. Chem. 2014, 79, 5097–5112. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, M.A.M.; Meanwell, N.A.; Kadow, J.F. Design strategies in the prodrugs of HIV-1 protease inhibitors to improve the pharmaceutical properties. Eur. J. Med. Chem. 2017, 139, 865–883. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Mandava, N.; Gokulgandhi, M.; Pal, D.; Mitra, A.K. Amino Acid Prodrugs: An Approach to Improve the Absorption of HIV-1 Protease Inhibitor, Lopinavir. Pharmaceuticals 2014, 7, 433–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [Green Version]
- Banoub, M.G.; Bade, A.N.; Lin, Z.; Cobb, D.; Gautam, N.; Dyavar Shetty, B.L.; Wojtkiewicz, M.; Alnouti, Y.; McMillan, J.; Gendelman, H.E.; et al. Synthesis and Characterization of Long-Acting Darunavir Prodrugs. Mol. Pharm. 2020, 17, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, M.A.M.; Ramar, T.; Subramani, L.; Desai, S.D.; Sinha, S.; Mandlekar, S.; Jenkins, S.M.; Krystal, M.R.; Subramanian, M.; Sridhar, S.; et al. (Carbonyl)oxyalkyl linker-based amino acid prodrugs of the HIV-1 protease inhibitor atazanavir that enhance oral bioavailability and plasma trough concentration. Eur. J. Med. Chem. 2020, 207, 112749. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Chu, Y.; Feng, W.; Fromont, C.; He, S.; Ali, J.; Lee, J.B.; Zgair, A.; Berton, M.; Bettonte, S.; et al. Targeted delivery of lopinavir to HIV reservoirs in the mesenteric lymphatic system by lipophilic ester prodrug approach. J. Control. Release 2021, 329, 1077–1089. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Nowicka-Sans, B.; Gong, Y.F.; McAuliffe, B.; Dicker, I.; Ho, H.T.; Zhou, N.; Eggers, B.; Lin, P.F.; Ray, N.; Wind-Rotolo, M.; et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob. Agents Chemother. 2012, 56, 3498–3507. [Google Scholar] [CrossRef] [Green Version]
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; et al. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2020, 382, 1232–1243. [Google Scholar] [CrossRef]
- Sloane, J.L.; Benner, N.L.; Keenan, K.N.; Zang, X.; Soliman, M.S.A.; Wu, X.; Dimapasoc, M.; Chun, T.W.; Marsden, M.D.; Zack, J.A.; et al. Prodrugs of PKC modulators show enhanced HIV latency reversal and an expanded therapeutic window. Proc. Natl. Acad. Sci. USA 2020, 117, 10688–10698. [Google Scholar] [CrossRef]
- Freeling, J.P.; Ho, R.J. Anti-HIV drug particles may overcome lymphatic drug insufficiency and associated HIV persistence. Proc. Natl. Acad. Sci. USA 2014, 111, E2512–E2513. [Google Scholar] [CrossRef] [Green Version]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS 2014, 28, 2625–2627. [Google Scholar] [CrossRef]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res. Hum. Retroviruses 2015, 31, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnachie, L.A.; Kinman, L.M.; Koehn, J.; Kraft, J.C.; Lane, S.; Lee, W.; Collier, A.C.; Ho, R.J.Y. Long-Acting Profile of 4 Drugs in 1 Anti-HIV Nanosuspension in Nonhuman Primates for 5 Weeks After a Single Subcutaneous Injection. J. Pharm. Sci. 2018, 107, 1787–1790. [Google Scholar] [CrossRef]
- Edagwa, B.; McMillan, J.; Sillman, B.; Gendelman, H.E. Long-acting slow effective release antiretroviral therapy. Expert Opin. Drug Deliv. 2017, 14, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Gautam, N.; McMillan, J.M.; Kumar, D.; Bade, A.N.; Pan, Q.; Kulkarni, T.A.; Li, W.; Sillman, B.; Smith, N.A.; Shetty, B.L.D.; et al. Lipophilic nanocrystal prodrug-release defines the extended pharmacokinetic profiles of a year-long cabotegravir. Nat. Commun. 2021, 12, 3453. [Google Scholar] [CrossRef]
- Lin, Z.; Gautam, N.; Alnouti, Y.; McMillan, J.; Bade, A.N.; Gendelman, H.E.; Edagwa, B. ProTide generated long-acting abacavir nanoformulations. Chem. Commun. 2018, 54, 8371–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, I.M.; Bade, A.N.; Lin, Z.; Soni, D.; Wojtkiewicz, M.; Dyavar Shetty, B.L.; Gautam, N.; McMillan, J.M.; Alnouti, Y.; Edagwa, B.J.; et al. Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation. Int. J. Nanomed. 2019, 14, 6231–6247. [Google Scholar] [CrossRef] [Green Version]
- Soni, D.; Bade, A.N.; Gautam, N.; Herskovitz, J.; Ibrahim, I.M.; Smith, N.; Wojtkiewicz, M.S.; Dyavar Shetty, B.L.; Alnouti, Y.; McMillan, J.; et al. Synthesis of a long acting nanoformulated emtricitabine ProTide. Biomaterials 2019, 222, 119441. [Google Scholar] [CrossRef]
- Smith, N.; Bade, A.N.; Soni, D.; Gautam, N.; Alnouti, Y.; Herskovitz, J.; Ibrahim, I.M.; Wojtkiewicz, M.S.; Dyavar Shetty, B.L.; McMillan, J.; et al. A long acting nanoformulated lamivudine ProTide. Biomaterials 2019, 223, 119476. [Google Scholar] [CrossRef] [PubMed]
- Cobb, D.A.; Smith, N.; Deodhar, S.; Bade, A.N.; Gautam, N.; Shetty, B.L.D.; McMillan, J.; Alnouti, Y.; Cohen, S.M.; Gendelman, H.E.; et al. Transformation of tenofovir into stable ProTide nanocrystals with long-acting pharmacokinetic profiles. Nat. Commum. 2021, 12, 5458. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.A.; Bade, A.N.; Sillman, B.; Shetty, B.L.D.; Wojtkiewicz, M.S.; Gautam, N.; Hilaire, J.R.; Sravanam, S.; Szlachetka, A.; Lamberty, B.G.; et al. A year-long extended release nanoformulated cabotegravir prodrug. Nat. Mater. 2020, 19, 910–920. [Google Scholar] [CrossRef] [PubMed]
- McMillan, J.; Szlachetka, A.; Slack, L.; Sillman, B.; Lamberty, B.; Morsey, B.; Callen, S.; Gautam, N.; Alnouti, Y.; Edagwa, B.; et al. Pharmacokinetics of a Long-Acting Nanoformulated Dolutegravir Prodrug in Rhesus Macaques. Antimicrob. Agents Chemother. 2018, 62, e01316-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillman, B.; Woldstad, C.; McMillan, J.; Gendelman, H.E. Neuropathogenesis of human immunodeficiency virus infection. Handb. Clin. Neurol. 2018, 152, 21–40. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Fogagnolo, M.; Ferraro, L.; Beggiato, S.; Hanuskova, M.; Maretti, E.; Sacchetti, F.; Leo, E.; Pavan, B. Bile salt-coating modulates the macrophage uptake of nanocores constituted by a zidovudine prodrug and enhances its nose-to-brain delivery. Eur. J. Pharm. Biopharm. 2019, 144, 91–100. [Google Scholar] [CrossRef]
- Custer, B.; Sullivan, S.D.; Hazlet, T.K.; Iloeje, U.; Veenstra, D.L.; Kowdley, K.V. Global epidemiology of hepatitis B virus. J. Clin. Gastroenterol. 2004, 38, S158–S168. [Google Scholar] [CrossRef]
- Childs-Kean, L.M.; Egelund, E.F.; Jourjy, J. Tenofovir alafenamide for the treatment of chronic hepatitis B monoinfection. Pharmacotherapy 2018, 38, 1051–1057. [Google Scholar] [CrossRef]
- Agarwal, K.; Brunetto, M.; Seto, W.K.; Lim, Y.S.; Fung, S.; Marcellin, P.; Ahn, S.H.; Izumi, N.; Chuang, W.L.; Bae, H.; et al. 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J. Hepatol. 2018, 68, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.R.; Alam, A.; Shukla, A.; Dashtseren, B.; Lesmana, C.R.A.; Duger, D.; Payawal, D.A.; Duy Cuong, D.; Jargalsaikhan, G.; Cua, I.H.Y.; et al. An expert review on the use of tenofovir alafenamide for the treatment of chronic hepatitis B virus infection in Asia. J. Gastroenterol. 2020, 55, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Matelich, M.C.; Ugarkar, B.G.; Gómez-Galeno, J.E.; DaRe, J.; Ollis, K.; Sun, Z.; Craigo, W.; Colby, T.J.; Fujitaki, J.M.; et al. Pradefovir: A prodrug that targets adefovir to the liver for the treatment of hepatitis B. J. Med. Chem. 2008, 51, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, M.; Zhu, X.; Li, C.; Li, X.; Jin, W.; Zhang, D.; Chen, H.; Liu, C.; Ding, Y.; et al. Safety, efficacy, and pharmacokinetics of pradefovir for the treatment of chronic hepatitis B infection. Antivir. Res. 2020, 174, 104693. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kong, F.; Song, X.; Shang, J.; Yao, L.; Xia, J.; Peng, Y.; Liu, W.; Gong, H.; Mu, M.; et al. Pradefovir Treatment in Patients with Chronic Hepatitis B: Week 24 Results from a Multicenter, Double-blind, Randomized, Noninferiority, Phase 2 Trial. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Clinical Trial NCT04543565. Available online: https://clinicaltrials.gov/ct2/show/NCT04543565 (accessed on 2 November 2021).
- Anthony, D.D.; Sulkowski, M.S.; Smeaton, L.M.; Damjanovska, S.; Shive, C.L.; Kowal, C.M.; Cohen, D.E.; Bhattacharya, D.; Alston-Smith, B.L.; Balagopal, A.; et al. Hepatitis C Virus (HCV) Direct-Acting Antiviral Therapy in Persons With Human Immunodeficiency Virus-HCV Genotype 1 Coinfection Resulting in High Rate of Sustained Virologic Response and Variable in Normalization of Soluble Markers of Immune Activation. J. Infect. Dis. 2020, 222, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Miller, R.H.; Purcell, R.H. Genetic heterogeneity of hepatitis C virus: Quasispecies and genotypes. Semin. Liver Dis. 1995, 15, 41–63. [Google Scholar] [CrossRef]
- Berliba, E.; Bogus, M.; Vanhoutte, F.; Berghmans, P.J.; Good, S.S.; Moussa, A.; Pietropaolo, K.; Murphy, R.L.; Zhou, X.J.; Sommadossi, J.P. Safety, pharmacokinetics and antiviral activity of AT-527, a novel purine nucleotide prodrug, in HCV-infected subjects with and without cirrhosis. Antimicrob. Agents Chemother. 2019, 63, 12. [Google Scholar] [CrossRef] [Green Version]
- Good, S.S.; Westover, J.; Jung, K.H.; Zhou, X.J.; Moussa, A.; La Colla, P.; Collu, G.; Canard, B.; Sommadossi, J.P. AT-527, a Double Prodrug of a Guanosine Nucleotide Analog, Is a Potent Inhibitor of SARS-CoV-2 In Vitro and a Promising Oral Antiviral for Treatment of COVID-19. Antimicrob. Agents Chemother. 2021, 65. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: Molnupiravir reduces risk of hospital admission or death by 50% in patients at risk, MSD reports. BMJ 2021, 375, n2422. [Google Scholar] [CrossRef]
- Eron, J., Jr.; Yeni, P.; Gathe, J., Jr.; Estrada, V.; DeJesus, E.; Staszewski, S.; Lackey, P.; Katlama, C.; Young, B.; Yau, L.; et al. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: A randomised non-inferiority trial. Lancet 2006, 368, 476–482. [Google Scholar] [CrossRef]
- Kevadiya, B.D.; Machhi, J.; Herskovitz, J.; Oleynikov, M.D.; Blomberg, W.R.; Bajwa, N.; Soni, D.; Das, S.; Hasan, M.; Patel, M.; et al. Pharmacotherapeutics of SARS-CoV-2 infections. J. Neuroimmune Pharmacol. 2021, 16, 12–37. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Covid-19: UK becomes first country to authorise antiviral molnupiravir. BMJ 2021, 375, n2697. [Google Scholar] [CrossRef] [PubMed]
- Mulangu, S.; Dodd, L.E.; Davey, R.T.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A randomized, controlled trial of ebola virus disease therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier drug targeting: The future of brain drug development. Mol. Interv. 2003, 3, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Pinnen, F.; Cacciatore, I.; Cornacchia, C.; Sozio, P.; Iannitelli, A.; Costa, M.; Pecci, L.; Nasuti, C.; Cantalamessa, F.; Di Stefano, A. Synthesis and study of L-DOPA−glutathione codrugs as new anti-Parkinson agents with free radical scavenging properties. J. Med. Chem. 2007, 50, 2506–2515. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| R13: 7,8-DHF prodrug [54] |  |
| Memit [46] |  |
| ALZ801 [63] |  |
| HIV | Hepatitis B | COVID-19 |
|---|---|---|
 |  |  |
 Tenofovir alafenamide (TAF) [146,147] |  AT-527 (Bemnifosbuvir) [156] |  Remdesivir [157] |
 Prodrug of abacavir (M3ABC) [136] | ||
 Prodrug of tenofovir (M1TFV) [140] | ||
 Prodrug of darunavir (M2DRV) [123] |  Pradefovir [149,150] |  Molnupiravir [158] |
 Fosamprenavir [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markovic, M.; Deodhar, S.; Machhi, J.; Yeapuri, P.; Saleh, M.; J. Edagwa, B.; Mosley, R.L.; Gendelman, H.E. Prodrug Therapies for Infectious and Neurodegenerative Diseases. Pharmaceutics 2022, 14, 518. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030518
Markovic M, Deodhar S, Machhi J, Yeapuri P, Saleh M, J. Edagwa B, Mosley RL, Gendelman HE. Prodrug Therapies for Infectious and Neurodegenerative Diseases. Pharmaceutics. 2022; 14(3):518. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030518
Chicago/Turabian StyleMarkovic, Milica, Suyash Deodhar, Jatin Machhi, Pravin Yeapuri, Maamoon Saleh, Benson J. Edagwa, Rodney Lee Mosley, and Howard E. Gendelman. 2022. "Prodrug Therapies for Infectious and Neurodegenerative Diseases" Pharmaceutics 14, no. 3: 518. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030518