
Pharmacokinetic/Pharmacodynamic Evaluation of a New Purine-2,6-Dione Derivative in Rodents with Experimental Autoimmune Diseases

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. Experimental Procedures
2.3.1. PDE Assay
2.3.2. Pharmacokinetic Study
2.3.3. Collagen-Induced Arthritis
2.3.4. Experimental Autoimmune Encephalomyelitis
2.3.5. ConA-Induced Hepatitis
2.4. Analytical Methods
2.5. Data Analysis
2.5.1. Pharmacokinetics
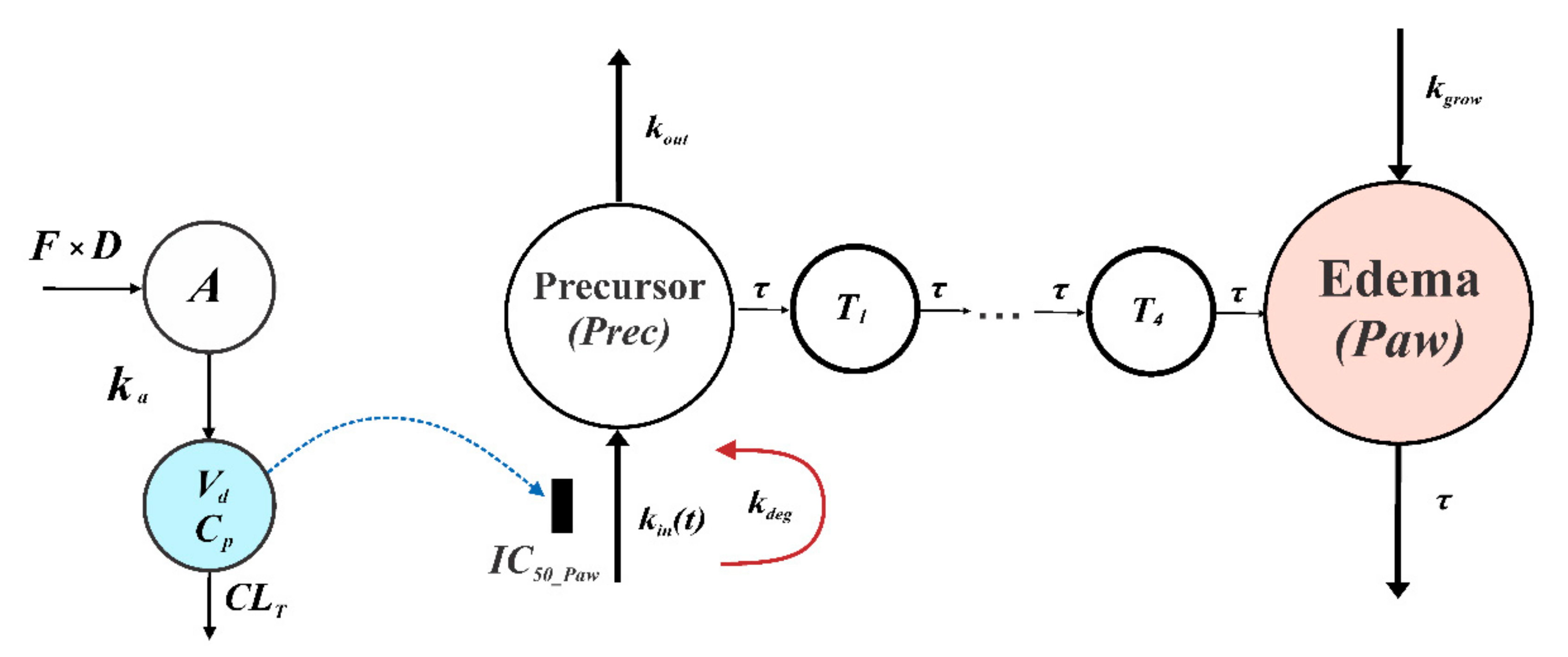
2.5.2. PK/PD CIA Progression Model of Compound 34
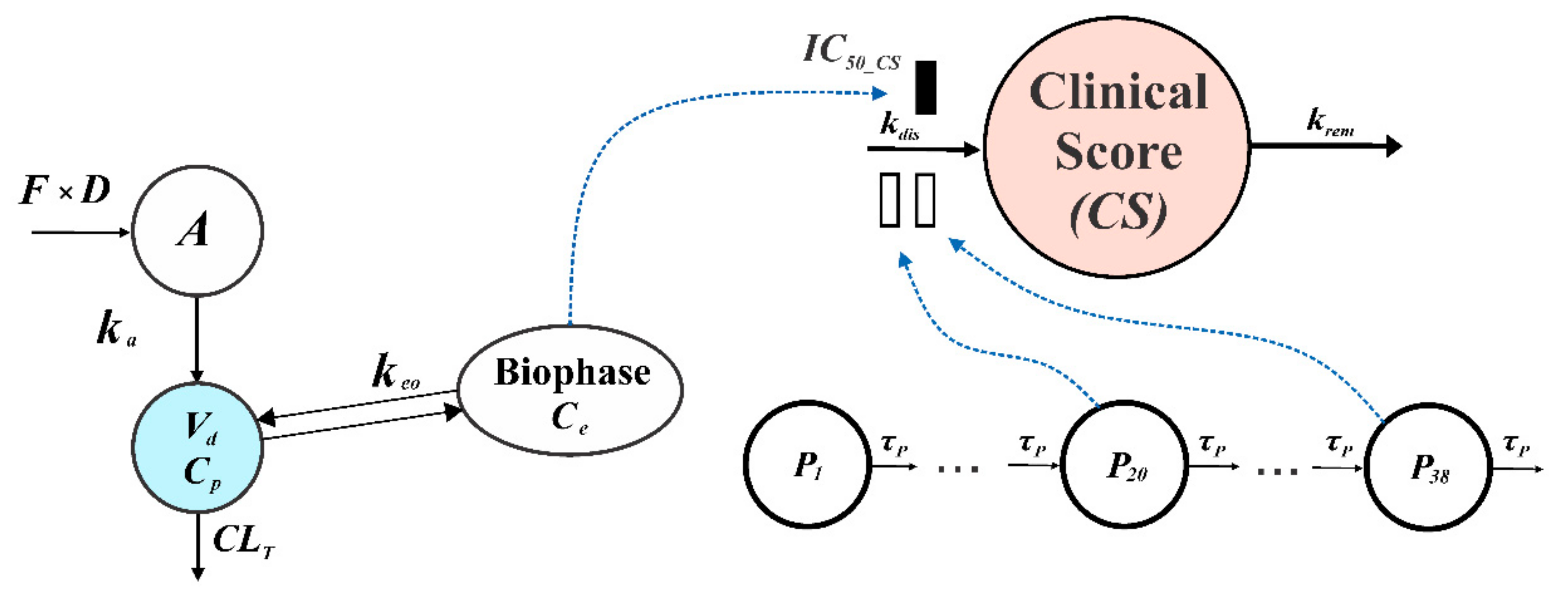
2.5.3. PK/PD EAE Progression Model of Compound 34
2.5.4. Computation
2.5.5. Statistical Analysis
3. Results
3.1. PDE Assay
3.2. Pharmacokinetics
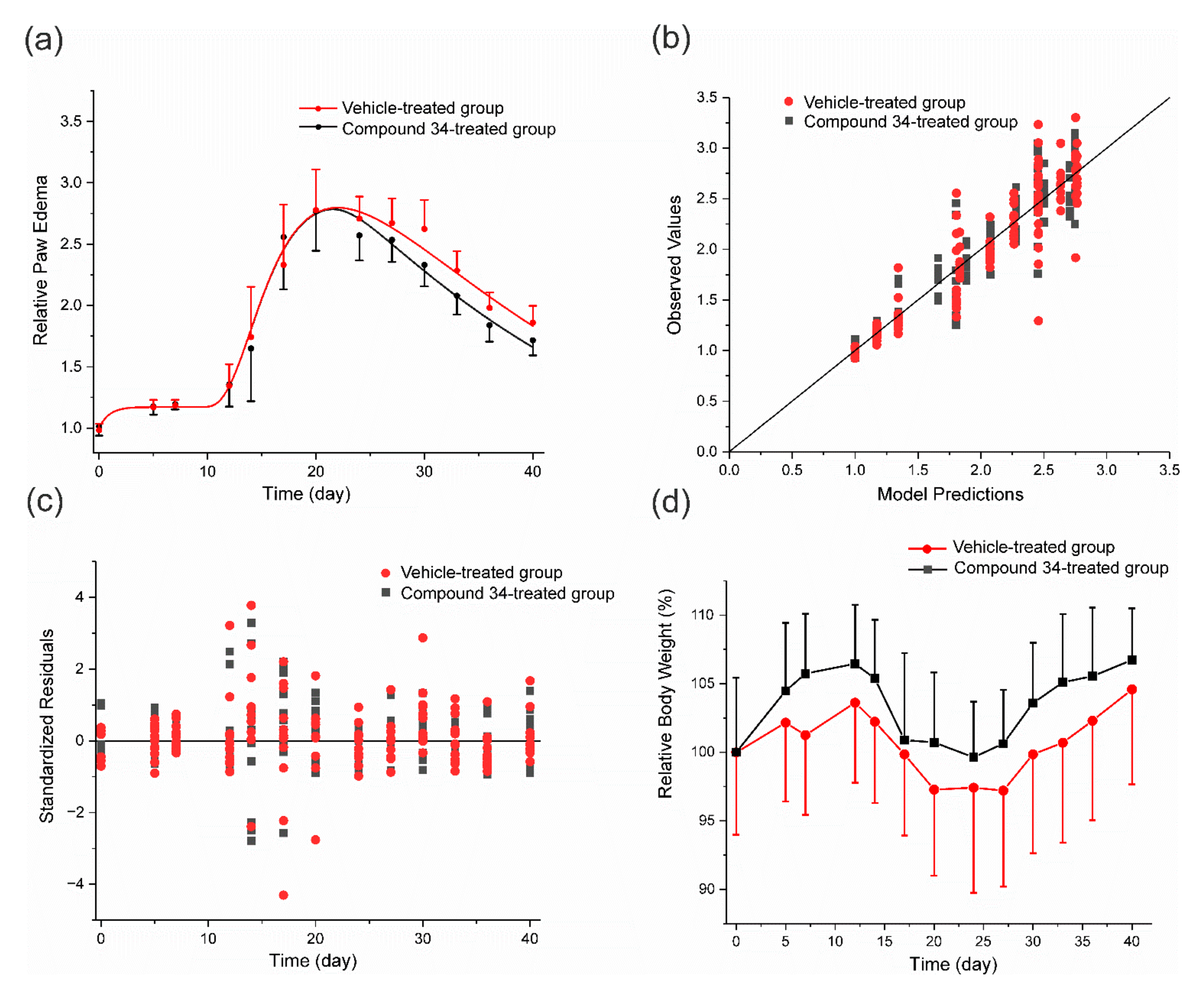
3.3. PK/PD Arthritis Progression Model
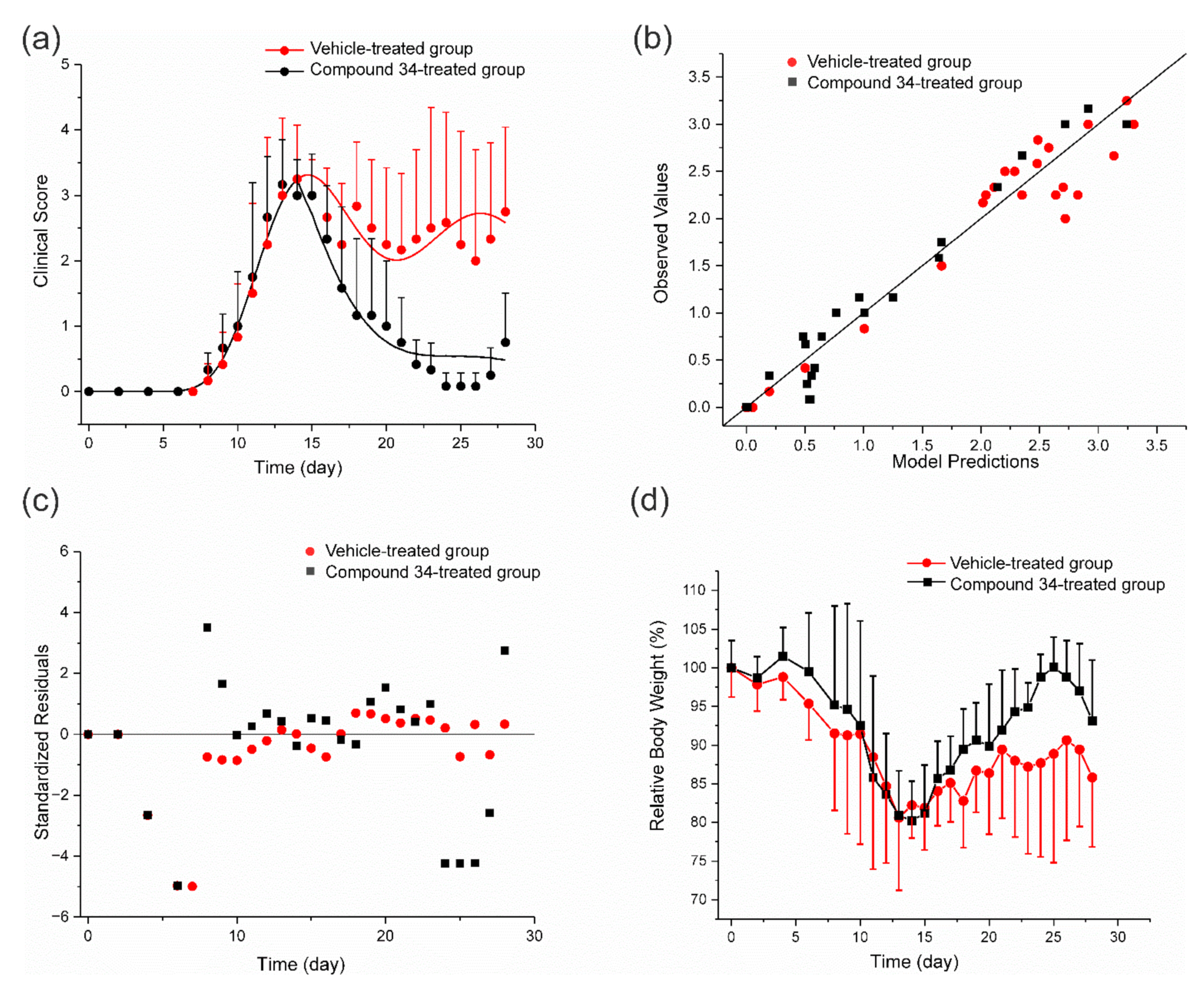
3.4. PK/PD EAE Progression Model
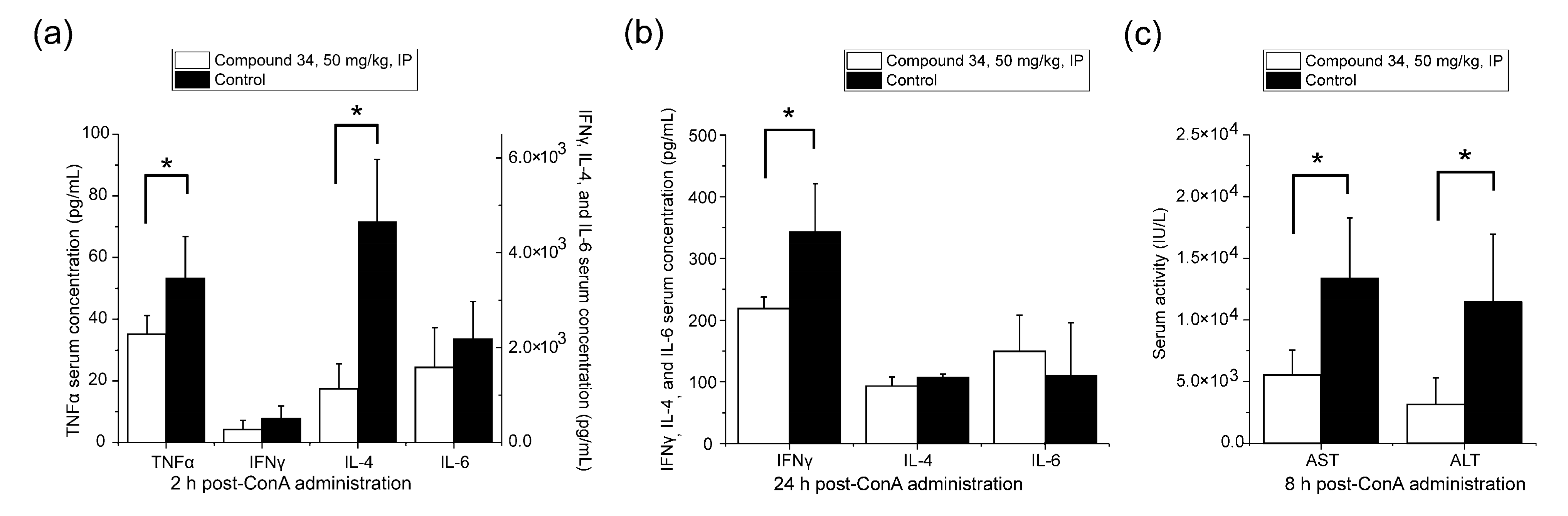
3.5. ConA-Induced Hepatitis Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soderling, S.H.; Beavo, J.A. Regulation of cAMP and cGMP Signaling: New Phosphodiesterases and New Functions. Curr. Opin. Cell Biol. 2000, 12, 174–179. [Google Scholar] [CrossRef]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master Regulator of Innate Immune Cell Function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.T.; Beavo, J.A. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Schett, G.; Sloan, V.S.; Stevens, R.M.; Schafer, P. Apremilast: A Novel PDE4 Inhibitor in the Treatment of Autoimmune and Inflammatory Diseases. Ther. Adv. Musculoskelet. Dis. 2010, 2, 271–278. [Google Scholar] [CrossRef]
- Mestre, L.; Redondo, M.; Carrillo-Salinas, F.J.; Morales-García, J.A.; Alonso-Gil, S.; Pérez-Castillo, A.; Gil, C.; Martínez, A.; Guaza, C. PDE7 Inhibitor TC3.6 Ameliorates Symptomatology in a Model of Primary Progressive Multiple Sclerosis. Br. J. Pharmacol. 2015, 172, 4277–4290. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van der Mei, I.; et al. Rising Prevalence of Multiple Sclerosis Worldwide: Insights from the Atlas of MS, Third Edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- González-García, C.; Bravo, B.; Ballester, A.; Gómez-Pérez, R.; Eguiluz, C.; Redondo, M.; Martínez, A.; Gil, C.; Ballester, S. Comparative Assessment of PDE 4 and 7 Inhibitors as Therapeutic Agents in Experimental Autoimmune Encephalomyelitis. Br. J. Pharmacol. 2013, 170, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Thakker, P.; Leach, M.W.; Kuang, W.; Benoit, S.E.; Leonard, J.P.; Marusic, S. IL-23 Is Critical in the Induction but Not in the Effector Phase of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2007, 178, 2589–2598. [Google Scholar] [CrossRef] [Green Version]
- Mendel, I.; De Rosbo, N.K.; Ben-Nun, A. A Myelin Oligodendrocyte Glycoprotein Peptide Induces Typical Chronic Experimental Autoimmune Encephalomyelitis in H-2b Mice: Fine Specificity and T Cell Receptor Vβ Expression of Encephalitogenic T Cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef]
- Sommer, N.; Löschmann, P.A.; Northoff, G.H.; Weller, M.; Steinbrecher, A.; Steinbach, J.P.; Lichtenfels, R.; Meyermann, R.; Riethmüller, A.; Fontana, A.; et al. The Antidepressant Rolipram Suppresses Cytokine Production and Prevents Autoimmune Encephalomyelitis. Nat. Med. 1995, 1, 244–248. [Google Scholar] [CrossRef]
- Martín-Álvarez, R.; Paúl-Fernández, N.; Palomo, V.; Gil, C.; Martínez, A.; Mengod, G. A Preliminary Investigation of Phoshodiesterase 7 Inhibitor VP3.15 as Therapeutic Agent for the Treatment of Experimental Autoimmune Encephalomyelitis Mice. J. Chem. Neuroanat. 2017, 80, 27–36. [Google Scholar] [CrossRef]
- García, A.M.; Brea, J.; Morales-García, J.A.; Perez, D.I.; González, A.; Alonso-Gil, S.; Gracia-Rubio, I.; Ros-Simó, C.; Conde, S.; Cadavid, M.I.; et al. Modulation of cAMP-Specific PDE without Emetogenic Activity: New Sulfide-like PDE7 Inhibitors. J. Med. Chem. 2014, 57, 8590–8607. [Google Scholar] [CrossRef] [Green Version]
- Robichaud, A.; Savoie, C.; Stamatiou, P.B.; Tattersall, F.D.; Chan, C.C. PDE4 Inhibitors Induce Emesis in Ferrets via a Noradrenergic Pathway. Neuropharmacology 2001, 40, 262–269. [Google Scholar] [CrossRef]
- Redondo, M.; Brea, J.; Perez, D.I.; Soteras, I.; Val, C.; Perez, C.; Morales-García, J.A.; Alonso-Gil, S.; Paul-Fernandez, N.; Martin-Alvarez, R.; et al. Effect of Phosphodiesterase 7 (PDE7) Inhibitors in Experimental Autoimmune Encephalomyelitis Mice. Discovery of a New Chemically Diverse Family of Compounds. J. Med. Chem. 2012, 55, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- Giembycz, M.A. Life after PDE4: Overcoming Adverse Events with Dual-Specificity Phosphodiesterase Inhibitors. Curr. Opin. Pharmacol. 2005, 5, 238–244. [Google Scholar] [CrossRef]
- Yamamoto, S.; Sugahara, S.; Ikeda, K.; Shimizu, Y. Amelioration of Collagen-Induced Arthritis in Mice by a Novel Phosphodiesterase 7 and 4 Dual Inhibitor, YM-393059. Eur. J. Pharmacol. 2007, 559, 219–226. [Google Scholar] [CrossRef]
- Ross, S.E.; Williams, R.O.; Mason, L.J.; Mauri, C.; Marinova-Mutafchieva, L.; Malfait, A.M.; Maini, R.N.; Feldmann, M. Suppression of TNF-Alpha Expression, Inhibition of Th1 Activity, and Amelioration of Collagen-Induced Arthritis by Rolipram. J. Immunol. 1997, 159, 6253–6259. [Google Scholar]
- Tagawa, Y.I.; Kakuta, S.; Iwakura, Y. Involvement of Fas/Fas Ligand System-Mediated Apoptosis in the Development of Concanavalin A-Induced Hepatitis. Eur. J. Immunol. 1998, 28, 4105–4113. [Google Scholar] [CrossRef]
- Wang, H.-X.; Liu, M.; Weng, S.-Y.; Li, J.-J.; Xie, C.; He, H.-L.; Guan, W.; Yuan, Y.-S.; Gao, J. Immune Mechanisms of Concanavalin A Model of Autoimmune Hepatitis. World J. Gastroenterol. 2012, 18, 119–125. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, H.; Hou, T. Concanavalin A-Induced Autoimmune Hepatitis Model in Mice: Mechanisms and Future Outlook. Open Life Sci. 2022, 17, 91–101. [Google Scholar] [CrossRef]
- Cook, S.F.; Bies, R.R. Disease Progression Modeling: Key Concepts and Recent Developments. Curr. Pharmacol. Rep. 2016, 2, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Ayyar, V.S.; Jusko, W.J. Transitioning from Basic toward Systems Pharmacodynamic Models: Lessons from Corticosteroids. Pharmacol. Rev. 2020, 72, 414–438. [Google Scholar] [CrossRef]
- Durie, F.H.; Fava, R.A.; Noelle, R.J. Collagen-Induced Arthritis as a Model of Rheumatoid Arthritis. Clin. Immunol. Immunopathol. 1994, 73, 11–18. [Google Scholar] [CrossRef]
- Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Świerczek, A.; Pociecha, K.; Wyska, E.; Bucki, A.; Gawalska, A.; Kołaczkowski, M.; Pawłowski, M. Novel Butanehydrazide Derivatives of Purine-2,6-Dione as Dual PDE4/7 Inhibitors with Potential Anti-Inflammatory Activity: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2018, 146, 381–394. [Google Scholar] [CrossRef]
- Earp, J.C.; Dubois, D.C.; Almon, R.R.; Jusko, W.J. Quantitative Dynamic Models of Arthritis Progression in the Rat. Pharm. Res. 2009, 26, 196–203. [Google Scholar] [CrossRef]
- Hooke Laboratories, Mouse EAE Scoring. Available online: https://hookelabs.com/services/cro/eae/MouseEAEscoring.html (accessed on 16 April 2022).
- Świerczek, A.; Pociecha, K.; Ślusarczyk, M.; Chłoń-Rzepa, G.; Baś, S.; Mlynarski, J.; Więckowski, K.; Zadrożna, M.; Nowak, B.; Wyska, E. Comparative Assessment of the New PDE7 Inhibitor—GRMS-55 and Lisofylline in Animal Models of Immune-Related Disorders: A PK/PD Modeling Approach. Pharm. Res. 2020, 37, 19. [Google Scholar] [CrossRef] [Green Version]
- Dayneka, N.L.; Garg, V.; Jusko, W.J. Comparison of Four Basic Models of Indirect Pharmacodynamic Responses. J. Pharmacokinet. Biopharm. 1993, 21, 457–478. [Google Scholar] [CrossRef]
- Marusic, S.; Leach, M.W.; Pelker, J.W.; Azoitei, M.L.; Uozumi, N.; Cui, J.; Shen, M.W.H.; DeClercq, C.M.; Miyashiro, J.S.; Carito, B.A.; et al. Cytosolic Phospholipase A2α-Deficient Mice Are Resistant to Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2005, 202, 841–851. [Google Scholar] [CrossRef]
- Smith, A.K.; Ropella, G.E.P.; McGill, M.R.; Krishnan, P.; Dutta, L.; Kennedy, R.C.; Jaeschke, H.; Anthony Hunt, C. Contrasting Model Mechanisms of Alanine Aminotransferase (ALT) Release from Damaged and Necrotic Hepatocytes as an Example of General Biomarker Mechanisms. PLoS Comput. Biol. 2020, 16, e1007622. [Google Scholar] [CrossRef]
- Maltby, V.E.; Lea, R.A.; Reeves, P.; Saugbjerg, B.; Lechner-Scott, J. Reduced Cognitive Function Contributes to Economic Burden of Multiple Sclerosis. Mult. Scler. Relat. Disord. 2022, 60, 103707. [Google Scholar] [CrossRef] [PubMed]
- Strehl, C.; Ehlers, L.; Gaber, T.; Buttgereit, F. Glucocorticoids-All-Rounders Tackling the Versatile Players of the Immune System. Front. Immunol. 2019, 10, 1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, A.C. Immunosuppressive Medications. Clin. J. Am. Soc. Nephrol. 2016, 11, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimani, B.; Murray, K.; Hunt, D. Established and Emerging Immunological Complications of Biological Therapeutics in Multiple Sclerosis. Drug Saf. 2019, 42, 941–956. [Google Scholar] [CrossRef]
- Jankowska, A.; Świerczek, A.; Chłoń-Rzepa, G.; Pawłowski, M.; Wyska, E. PDE7-Selective and Dual Inhibitors: Advances in Chemical and Biological Research. Curr. Med. Chem. 2017, 24, 673–700. [Google Scholar] [CrossRef]
- O’Brien, J.J.; O’Callaghan, J.P.; Miller, D.B.; Chalgeri, S.; Wennogle, L.P.; Davis, R.E.; Snyder, G.L.; Hendrick, J.P. Inhibition of Calcium-Calmodulin-Dependent Phosphodiesterase (PDE1) Suppresses Inflammatory Responses. Mol. Cell. Neurosci. 2020, 102, 103449. [Google Scholar] [CrossRef]
- Goto, M.; Tanaka, Y.; Murakawa, M.; Kadoshima-Yamaoka, K.; Inoue, H.; Murafuji, H.; Nagahira, A.; Kanki, S.; Hayashi, Y.; Nagahira, K.; et al. Inhibition of Phosphodiesterase 7A Ameliorates Concanavalin A-Induced Hepatitis in Mice. Int. Immunopharmacol. 2009, 9, 1347–1351. [Google Scholar] [CrossRef]
- Xiang, M.; Zaccone, P.; Di Marco, R.; Magro, G.; Di Mauro, M.; Beltrami, B.; Meroni, P.; Nicoletti, F. Prevention by Rolipram of Concanavalin A-Induced T-Cell-Dependent Hepatitis in Mice. Eur. J. Pharmacol. 1999, 367, 399–404. [Google Scholar] [CrossRef]
- Świerczek, A.; Pomierny, B.; Wyska, E.; Jusko, W.J. Pharmacokinetic/Pharmacodynamic Assessment of Selective Phosphodiesterase Inhibitors in a Mouse Model of Autoimmune Hepatitis. J. Pharmacol. Exp. Ther. 2022, 381, 151–163. [Google Scholar] [CrossRef]
- Świerczek, A.; Plutecka, H.; Ślusarczyk, M.; Chłoń-Rzepa, G.; Wyska, E. PK/PD Modeling of the PDE7 Inhibitor—GRMS-55 in a Mouse Model of Autoimmune Hepatitis. Pharmaceutics 2021, 13, 597. [Google Scholar] [CrossRef]
- Giembycz, M.A.; Corrigan, C.J.; Seybold, J.; Newton, R.; Barnes, P.J. Identification of Cyclic AMP Phosphodiesterases 3, 4 and 7 in Human CD4+ and CD8+ T-Lymphocytes: Role in Regulating Proliferation and the Biosynthesis of Interleukin-2. Br. J. Pharmacol. 1996, 118, 1945–1958. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.T.; Ostenson, C.L.; Wang, E.H.; Beavo, J.A. Selective Up-Regulation of PDE1B2 upon Monocyte-to-Macrophage Differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-Y.Y.; Lon, H.-K.K.; Wang, Y.-L.L.; Dubois, D.C.; Almon, R.R.; Jusko, W.J. Pharmacokinetics, Pharmacodynamics and Toxicities of Methotrexate in Healthy and Collagen-Induced Arthritic Rats. Biopharm. Drug Dispos. 2013, 34, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Sugahara, S.; Ikeda, K.; Shimizu, Y. Pharmacological Profile of a Novel Phosphodiesterase 7A and -4 Dual Inhibitor, YM-393059, on Acute and Chronic Inflammation Models. Eur. J. Pharmacol. 2006, 550, 166–172. [Google Scholar] [CrossRef]
- Wehbi, V.L.; Taskén, K. Molecular Mechanisms for cAMP-Mediated Immunoregulation in T Cells—Role of Anchored Protein Kinase a Signaling Units. Front. Immunol. 2016, 7, 222. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, S.J.; Houslay, M.D. Action of Rolipram on Specific PDE4 cAMP Phosphodiesterase Isoforms and on the Phosphorylation of cAMP-Response-Element-Binding Protein (CREB) and P38 Mitogen-Activated Protein (MAP) Kinase in U937 Monocytic Cells. Biochem. J. 2000, 347, 571–578. [Google Scholar] [CrossRef]
- Pilla Reddy, V.; Kozielska, M.; Suleiman, A.A.; Johnson, M.; Vermeulen, A.; Liu, J.; De Greef, R.; Groothuis, G.M.M.; Danhof, M.; Proost, J.H. Pharmacokinetic-Pharmacodynamic Modeling of Antipsychotic Drugs in Patients with Schizophrenia Part I: The Use of PANSS Total Score and Clinical Utility. Schizophr. Res. 2013, 146, 144–152. [Google Scholar] [CrossRef]
- Bastida, C.; Soy, D.; Ruiz-Esquide, V.; Sanmartí, R.; Huitema, A.D.R. Exposure-Response Modeling of Tocilizumab in Rheumatoid Arthritis Using Continuous Composite Measures and Their Individual Components. Br. J. Clin. Pharmacol. 2019, 85, 1710–1718. [Google Scholar] [CrossRef]
- Hofstetter, H.H.; Karulin, A.Y.; Forsthuber, T.G.; Ott, P.A.; Tary-Lehmann, M.; Lehmann, P.v. The Cytokine Signature of MOG-Specific CD4 Cells in the EAE of C57BL/6 Mice. J. Neuroimmunol. 2005, 170, 105–114. [Google Scholar] [CrossRef]
- Quintero, O.L.; Amador-Patarroyo, M.J.; Montoya-Ortiz, G.; Rojas-Villarraga, A.; Anaya, J.M. Autoimmune Disease and Gender: Plausible Mechanisms for the Female Predominance of Autoimmunity. J. Autoimmun. 2012, 38, J109–J119. [Google Scholar] [CrossRef]
- Gantner, F.; Kusters, S.; Wendel, A.; Hatzelmann, A.; Schudt, C.; Tiegs, G. Protection from T Cell-Mediated Murine Liver Failure by Phosphodiesterase Inhibitors. J. Pharmacol. Exp. Ther. 1997, 280, 53–60. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
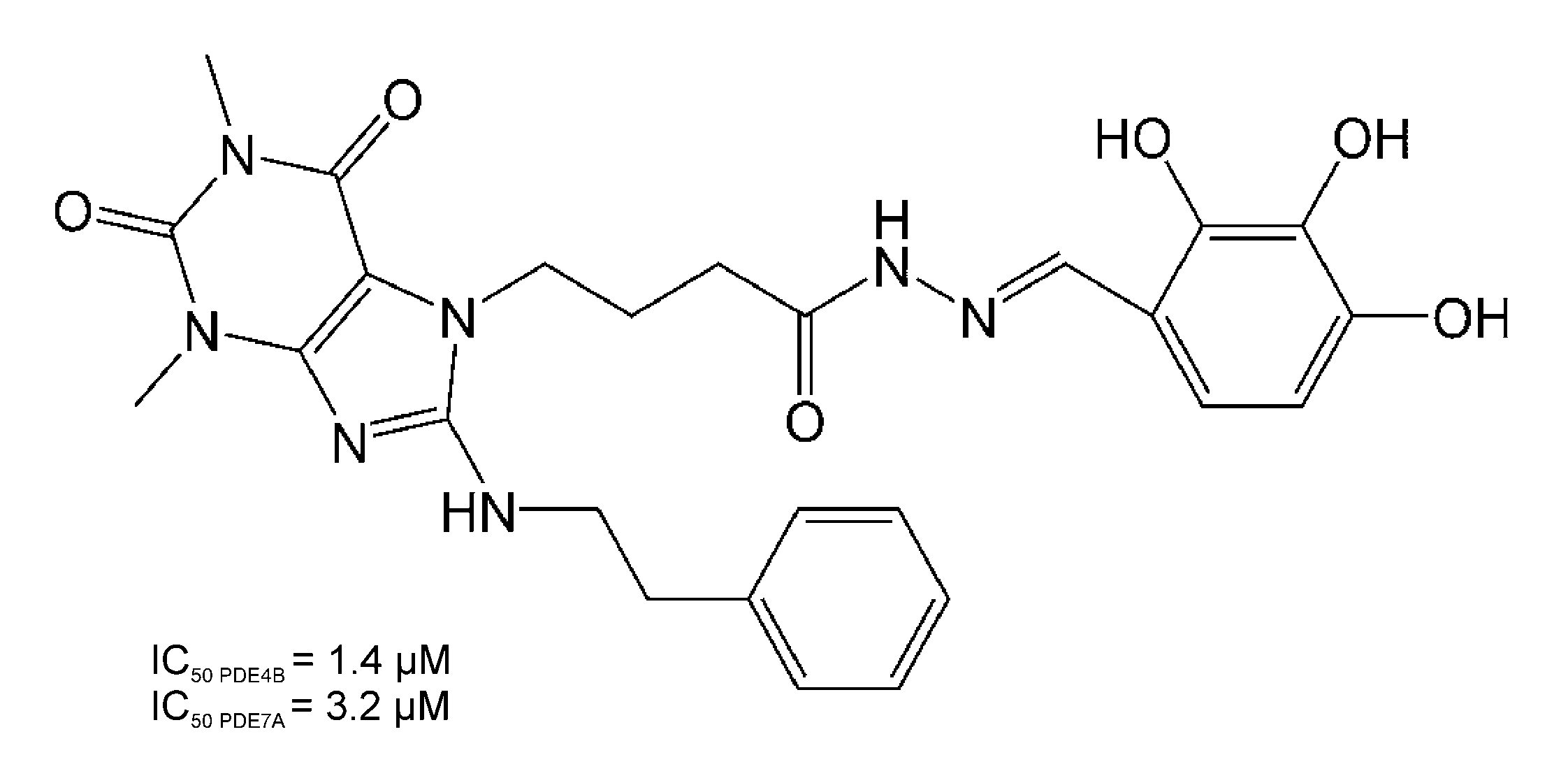
| Compound | IC50 (µM) Against PDE Isoform (CV%) | |||
|---|---|---|---|---|
| PDE1B | PDE3A | PDE4B | PDE7A | |
| Compound 34 | 0.21 (29.2) | 0.78 (8.3) | 1.4 c | 3.2 c |
| GRMS-55 | 2.5 b | 13.4 b | 26.9 b | 7.3 b |
| Reference compound a | 7.5 b | 0.2 b | 1.1 b | 2.1 b |
| Parameter | Brief Description | Species | |
|---|---|---|---|
| Rat | Mouse | ||
| Final Estimate (CV%) | Final Estimate (CV%) | ||
| Vd/F (L·kg−1) | Apparent volume of distribution | 8.70 (18.0) | 21.69 (34.6) |
| ka (min−1) | Absorption rate constant | 0.58 (159.2) | 0.15 (65.6) |
| AUC (mg·L−1·min) | Area under the concentration-time curve | 45.1 (12.6) | 49.1 (12.3) |
| t0.5 (min) | Elimination half-life | 13.6 (13.5) | 14.7 (31.2) |
| Cmax (mg·L−1) | Maximum serum concentration | 1.82 (28.9) | 1.37 (14.2) |
| CLT/F (L·min−1·kg−1) | Apparent clearance | 0.44 (12.6) | 1.02 (12.3) |
| Parameter | Brief Description | Final Estimate (CV%) |
|---|---|---|
| kout (day−1) | Degradation of precursor rate constant | 0.207(37.9) |
| kdeg (day−1) | Loss of production rate constant | 0.035 (18.4) |
| tonset (day) | Time of disease onset | 8.70 (9.5) |
| τ (day) | Mean transit time | 0.807 (34.5) |
| IC50_Paw (mg·L−1) | Concentration producing 50% of maximum inhibition | 0.001 (354.5) |
| kgrow (day−1) | Natural growth rate constant | 0.212 (37.8) |
| kin(0)C (day−1) | Initial value of precursor production variable in the compound 34-treated group | 0.748 (28.2) |
| kin(0)S (day−1) | Initial value of precursor production variable in vehicle-treated group | 0.752 (28.9) |
| Parameter | Brief Description | Estimate (CV%) |
| τP (day) | Mean transit time of disease progression precursor | 0.6564 (1.6) |
| kdis (day−1) | Disease progression rate constant | 16.46 (15.1) |
| krem (day−1) | Disease remission rate constant | 0.3654 (17.6) |
| keo (day−1) | Serum–effect-site equilibration rate constant | 0.3287 (95.2) |
| P1(0) | Initial value of the first disease progression precursor | 1 (fix) |
| IC50_CS (mg·L−1) | Compound 34 concentration resulting in 50% inhibition of disease progression | 0.0069 (17.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świerczek, A.; Pociecha, K.; Plutecka, H.; Ślusarczyk, M.; Chłoń-Rzepa, G.; Wyska, E. Pharmacokinetic/Pharmacodynamic Evaluation of a New Purine-2,6-Dione Derivative in Rodents with Experimental Autoimmune Diseases. Pharmaceutics 2022, 14, 1090. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051090
Świerczek A, Pociecha K, Plutecka H, Ślusarczyk M, Chłoń-Rzepa G, Wyska E. Pharmacokinetic/Pharmacodynamic Evaluation of a New Purine-2,6-Dione Derivative in Rodents with Experimental Autoimmune Diseases. Pharmaceutics. 2022; 14(5):1090. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051090
Chicago/Turabian StyleŚwierczek, Artur, Krzysztof Pociecha, Hanna Plutecka, Marietta Ślusarczyk, Grażyna Chłoń-Rzepa, and Elżbieta Wyska. 2022. "Pharmacokinetic/Pharmacodynamic Evaluation of a New Purine-2,6-Dione Derivative in Rodents with Experimental Autoimmune Diseases" Pharmaceutics 14, no. 5: 1090. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14051090