
The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K–Akt Pathways

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
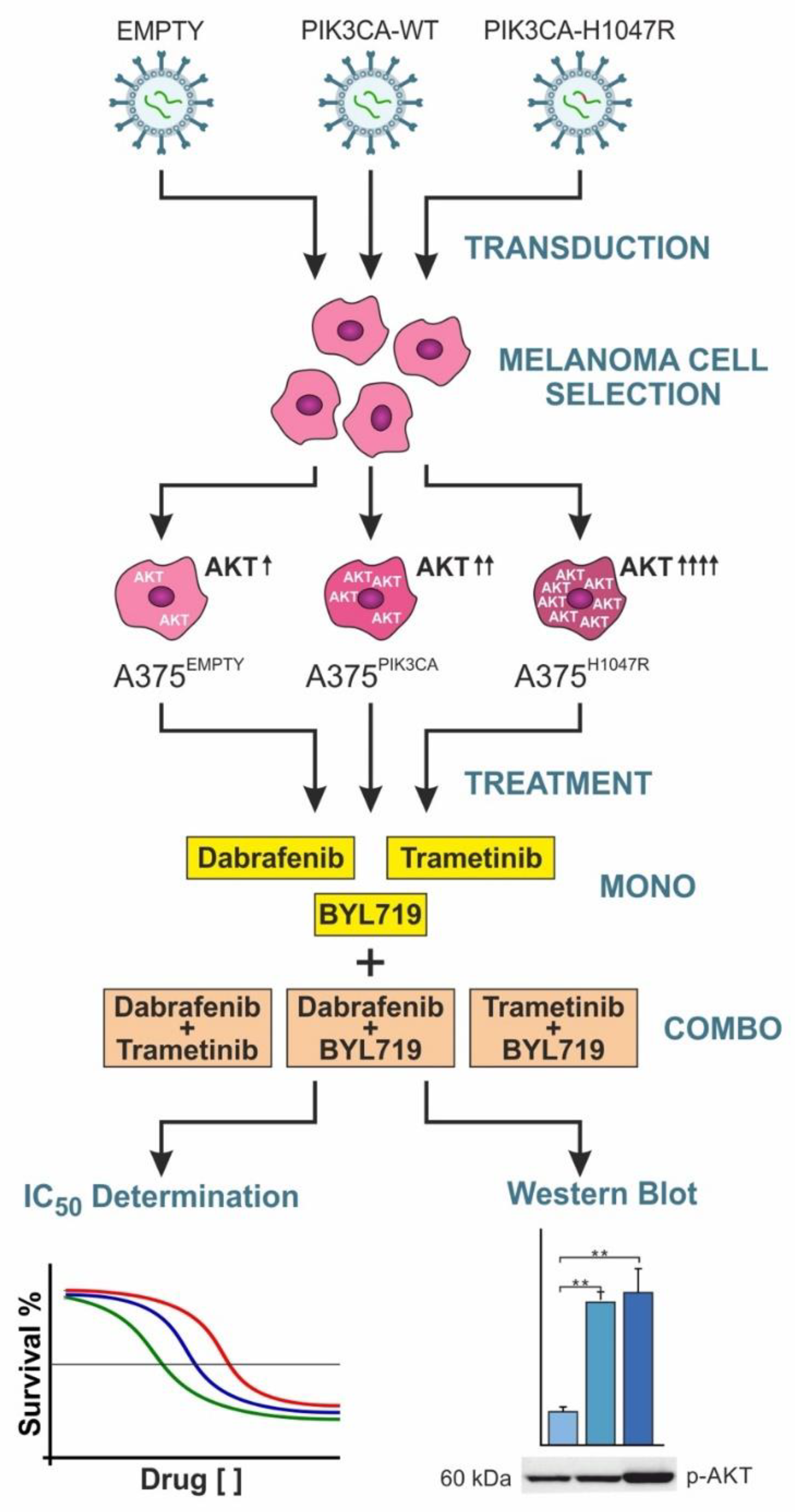
2.1. Cell Lines and Retroviral Transduction
2.2. Cell Growth and Cell Viability Assays
2.3. Treatment of the Transduced A375 Cells
2.4. Western Blot
2.5. Statistical Analysis
3. Results
3.1. PIK3CA H1047R Mutation Induces PI3K–Akt Pathway Activation and A375 Proliferation
3.2. PIK3CA H1047R Mutation in A375 Cells Induces Resistance to BRAF and MEK Inhibitors
3.3. The Inhibition of PIK3CA H1047R Mutation Induces Dabrafenib and Trametinib Sensitivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonardi, G.C.; Candido, S.; Falzone, L.; Spandidos, D.A.; Libra, M. Cutaneous melanoma and the immunotherapy revolution (Review). Int. J. Oncol. 2020, 57, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Christofi, T.; Baritaki, S.; Falzone, L.; Libra, M.; Zaravinos, A. Current Perspectives in Cancer Immunotherapy. Cancers 2019, 11, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavò, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NF-κB inhibition is associated with OPN/MMP-9 downregulation in cutaneous melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Daphu, I.; Horn, S.; Stieber, D.; Varughese, J.K.; Spriet, E.; Dale, H.A.; Skaftnesmo, K.O.; Bjerkvig, R.; Thorsen, F. In vitro treatment of melanoma brain metastasis by simultaneously targeting the MAPK and PI3K signaling pathways. Int. J. Mol. Sci. 2014, 15, 8773–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yang, X.; Liu, J.; Zhao, B.; Cai, W.; Li, Y.; Hu, D. Efficacy and safety of BRAF inhibition alone versus combined BRAF and MEK inhibition in melanoma: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 32258–32269. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Weber, J.S.; Infante, J.R.; Kim, K.B.; Daud, A.; Gonzalez, R.; Sosman, J.A.; Hamid, O.; Schuchter, L.; Cebon, J.; et al. Overall Survival and Durable Responses in Patients with BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined with Trametinib. J. Clin. Oncol. 2016, 34, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients with BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Chi, M.; Ye, Y.; Zhang, X.D.; Chen, J. Insulin induces drug resistance in melanoma through activation of the PI3K/Akt pathway. Drug Des. Dev. Ther. 2014, 8, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Irvine, M.; Stewart, A.; Pedersen, B.; Boyd, S.; Kefford, R.; Rizos, H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.L.; Figueroa, C.; Brtva, T.R.; Zhu, T.; Taylor, J.; Barber, T.D.; Vojtek, A.B. Negative regulation of the serine/threonine kinase B-Raf by Akt. J. Biol. Chem. 2000, 275, 27354–27359. [Google Scholar] [CrossRef]
- Guri, Y.; Hall, M.N. mTOR Signaling Confers Resistance to Targeted Cancer Drugs. Trends Cancer 2016, 2, 688–697. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Tran, K.B.; Kolekar, S.; Jabed, A.; Jaynes, P.; Shih, J.H.; Wang, Q.; Flanagan, J.U.; Rewcastle, G.W.; Baguley, B.C.; Shepherd, P.R. Diverse mechanisms activate the PI 3-kinase/mTOR pathway in melanomas: Implications for the use of PI 3-kinase inhibitors to overcome resistance to inhibitors of BRAF and MEK. BMC Cancer 2021, 21, 136. [Google Scholar] [CrossRef]
- Silva, J.M.; Deuker, M.M.; Baguley, B.C.; McMahon, M. PIK3CA-mutated melanoma cells rely on cooperative signaling through mTORC1/2 for sustained proliferation. Pigment Cell Melanoma Res. 2017, 30, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.Y.; Ma, W.L.; Lu, Y.S. Role of Alpelisib in the Treatment of PIK3CA-Mutated Breast Cancer: Patient Selection and Clinical Perspectives. Ther. Clin. Risk Manag. 2021, 17, 193–207. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Dabrafenib | Trametinib | BYL719 |
|---|---|---|---|
| Dabrafenib (0.01–0.1–1–10–100–1000 nM) | / | 0.5 nM | 1 μM |
| Trametinib (0.001–0.01–0.1–1–10–100 nM) | 0.5 nM | / | 1 μM |
| BYL719 (0.001–0.01–0.1–1–10–100 μM) | 1 nM | 0.5 nM | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candido, S.; Salemi, R.; Piccinin, S.; Falzone, L.; Libra, M. The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K–Akt Pathways. Pharmaceutics 2022, 14, 590. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030590
Candido S, Salemi R, Piccinin S, Falzone L, Libra M. The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K–Akt Pathways. Pharmaceutics. 2022; 14(3):590. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030590
Chicago/Turabian StyleCandido, Saverio, Rossella Salemi, Sara Piccinin, Luca Falzone, and Massimo Libra. 2022. "The PIK3CA H1047R Mutation Confers Resistance to BRAF and MEK Inhibitors in A375 Melanoma Cells through the Cross-Activation of MAPK and PI3K–Akt Pathways" Pharmaceutics 14, no. 3: 590. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14030590