
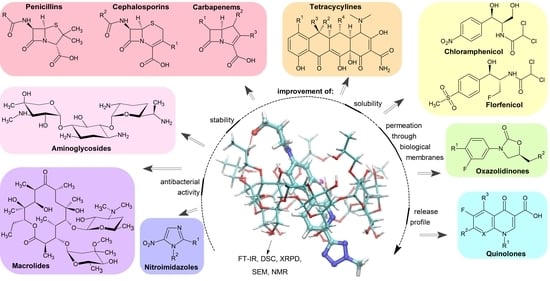
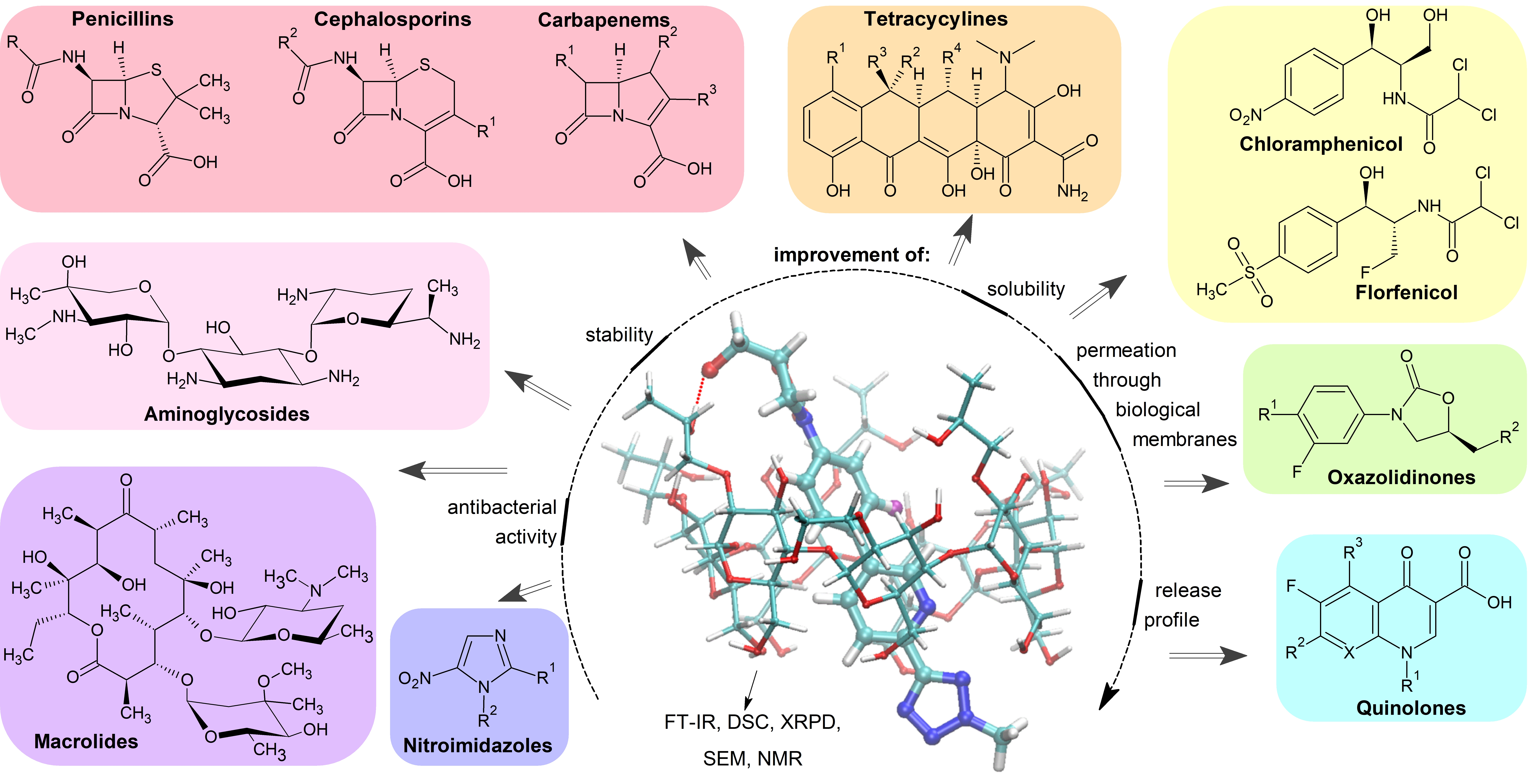
Cyclodextrin Inclusion Complexes with Antibiotics and Antibacterial Agents as Drug-Delivery Systems—A Pharmaceutical Perspective


Abstract
:
1. Introduction
1.1. The Use of CDs and Their Derivatives as Drug Carriers
1.2. New Possibilities of Using CDs
1.3. Expectations for the Drug-Delivery Systems Containing Antibiotics and Antibacterial Agents
2. Beta-Lactam Antibiotics
2.1. Penicillins
2.2. Cephalosporins
2.3. Carbapenems
3. Tetracyclines
4. Chloramphenicol and Florfenicol

{kind=link}
{kind=link}
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| chlorampehnicol | β-CD, HP-β-CD | 1:1, 1:2 | binary | — | spray-drying | PSS, dissolution, HPLC, UV-VIS, DSC, XRPD, NMR, in vivo studies | 120 (HP-β-CD), 170 (β-CD) | PSS 6 | [117] |
| β-CD | 1:1 | binary | — | in solution | NMR | n/a | n/a | [118] | |
| β-CD | 1:1 | binary | — | in silico | quantum mechanical calculations | n/a | n/a | [119] | |
| β-CD | 1:1 | binary | — | in solution | resonance Rayleigh scattering, UV-VIS | dependent on the method and temp. | n/a | [120] | |
| HP-β-CD, M-β-CD | 1:1 | binary | — | kneading | PSS, DSC, UV-VIS, antimicrobial activity | 86.3 (HP-β-CD), 259.5 (M-β-CD) | 4.4 → 30.7 (HP-β-CD), 42.0 g/L (M-β-CD) | [121] | |
| Dimeb | 1:1 | binary | — | co-evaporation | PSS, UV-VIS, circular dichroism spectroscopy, FT-IR, NMR | 493 | 2.24-fold (in 20 mM Dimeb solution) | [122] | |
| β-CD, Trimeb | 1:1 | binary | — | co-dissolution, co-grinding | single-crystal X-ray diffraction, XRPD, TGA, FT-IR, solid-state NMR, SEM, DFT, antibacterial activity | n/a | n/a | [123] | |
| β-CD | 1:1 | ternary | glycine, cysteine | freeze-drying | PSS, HPLC, NMR, FT-IR, DSC, TGA, XRPD, SEM, antimicrobial activity, chemiluminescence | 180 (binary), 107 (with glycine), 101 (with cysteine) | 1.5-fold | [23] | |
| β-CD | 1:1 | ternary | NAC | freeze-drying | PSS, HPLC, NMR, XRPD, SEM, FL, chemiluminescence, microbiological studies | 75 | 1.5-fold | [124] | |
| γ-CD | 1:1 | ternary | NAC | freeze-drying | PSS, ITC, NMR, SEM, DSC, TGA, XRPD, MM, FL, electron probe microanalysis, antibacterial activity | 68 (without NAC), 165 (with NAC) | 2.8 → 7.6 (binary), 14.4 mg/mL (with NAC) | [125] | |
| γ-CD | 1:1 | NPs | silver NPs | freeze-drying | XRPD , FT-IR, DSC, TGA, SEM, EDX, TEM, NMR, UV-VIS, SERS, MM, zeta potential, antibacterial activity | n/a | n/a | [126] | |
| SBE-β-CD | 1:1 | eye drop formulation | PVP, PVA, PVP, HPMC | freeze-drying | PSS, UV-VIS, DSC, XRPD, MM, NMR, SEM, EDS, HPLC-MS/MS, in vitro release, in vitro permeabilization tests, in vivo studies | n/a | PSS 6 | [127] | |
| florfenicol | HP-β-CD | n/a | binary | — | freeze-drying | SEM, DSC, XRPD, FT-IR, NMR, muscle irritation test, in vivo pharmacokinetic studies | n/a | 2.23 → 78.9 mg/mL | [128] |
| β-, HP-β-, γ-CD, Captisol | n/a | ternary | PEG-300 | freeze-drying | PSS, HPLC | 1430 (β-CD), 612 (γ-CD), 817 (HP-β-CD), 1021 (Captisol) | PSS 6 | [129] | |
| HP-β-CD | 1:1 | microparticles | chitosan | evaporation, freeze-drying, spray-drying | PSS, HPLC, DSC, SEM, dissolution | 181.4 (without addit.) | PSS 6 | [130] | |
| HP-β-CD | n/a | microparticles | PVP, HPMC | as described 4 | SEM, UV-VIS, HPLC, particle size analysis, stability, in vitro release, pharmacokinetic study antibacterial activity | n/a | n/a | [110] | |
| γ-CD | n/a | MOF | KOH, CH3OH | as described 4 | SEM, XRPD, UV-VIS, FL, HPLC, in vitro release, antibacterial activity | n/a | 9.12 → 76.11 mg/L | [131] |
5. Quinolones
5.1. First-Generation Quinolones
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| nalidixic acid | α-, β-CD | 1:1 | binary | — | in solution | PSS, UV-VIS, MM, electrochemistry: differential pulse stripping voltammetry, cyclic voltammetry | 398 (α-CD), 1585 (β-CD) | n/a | [135] |
| β-CD | n/a | binary | — | kneading | SEM, XRPD, dissolution | n/a | n/a | [136] | |
| γ-CD | 1:1 | binary | — | in solution | UV-VIS, NMR | 3480 (in 0.03 M HCl), 3760 (in 0.03 M NaOH) | n/a | [137] | |
| piromidic acid | DM-β-CD | 1:2 | binary | — | neutralisation method | PSS, UV-VIS, DSC, NMR, dissolution | 244 | PSS 6 | [138] |
| β-CD, DM-β-CD | 1:2 | binary | — | neutralisation, co-precipitation | PSS, UV-VIS, DSC, NMR, XRPD, dissolution | 77.5 (β-CD), 244 (DM-β-CD) | PSS 6 | [139] | |
| pipemidic acid | β-CD | 1:1 | binary | — | kneading | PSS, UV-VIS, FT-IR, NMR, MM, bioactivity evaluation | 250.8 (pH 4.6), 88.5 (pH 6.8), 86.7 (pH 8.6) | PSS 6 | [140] |
| oxolinic acid | HP-β-CD | 1:1 | binary | — | in solution | UV-VIS, HPLC (photodegradation), NMR | 2.65 × 103 | n/a | [141] |
| γ-CD | 1:1 | binary | — | in solution | UV-VIS, NMR | 1616 (in 0.03 M HCl), 1765 (in 0.03 M NaOH) | n/a | [137] | |
| ofloxacin | β-CD | n/a | binary | — | in solution | solubility studies, DSC, NMR, HPLC (photostability), MM | 152 (pH 8.3) | n/a | [142] |
| β-CD, HP-β-CD | 1:1 | binary | — | in solution | UV-VIS, FL, NMR | dependent on the type of CD and pH | n/a | [143] | |
| β-CD, HP-β-CD | 1:1 | binary | — | freeze-drying | FT-IR, TGA, DTA, NMR, ITC, DLS, antitumour and antibacterial activity | 880 (β-CD), 65.2 (HP-β-CD) | n/a | [144] | |
| HP-β-CD | 1:1 | binary | — | kneading | PSS, UV-VIS, FT-IR, MM | 1.28 × 103 | 0.3 → 1.1 mg/mL | [145] | |
| α-, β-CD | 1:1 | binary | — | as described 4 | UV-VIS, FT-IR, NMR, SEM, MM, time-resolved fluorescence | as described 4 | n/a | [146] | |
| β-CD | 1:1 | binary | — | in solution | fluorescence quenching method | 1.02 × 106 (neutral pH), 0.99 × 106 (acidic pH) | n/a | [147] | |
| M-β-CD | 1:1 | binary | — | co-precipitation | UV-VIS, FL, FT-IR | 7.8 × 103 | n/a | [148] | |
| M-β-CD | 1:1 | binary | — | solid-state | solubility studies, FL, FT-IR, SEM, NMR, MM | 167 (pH 3.05), 1000 (pH 7.53), 200 (pH 10.53) | 1.28 → 4.26 mg/mL | [149] | |
| α-CD | 1:1 | NFs | PEG, PVA | as described 4 | FT-IR, NMR, SEM, EDX, UV-VIS, antibacterial activity | n/a | n/a | [150] | |
| ciprofloxacin | β-CD | 1:1 | binary | — | co-precipitation | NMR, FL, IR, DSC, SEM | 278 | n/a | [151] |
| HP-β-CD | 1:1 | binary | — | co-precipitation | NMR, FL, IR, DSC, SEM | 343 | n/a | [152] | |
| α-, β-CD | 1:1 | binary | — | precipitation | UV-VIS, FT-IR, NMR, SEM, MM, time-resolved fluorescence | as described 4 | n/a | [146] | |
| HP-β-CD, M-β-CD | n/a | hydrogel | agar, ethyleneglycol diglycidylether | as described 4 | FT-IR, UV-VIS, drug release, microbiological tests | n/a | n/a | [153] | |
| β-CD | n/a | hydrogel | sterculia gum, carbopol 940, N,N′-methylenebisacrylamide ammoniumpersulphate | as described 4 | cryo-SEM, FT-IR, solid-state NMR, drug release, UV-VIS, biomedical properties of hydrogels | n/a | n/a | [154] | |
| poly(cyclodextrin citrate) | n/a | hydrogel | chitosan | as described 4 | SEM, rheological parameters, degradation studies, drug release, HPLC, antibacterial activity, cytotoxicity | n/a | n/a | [26] | |
| α-CD | n/a | hydrogel | poloxamer 407 | as described 4 | rheological parameters, in vitro release, HPLC, permeation, antibacterial studies | n/a | n/a | [155] | |
| HP-β-CD | 1:1 | eye drop formulation | HPMC, PVP | as described 4 | PSS, UV-VIS, stability, drug release | 175 (pH 5.5), 83 (pH 7.4) | 3-fold (pH 5.5), 2-fold (pH 7.4) | [156] | |
| HP-β-CD | 1:1 | eye drop formulation | Carbopol 934 and 940, Poloxamer 407 and 188, HPMC | as described 4 | FT-IR, DSC, NMR | n/a | n/a | [157] | |
| β-CD | n/a | polyurethane composite | poly(butyleneadipate), 4,4′-diphenylmethane-diisocyanate | as described 4 | FT-IR, NMR, wide-angle XRD, TGA, DSC, SEM, EDX, antimicrobial activity | n/a | n/a | [158] | |
| β-CD | n/a | polyurethane composite | HDI, calcium β-glycerophosphate | kneading | FT-IR, XRPD, TGA, UV-VIS, solid-state NMR | n/a | n/a | [159] | |
| α-, β-CD | 1:1 | NFs | PCL | solvent evaporation, ultrasonic | solubility study, solid-state NMR, FT-IR, XRPD, SEM, UV-VIS | n/a | PSS 6 | [160] | |
| β-CD | 1:1 | NFs | gelatine | freeze-drying | PSS, UV-VIS, NMR, FT-IR, XRPD, TGA, SEM, MM, dissolution | n/a | PSS 6 | [161] | |
| β-CD | n/a | NFs | polylactic acid | co-precipitation | solid-state NMR, SEM, TGA, Raman spectroscopy, UV-VIS | n/a | n/a | [162] | |
| mono-6-prop- argylamino- 6-deoxy-β-CD | n/a | NFs | azidated cellulose fibres | as described 4 | FT-IR, SEM, XPS, XRPD, drug-release study, antibacterial assay | n/a | n/a | [163] | |
| ciprofloxacin | β-, γ-CD | n/a | macromolecule | chloroacetyl chloride | as described 4 | NMR, FT-IR, MS, UV-VIS, drug release, antibacterial activity (MTT assay) | n/a | n/a | [164] |
| O-p-toluene- sulfonyl-β-CD | n/a | NPs | chitosan, 3-chloro-2-hydroxypropyl trimethyl ammonium chloride | as described 4 | solubility studies, UV-VIS, DLS, FT-IR, XRPD, SEM, EDX, NMR, antibacterial and anti-fungal activity, in vitro release | n/a | PSS 6 | [165] | |
| M-β-CD, poly-M-β-CD | 1:1 | vascular grafts | PET, citric acid | as described 4 | solubility study, UV-VIS, NMR, in vitro release | 55.9 (M-β-CD), 793.8 (poly-M-β-CD) | PSS 6 | [166] | |
| HP-γ-CD | n/a | vascular prosthesis | PET, citric acid | as described 4 | UV-VIS, SEM, microbiological tests, in vitro cell proliferation | 38 | n/a | [167] | |
| pefloxacin | α-, β-, HP-β-CD | 1:1 | binary | — | co-precipitation | NMR, FL | 730 (α-CD), 140 (β-CD), 760 (HP-β-CD) | n/a | [168] |
| lomefloxacin | HP-β-CD | 1:1 | binary | — | solvent evaporation | FT-IR, XRPD, UV-VIS, in vitro dissolution, in vivo absorption studies, stability | n/a | n/a | [169] |
| rufloxacin | β-CD, HP-β-CD, γ-CD | 1:1 | ternary | HPMC | in solution | PSS, UV-VIS, HPLC, bioavailability studies | 139 (β-CD), 95 (HP-β-CD), 48 (γ-CD), 111 (HP-β-CD, HPMC, pH 7.4) | PSS 6 | [170] |
| norfloxacin BI and C | β-CD | 1:1 | binary | — | kneading | solid-state NMR, XRPD, FT-IR | n/a | n/a | [171] |
| norfloxacin A | β-CD | 1:1 | binary | — | kneading, freeze-drying | PSS, DSC, TGA, FT-IR, XRPD, solid-state NMR, in vitro dissolution, microbiological studies | 14 (in water) 58 (pH 6.0) 72 (pH 8.0) | 1.2-fold (in water) 1.9-fold (pH 6.0) 2.4-fold (pH 8.0) | [172] |
| norfloxacin BI | β-CD | 1:1 | binary | — | kneading, freeze-drying | PSS, HPLC (stability), dissolution, microbiological studies | n/a (in water) 40 (pH 6.0) 33 (pH 8.0) | 1.2-fold (in water) 1.5-fold (pH 6.0) 1.3-fold (pH 8.0) | [173] |
| norfloxacin C | β-CD | 1:1 | binary | — | kneading | PSS, HPLC (stability), dissolution, solid-state NMR, FT-IR, XRPD, SEM | n/a | 0.29 → 0.34 mg/mL (in water), decrease at pH 6.0 and 8.0 | [174] |
| norfloxacin | β-CD, HP-β-CD | n/a | binary | — | freeze-drying | PSS, UV-VIS, XRPD, DSC, dissolution | n/a | PSS 6 | [175] |
| β-CD, HP-β-CD | n/a | binary | — | physical trituration, kneading, solvent evaporation | PSS, UV-VIS, in vitro dissolution, SEM, FT-IR, DSC, XRPD | 103.5 (β-CD) 642.7 (HP-β-CD) | 0.39 → 10.90 mg/mL | [176] | |
| β-CD | 1:1 | binary | — | in silico | DFT | n/a | n/a | [177] | |
| 2-methyl-β-CD | 1:1 | binary | — | in solution | UV-VIS, FL, NMR | 2.075 × 104 (pH 3.05) 1.315 × 104 (pH 6.53) 1.425 × 103 (pH 10.53) | n/a | [178] | |
| β-CD, HP-β-CD, γ-CD | 1:1 | binary | — | solvent evaporation, co-evaporation, kneading, freeze-drying, spray-drying | PSS, potentiometric titration, DSC, FT-IR, XRPD, SEM, hot-stage microscopy, in vitro dissolution, antimicrobial assay | 121.1 (β-CD) 65.9 (HP-β-CD) 84.6 (γ-CD) | up to 2.4-fold | [179] | |
| β-CD | 1:1 | binary | — | co-evaporation, kneading followed by freeze-drying | potentiometric titrations, NMR, HPLC (stability), DSC, TGA, FT-IR, XRPD, antimicrobial assay | n/a | up to 2.4-fold | [180] | |
| β-CD | n/a | ternary | HPMC | solvent evaporation | PSS, UV-VIS, SEM, DSC, FT-IR, XRPD, in vitro dissolution | 103.5 (binary system) 253.3 (2.5% w/v HPMC) 307.5 (5% w/v HPMC) | 0.39 → 4.23 (1:1), up to 6.92 mg/mL (1:1, 5% w/w HPMC) | [181] | |
| β-CD | n/a | ternary | citric acid, ascorbic acid | kneading, solvent evaporation | PSS, UV-VIS, IR, DSC, particle size analysis, in vitro dissolution, microbiological studies | 22.4 | PSS 6 | [182] | |
| HP-β-CD | 1:1 | ternary | glutamic acid, proline, lysine | kneading, freeze-drying | PSS, UV-VIS, NMR, DSC, TGA, in vitro release | n/a | as described 4 | [183] | |
| β-CD | 1:1 | liposome | soybean phospholipids and cholesterol | freeze-drying | FT-IR, XRPD, NMR, MM, TEM | 11 | n/a | [184] | |
| β-CD | n/a | nanosponges | diphenyl carbonate | as described 4 | HPLC, DLS, DSC, FT-IR, TEM, zeta potential, permeability (Ussing chamber experiments), in vitro release, antimicrobial in vivo experiments | n/a | n/a | [185] | |
| β-CD | n/a | tablet formulation | PVP, HPMC | kneading | PSS, UV-VIS, in vitro dissolution | 333 | PSS 6 | [186] | |
| enrofloxacin | α-, β-, γ-, HP-β-CD | 1:1 | ternary | citric acid | kneading | PSS, UV-VIS, DSC, TGA | 20.5 (α-CD), 35.6 (β-CD), 14.0 (γ-CD), 29.5 (HP-β-CD) | 255% (α-CD), 38% (β-CD), 232% (γ-CD), 1258% (HP-β-CD) | [187] |
| HP-β-CD | 1:1 | binary | — | as described 4 | UV-VIS, NMR, FT-IR, HPLC, SEM, dissolution, pharmacokinetic studies | n/a | 916-fold | [188] | |
| γ-CD | n/a | MOF | KOH, CH3OH | as described 4 | SEM, XRPD, UV-VIS, FL, HPLC, in vitro release, antibacterial activity | n/a | 158.45 → 372.14 mg/L | [131] | |
| β-CD | n/a | covalent organic framework | tetraphtaladehyde | as described 4 | SEM, TEM, FT-IR, HPLC, drug release, cytotoxicity test, antibacterial ability | n/a | n/a | [189] | |
| sparfloxacin | HP-β-CD | 1:1 | binary | — | as described 4 | PSS, UV-VIS, FL, FT-IR, potentiometric titration, dissolution | 248.8 | PSS 6 | [190] |
| α-, β-CD | 1:1 | binary | — | as described 4 | UV-VIS, FT-IR, NMR, SEM, MM, time-resolved fluorescence | as described 4 | n/a | [146] | |
| β-CD | 1:1 | binary | — | co-precipitation | FL, NMR, FT-IR, DSC, SEM | 0.5 × 102 | n/a | [191] | |
| levofloxacin | HP-β-CD | n/a | NPs | chitosan, tripolyphosphate | co-precipitation | PSS, in vitro release, UV-VIS, SEM particle size analysis, zeta potential, accelerated stability studies | n/a | n/a | [192] |
| SBE-β-CD | 1:1 | NPs | chitosan | ionotropic gelation method | UV-VIS, NMR, HPLC (stability), in vitro release, antibacterial activity | n/a | n/a | [193] | |
| β-CD | 1:1 | NPs | curdlan, epi- chlorohydrin | freeze-drying | SEM, FT-IR, DLS, in vitro release, antibacterial activity, cell culture studies | n/a | n/a | [194] | |
| β-CD | n/a | dendrimers | polyamidoamine | as described 4 | NMR, FT-IR, FL, MM, dialysis experiments | n/a | n/a | [195] | |
| β-CD | n/a | polypropylene mesh devices | HDI | as described 4 | SEM, EDX, FT-IR, antibacterial activity, drug release | n/a | n/a | [196] | |
| tosufloxacin tosylate | HP-β-CD | 1:1 | binary | — | solvent evaporation | PSS, UV-VIS, in vitro dissolution, XRPD, SEM, DSC, FT-IR, NMR | 2461 | 42 times (0.246 → 10.368 mg/mL) | [197] |
| HP-β-CD | 1:1 | binary | — | supercritical antisolvent method | UV-VIS, FT-IR, XRPD, SEM, EDX, dissolution | n/a | n/a | [198] | |
| HP-β-CD | n/a | binary | — | solution-enhanced dispersion with supercritical CO2 | UV-VIS, SEM, particle size analysis, DSC, TGA, XRPD, FT-IR, NMR, MM, solubility, in vitro dissolution, antibacterial activity | n/a | 6.6 times (up to 489.87 μg/mL) | [199] | |
| moxifloxacin | β-CD | 1:1 | binary | — | freeze-drying | FL, UV-VIS, FT-IR, SEM, NMR | 395 | n/a | [200] |
| β-CD | 1:1 | binary | — | freeze-drying | NMR, capillary electrophoresis, MS, FT-IR, DSC, MM, antibacterial activity | 324 | n/a | [201] | |
| M-β-CD | n/a | binary | — | in solution | UV-VIS, FT-IR, MM, drug release | 2.5 × 104 | n/a | [202] | |
| HP-β-CD | 1:1 | binary | — | rapid expansion of supercritical solutions | SEM, IR, XRPD, circular dichroism, equilibrium dialysis | n/a | n/a | [203] | |
| SBE-β-CD and its oligomer | n/a | NPs | HDI | freeze-drying | UV-VIS, FT-IR, NP tracking analysis | 104 (SBE-β-CD) 2 × 105 (SBE-β-CD oligomer) | n/a | [204] | |
| HP-β-CD, M-β-CD, SBE-β-CD oligomers | n/a | NPs | HDI | freeze-drying | UV-VIS, NMR, FT-IR, DLS, equilibrium dialysis, circular dichroism, antibacterial activity, NP tracking analysis | dependent on the type of CD and molar excess of the cross-linking agent | n/a | [205] | |
| SBE-β-CD | 1:1 | NPs | HDI | freeze-drying | UV-VIS, NMR, FT-IR, DLS, TEM, equilibrium dialysis, circular dichroism, antibacterial activity, NP tracking analysis | n/a | n/a | [206] | |
| gemifloxacin | HP-β-CD | 1:1 | binary | — | freeze-drying | FL, UV-VIS, FT-IR, NMR, HPLC/MS, MM | 2.7 × 102 | n/a | [207] |
5.2. Fluoroquinolones
6. Macrolides
7. Other Antibacterial Drugs
7.1. Aminoglycosides
7.2. Glycopeptide and Polypeptide Antibiotics
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| gentamicin sulphate | β-CD | n/a | NFs | polyurethane, nanochitosan | electrospinning | SEM, FT-IR, TGA, tensile strength, contact angle, vapour transmission rate, sorption, in vitro degradation, haemolysis assay, in vitro cytotoxicity, cell proliferation, in vitro release, antibacterial activity | n/a | n/a | [231] |
| amikacin sulphate | β-CD | n/a | microspheres | terephtaloyle chloride, sorbitan 85 trioleate | freeze-drying | laser diffraction, FT-IR, DSC | n/a | n/a | [232] |
| paromomycin | HP-β-CD | n/a | microspheres | glyceryl monostearate, soya lecithin, PVA, PEG 400, Tween 80, trehalose | emulsion/solvent evaporation + freeze-drying | particle size analysis, zeta potential, UV-VIS, FL, SEM, TEM, FT-IR, XRPD, in vitro release, in vitro cytotoxicity, in vitro and in vivo antileishmanial activity, in vivo toxicity study | n/a | n/a | [233] |
| vancomycin | triacetyl-α-CD, triacetyl-β-CD, triacetyl-γ-CD | 1:1 | binary | — | kneading, co-evaporation, spray-drying | DSC, TGA, particle size analysis, in vitro release, HPLC, antimicrobial activity | n/a | n/a | [234] |
| β-CD | 1:1 | binary | — | freeze-drying kneading | XRPD, TGA, SEM, FT-IR, dissolution, HPLC | n/a | n/a | [235] | |
| β-CD | n/a | binary | — | in solution | cytotoxicity | n/a | n/a | [236] | |
| β-CD | n/a | hydrogel | 2-isocyanatoethyl 2,6-diisocyanatohexanoate, 1,6-diisocyanatohexane | ss described 4 | FT-IR, XRPD, SEM, drug release, antibacterial activity | n/a | n/a | [237] | |
| β-CD | n/a | pseudopolyrotaxane | PEG diglycidyl ether | ss described 4 | NMR, DSC, XRPD, SEM, rheological properties, in vitro release, UV-VIS, antibacterial activity, cell adhesion and proliferation | n/a | n/a | [238] | |
| β-CD | 1:1 | supramolecular amphiphile | oleyamine | ss described 4 | FT-IR, NMR, DLS, TEM, DSC, MM, in vitro release, antibacterial activity, in vitro cytotoxicity assay | n/a | n/a | [239] | |
| α-CD | n/a | supramolecular gel | Pluronic F127 | ss described 4 | π-A isotherms, XRPD, FT-IR, rheological properties, release study, antibacterial activity | n/a | n/a | [240] | |
| vancomycin + ceftazidime | HP-β-CD, HP-γ-CD | 1:1 | two drugs + CD | — | in solution | HPLC, NMR, UV-VIS, turbidimetry, solubility, microbiological studies | n/a | PSS 6 | [77] |
| teicoplanin | triacetyl-α-CD, triacetyl-β-CD, triacetyl-γ-CD | n/a | binary | hydroacetonic solution | kneading, evaporative crystallisation under microwave irradiation | DSC, TGA, dissolution | n/a | n/a | [241] |
| polymyxin B | β-CD | 1:1 | binary | — | in solution | surface tension measurement | 3.0 × 103 | n/a | [242] |
| metronidazole benzoate | β-CD | 1:1 | binary | — | as described 4 | PSS, stability, HPLC, UV-VIS, DSC | 1.3 × 103 | PSS 6 | [243] |
| β-CD | 1:1 | binary | — | in solution | PSS, TLC, NMR, UV-VIS | 251 | 0.14 → 1.39 mg/mL | [244] | |
| metronidazole | β-CD, low methylated-β-CD (CRYSMEB), RM-β-CD | 1:1 | binary | — | in solution | PSS, HPLC, antibacterial activity | n/a | 1.02 (β-CD) 2.14 (CRYSMEB) 3.65 (RM-β-CD) | [245] |
| HP-β-CD | n/a | NFs | — | electrospinning | PSS, rheological properties, SEM, NMR, FT-IR, DSC, TGA, XRPD, in vitro dissolution tests | 30 | ~2 times (in 120 mM HP-β-CD) | [246] | |
| β-CD, HP-β-CD | 1:1 | NPs | chitosan | freeze-drying | PSS, UV-VIS, NMR, FT-IR, SEM, DSC, TGA, differential thermogravimetric analysis, in vitro release, antibacterial activity | n/a | PSS 6 | [247] | |
| hexanoyl-β-CD ester | n/a | nanospheres | Pluronic PE/F68 | as described 4 | particle size analysis, drug loading, HPLC, in vitro release | n/a | n/a | [248] | |
| metronidazole, ornidazole, tinidazole, secnidazole | β-CD, HP-β-CD, M-β-CD | 1:1 | ternary | PVP | in solution | solubility studies, calorimetry, NMR | dependent on the drug, type of CD and addition of PVP | [249] | |
| tinidazole | β-CD | 1:0.5 | formulation | PEG-6000, urea, PVP, gum acacia | solvent-free, microwave-assisted | solubility studies, UV-VIS (stability), FT-IR, XRPD, DSC, MM, microscopy, in vitro release | n/a | 3.76 → 36.89 mg/L | [250] |
| ornidazole | β-CD | n/a | microspheres | epichlorohydrin | as described 4 | UV-VIS, SEM, FT-IR, NMR, solid state NMR, XRPD, DSC, TGA, MM, in vitro release, antibacterial test | n/a | n/a | [251] |
| linezolid | β-CD | 1:1 | binary | — | solvent evaporation | UV-VIS, FL, NMR, MM | 351 | n/a | [252] |
| β-CD | 1:1 | ternary | HPMC e5LV | kneading, co-evaporation, microwave method | PSS, UV-VIS, XRPD, DSC, FT-IR, MM, in vitro dissolution | 1024 (binary), 1393 (5% w/v HPMC) | PSS 6 | [253] | |
| tedizolid | β-CD, γ-CD | 1:1, 1:2 | binary | — | in solution | NMR | dependent on CD and technique | n/a | [254] |
| HP-β-CD | 1:1 | binary | — | kneading | DSC, XRPD, FT-IR, dissolution, HPLC, MM, permeability, antibacterial activity | n/a | n/a | [255] | |
| fusidic acid sodium salt | β-CD, γ-CD | 1:2, 1:1 | binary | — | in solution | NMR | 3.5 × 103 (1:1) | n/a | [256] |
| fusidic acid | β-CD | 1:1 | binary | — | kneading co-precipitation freeze-drying | FT-IR, XRPD, SEM, thermal analysis, antimicrobial test | n/a | n/a | [257] |
| furazolidone | β-CD | n/a | binary | — | kneading, freeze-drying | PSS, UV-VIS, SEM, FT-IR, DSC, TGA, NMR, biological in vitro assays | 220 | 1.68-fold in 12 mM β-CD | [258] |
| β-CD, HP-β-CD | n/a | binary | — | kneading, freeze-drying | DSC, TGA, SEM. XRPD, solid-state NMR, MM, Raman chemical imaging, cytotoxic, antibacterial activity | n/a | n/a | [259] | |
| novobiocin | β-CD | n/a | hydrogel | 2-isocyanatoethyl 2,6-diisocyanatohexanoate, 1,6-diisocyanatohexane | as described 4 | FT-IR, XRPD, SEM, drug release, antibacterial activity | n/a | n/a | [237] |
| β-CD | n/a | pseudo- polyrotaxane | PEG diglycidyl ether | as described 4 | NMR, DSC, XRPD, SEM, rheological properties, in vitro release, UV-VIS, antibacterial activity, cell adhesion and proliferation | n/a | n/a | [238] | |
7.3. Nitroimidazole Derivatives
7.4. Oxazolidinones
7.5. Fusidic Acid and Furazolidone
8. Modern Drug-Delivery Systems
8.1. Lipid-Based Nanocarriers
8.2. Polymeric Nanocarriers
8.2.1. Natural Polymer-Based
8.2.2. Synthetic Polymer-Based
8.3. Polymeric Nanosystems Based on CDs
8.4. Graphene Derivatives
8.5. Inorganic NPs
8.6. Nanofibres
8.7. Hydrogels
8.8. Other Nanosystems
8.9. Formulations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Cyclodextrins | |
| α-CD | α-cylodextrin |
| β-CD | β-cyclodextrin |
| γ-CD | γ-cyclodextrin |
| HP-β-CD | hydroxypropyl-β-cyclodextrin |
| SBE-β-CD | sulfobutyl ether-β-cyclodextrin |
| M-β-CD | methyl-β-cyclodextrin |
| DM-β-CD | dimethyl-β-cyclodextrin |
| RM-β-CD | randomly-methylated-β-cyclodextrin |
| Dimeb | heptakis(2,6-di-O-methyl)-β-cyclodextrin |
| Trimeb | heptakis(2,3,6-tri-O-methyl) β-cyclodextrin |
| Trimeg | octakis(2,3,6-tri-O-methyl)-γ-cyclodextrin |
| Additives | |
| CA | citric acid |
| CMC | carboxymethylcellulose |
| HDI | 1,6-hexamethylene diisocyanate |
| HPMC | hydroxypropylmethylcellulose |
| NAC | N-acetylcysteine |
| PCL | polycaprolactone |
| PEG | poly(ethylene glycol) |
| PET | poly(ethylene terephthalate) |
| PLA | polylactic acid |
| PLGA | poly(lactic-co-glycolic acid) |
| PVA | poly(vinyl alcohol) |
| PVP | polyvinylpyrrolidone |
| SSG | sodium starch glycolate |
| Characterisation techniques | |
| AFM | Atomic force microscopy |
| ATR | Attenuated total reflectance (in conjunction with infrared spectroscopy) |
| COSY | COrrelation SpectroscopY (NMR technique) |
| DFT | Density-functional theory |
| DLS | Dynamic light scattering |
| DSC | Differential scanning calorimetry |
| DTA | Differential thermal analysis |
| EDX | Energy-dispersive X-ray spectroscopy |
| FL | Fluorescence spectroscopy (spectrofluorimetry) |
| FT-IR | Fourier-transform infrared spectroscopy |
| IMC | Isothermal microcalorimetry |
| IR | Infrared spectroscopy |
| ITC | Isothermal titration calorimetry |
| HPLC | High-performance liquid chromatography |
| MM | Molecular modelling |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| PAMPA | Parallel artificial membrane permeability assay |
| PSS | Phase solubility study |
| ROESY | Rotation Frame Nuclear Overhauser Effect SpectroscopY |
| SEM | Scanning electron microscopy |
| SERS | Surface-enhanced Raman spectroscopy |
| TEM | Transmission electron microscopy |
| TGA | Thermogravimetric analysis |
| UV-VIS | Ultraviolet-visible spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRPD | X-ray powder diffractometry |
| Other | |
| API | Active pharmaceutical ingredient |
| BCS | Biopharmaceuticals Classification System |
| BP | Benzylpenicillin |
| CPP | Cefpodoxime proxetil |
| FF | Florfenicol |
| IC | Inclusion complex |
| MIC | Minimum inhibitory concentration |
| MOF | Metal-organic framework |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| n/a | Not available |
| NF | Nanofiber |
| NP | Nanoparticle |
| NS | Nanosponge |
| PMP | Phenoxymethylpenicillin |
| XTT | 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide |
References
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, P.; Bhatia, M. Pharmaceutical applications of cyclodextrins and their derivatives. J. Incl. Phenom. Macrocycl. Chem. 2020, 98, 171–186. [Google Scholar] [CrossRef]
- Morin-Crini, N.; Fourmentin, S.; Fenyvesi, É.; Lichtfouse, E.; Torri, G.; Fourmentin, M.; Crini, G. 130 years of cyclodextrin discovery for health, food, agriculture, and the industry: A review. Environ. Chem. Lett. 2021, 19, 2581–2617. [Google Scholar] [CrossRef]
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1–31. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef]
- Rincón-López, J.; Almanza-Arjona, Y.C.; Riascos, A.P.; Rojas-Aguirre, Y. Technological evolution of cyclodextrins in the pharmaceutical field. J. Drug Deliv. Sci. Technol. 2021, 61, 102156. [Google Scholar] [CrossRef]
- Rincón-López, J.; Almanza-Arjona, Y.C.; Riascos, A.P.; Rojas-Aguirre, Y. When Cyclodextrins Met Data Science: Unveiling Their Pharmaceutical Applications through Network Science and Text-Mining. Pharmaceutics 2021, 13, 1297. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Szejtli, J. Past, Present, and Future of Cyclodextrin Research. ChemInform 2005, 36. [Google Scholar] [CrossRef]
- European Medicines Agency. Cyclodextrins Used as Excipients. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-cyclodextrins-used-excipients-medicinal-products-human-use_en.pdf (accessed on 13 April 2022).
- Imperiale, J.C.; Sosnik, A.D. Cyclodextrin complexes for treatment improvement in infectious diseases. Nanomedicine 2015, 10, 1621–1641. [Google Scholar] [CrossRef]
- Wong, C.E.; Dolzhenko, A.V.; Lee, S.M.; Young, D.J. Cyclodextrins: A Weapon in the Fight against Antimicrobial Resistance. J. Mol. Eng. Mater. 2017, 5, 1740006. [Google Scholar] [CrossRef]
- Loftsson, T. Cyclodextrins and the Biopharmaceutics Classification System of Drugs. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 63–67. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef]
- Haimhoffer, Á.; Rusznyák, Á.; Réti-Nagy, K.; Vasvári, G.; Váradi, J.; Vecsernyés, M.; Bácskay, I.; Fehér, P.; Ujhelyi, Z.; Fenyvesi, F. Cyclodextrins in Drug Delivery Systems and Their Effects on Biological Barriers. Sci. Pharm. 2019, 87, 33. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Gao, H.; Cao, Y.; Jia, L. In Silico understanding of the cyclodextrin–phenanthrene hybrid assemblies in both aqueous medium and bacterial membranes. J. Hazard. Mater. 2015, 285, 148–156. [Google Scholar] [CrossRef]
- Erdoğar, N.; Bilensoy, E. Cyclodextrins in Drug Delivery. In Nanotechnology and Drug Delivery, Volume One: Nanoplatforms in Drug Delivery; CRC Press: Boca Raton, FL, USA, 2014; pp. 178–209. [Google Scholar]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of Cyclodextrins on Drug Delivery through Biological Membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef]
- Real, D.A.; Bolaños, K.; Priotti, J.; Yutronic, N.; Kogan, M.J.; Sierpe, R.; Donoso-González, O. Cyclodextrin-Modified Nanomaterials for Drug Delivery: Classification and Advances in Controlled Release and Bioavailability. Pharmaceutics 2021, 13, 2131. [Google Scholar] [CrossRef]
- Bruschi, M.L. 5—Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Bruschi, M.L., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Aiassa, V.; Garnero, C.; Longhi, M.R.; Zoppi, A. Cyclodextrin Multicomponent Complexes: Pharmaceutical Applications. Pharmaceutics 2021, 13, 1099. [Google Scholar] [CrossRef]
- Aiassa, V.; Zoppi, A.; Albesa, I.; Longhi, M.R. Inclusion complexes of chloramphenicol with β-cyclodextrin and aminoacids as a way to increase drug solubility and modulate ROS production. Carbohydr. Polym. 2015, 121, 320–327. [Google Scholar] [CrossRef]
- Ndayishimiye, J.; Kumeria, T.; Popat, A.; Blaskovich, M.A.T.; Falconer, J.R. Understanding the relationship between solubility and permeability of γ-cyclodextrin-based systems embedded with poorly aqueous soluble benznidazole. Int. J. Pharm. 2022, 616, 121487. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef] [PubMed]
- Amiel, A.G.; Palomino-Durand, C.; Maton, M.; Lopez, M.; Cazaux, F.; Chai, F.; Neut, C.; Foligné, B.; Martel, B.; Blanchemain, N. Designed sponges based on chitosan and cyclodextrin polymer for a local release of ciprofloxacin in diabetic foot infections. Int. J. Pharm. 2020, 587, 119677. [Google Scholar] [CrossRef]
- Popielec, A.; Loftsson, T. Effects of cyclodextrins on the chemical stability of drugs. Int. J. Pharm. 2017, 531, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Alsarra, I.A.; Ahmed, M.O.; El-Badry, M.; Alanazi, F.K.; Al-Mohizea, A.M.; Ahmed, S.M. Effect of β-cyclodextrin derivatives on the kinetics of degradation of cefotaxime sodium in solution state. J. Drug Deliv. Sci. Technol. 2007, 17, 353–357. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshioka, S.; Takeda, Y. Epimerization and Racemization of Some Chiral Drugs in the Presence of Cyclodextrin and Liposomes. Chem. Pharm. Bull. 1989, 37, 2786–2789. [Google Scholar] [CrossRef] [Green Version]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Chen, S.; Li, Z.; Gu, Z.; Xu, S.; Ban, X.; Hong, Y.; Cheng, L.; Li, C. A review of controlled release from cyclodextrins: Release methods, release systems and application. Crit. Rev. Food Sci. Nutr. 2021, 1–13. [Google Scholar] [CrossRef]
- Suárez, D.F.; Consuegra, J.; Trajano, V.C.; Gontijo, S.M.L.; Guimarães, P.P.G.; Cortés, M.E.; Denadai, Â.L.; Sinisterra, R.D. Structural and thermodynamic characterization of doxycycline/β-cyclodextrin supramolecular complex and its bacterial membrane interactions. Colloids Surf. B Biointerfaces 2014, 118, 194–201. [Google Scholar] [CrossRef]
- Athanassiou, G.; Michaleas, S.; Lada-Chitiroglou, E.; Tsitsa, T.; Antoniadou-Vyza, E. Antimicrobial activity of β-lactam antibiotics against clinical pathogens after molecular inclusion in several cyclodextrins. A novel approach to bacterial resistance. J. Pharm. Pharmacol. 2003, 55, 291–300. [Google Scholar] [CrossRef]
- Maffeo, D.; Leondiadis, L.; Mavridis, I.M.; Yannakopoulou, K. Positive effect of natural and negatively charged cyclodextrins on the stabilization of penicillins towards β-lactamase degradation due to inclusion and external guest–host association. An NMR and MS study. Org. Biomol. Chem. 2006, 4, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, H.; Miyagawa, A.; Sugiyama, H.; Murata, K.; Mabuti, T.; Mitsuhashi, R.; Hagiwara, T.; Nonaka, M.; Tanimoto, K.; Tomita, H. Rule of Hydrophobicity/Hydrophilicity Balance in Membrane-Disrupting Antimicrobial Activity of Polyalkylamino Cyclodextrins Synthesized via Click Chemistry. ChemistrySelect 2016, 1, 469–472. [Google Scholar] [CrossRef]
- Page, M.G.P. Beta-Lactam Antibiotics. In Antibiotic Discovery and Development; Dougherty, T.J., Pucci, M.J., Eds.; Springer: Boston, MA, USA, 2012; pp. 79–117. [Google Scholar]
- Macielag, M.J. Chemical Properties of Antimicrobials and Their Uniqueness. In Antibiotic Discovery and Development; Dougherty, T.J., Pucci, M.J., Eds.; Springer: Boston, MA, USA, 2012; pp. 793–820. [Google Scholar]
- Pop, E.; Loftsson, T.; Bodor, N. Solubilization and Stabilization of a Benzylpenicillin Chemical Delivery System by 2-Hydroxypropyl-β-cyclodextrin. Pharm. Res. 1991, 8, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.K.; Sunderland, V.B.; McDonald, C. Influence of Hydroxypropyl β-Cyclodextrin on the Stability of Benzylpenicillin in Chloroacetate Buffer. J. Pharm. Pharmacol. 1997, 49, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Hada, S.; Neya, S.; Funasaki, N. Acceleration and Inhibition of the Hydrolysis of Penicillin G by Dimerization and Cyclodextrin Inclusion. Chem. Pharm. Bull. 1997, 45, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Aki, H.; Ikeda, H.; Yukawa, M.; Iwase, Y.; Mibu, N. Effect of pH on the formation of inclusion complexes between β-lactam antibiotics and 2-hydroxypropyl-β-cyclodextrin in aqueous solution. J. Therm. Anal. Calorim. 2009, 95, 421–426. [Google Scholar] [CrossRef]
- Popielec, A.; Fenyvesi, É.; Yannakopoulou, K.; Loftsson, T. Effect of cyclodextrins on the degradation rate of benzylpenicillin. Die Pharm. Int. J. Pharm. Sci. 2016, 71, 68–75. [Google Scholar] [CrossRef]
- Popielec, A.; Agnes, M.; Yannakopoulou, K.; Fenyvesi, É.; Loftsson, T. Effect of β- and γ-cyclodextrins and their methylated derivatives on the degradation rate of benzylpenicillin. J. Incl. Phenom. Macrocycl. Chem. 2018, 91, 199–209. [Google Scholar] [CrossRef]
- Szaruga, K.; Fuz, M.; Wszelaka-Rylik, M.; Gierycz, P. Thermodynamics of antibiotics: Natural cyclodextrin inclusion complex formation and covering of nano-metric calcite with these substances. J. Therm. Anal. Calorim. 2021, 146, 1283–1296. [Google Scholar] [CrossRef]
- Wu, X.; Yang, S.; Yang, S.; Yang, X.; Wang, Y.; Liu, Y.; Liu, M.; Zhang, Q.; Peng, L.; Chen, L.; et al. Preparation method of penicillin V potassium β-cyclodextrin inclusion. Pak. J. Pharm. Sci. 2018, 31, 1633–1638. [Google Scholar]
- Aki, H.; Yamamoto, K.; Sawai, N.; Yamamoto, M. Inhibitory effect of beta-cyclodextrin on ampicillin polymerization in aqueous solution. Drug Des. Deliv. 1990, 7, 59–63. [Google Scholar] [PubMed]
- Ammar, H.O.; el-Nahhas, S.A.; Ghorab, M.M. Improvement of some pharmaceutical properties of drugs by cyclodextrin complexation. Part 6: Ampicillin. Pharmazie 1996, 51, 568–570. [Google Scholar] [PubMed]
- Aki, H.; Niiya, T.; Iwase, Y.; Kawasaki, Y.; Kumai, K.; Kimura, T. Multimodal inclusion complexes of ampicillin with β-cyclodextrins in aqueous solution. Thermochim. Acta 2004, 416, 87–92. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Ali, S.M. NMR investigation on the interaction of β-cyclodextrin and ampicillin. J. Struct. Chem. 2010, 51, 972–975. [Google Scholar] [CrossRef]
- Namazi, H.; Kanani, A. Synthesis of New Prodrugs Based on β-CD as the Natural Compounds Containing β-lactam Antibiotics. J. Bioact. Compat. Polym. 2007, 22, 77–88. [Google Scholar] [CrossRef]
- Lampropoulou, M.; Misiakos, K.; Paravatou, M.; Mavridis, I.M.; Yannakopoulou, K. Synthesis of cyclodextrin derivatives with monosaccharides and their binding with ampicillin and selected lectins. ARKIVOC 2015, 2015, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Akbar, N.; Aslam, Z.; Siddiqui, R.; Shah, M.R.; Khan, N.A. Zinc oxide nanoparticles conjugated with clinically-approved medicines as potential antibacterial molecules. AMB Express 2021, 11, 104. [Google Scholar] [CrossRef]
- Mizera, M.; Lewandowska, K.; Miklaszewski, A.; Cielecka-Piontek, J. Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis. Molecules 2019, 24, 743. [Google Scholar] [CrossRef] [Green Version]
- Hidaka, S.; Tokumura, T.; Tomono, K.; Suzuki, T.; Ueda, H.; Nagai, T.; Nagaoka, M.; Nakane, R.; Machida, Y. Effect of β-Cyclodextrin on the Degradation Rate of Amoxicillin in Acidic Solution. Yakugaku Zasshi-J. Pharm. Soc. Jpn. Yakugaku Zasshi-J. Pharm. Soc. J. 2010, 130, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Bisson-Boutelliez, C.; Fontanay, S.; Finance, C.; Kedzierewicz, F. Preparation and Physicochemical Characterization of Amoxicillin β-cyclodextrin Complexes. AAPS PharmSciTech 2010, 11, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Aki, H.; Niiya, T.; Iwase, Y.; Goto, M.; Kimura, T. Mechanism for the inhibition of the acid degradation of ampicillin by 2-hydroxypropyl-β-cyclodextrin. J. Therm. Anal. Calorim. 2004, 77, 423–435. [Google Scholar] [CrossRef]
- Aki, H.; Nakashima, Y.; Kawasaki, Y.; Niiya, T. Thermodynamic evaluation of antibacterial activity for inclusion complexes of amoxicillin with cyclodextrins. J. Therm. Anal. Calorim. 2006, 85, 685–688. [Google Scholar] [CrossRef]
- Khushbu; Jindal, R. Comparative Evaluation for Controlled Release of Amoxicillin from RSM-CCD-Optimized Nanocomposites Based on Sodium Alginate and Chitosan-Containing Inclusion Complexes. Mol. Pharm. 2021, 18, 3795–3810. [Google Scholar] [CrossRef]
- Echezarreta Lopez, M.M.; Otero-Mazoy, I.; Ramirez, H.; Villalonga, R.; Torres-Labandeira, J. Solubilization and Stabilization of Sodium Dicloxacillin by Cyclodextrin Inclusion. Curr. Drug Discov. Technol. 2008, 5, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.-B.; Zhang, B.-T. Preparation and study on the solid inclusion complex of cloxacillin sodium with β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 68, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.-Z. Methicillin/per-6-(4-methoxylbenzyl)-amino-6-deoxy-β-cyclodextrin 1:1 complex and its potentiation in vitro against methicillin-resistant Staphylococcus aureus. J. Antibiot. 2013, 66, 517–521. [Google Scholar] [CrossRef]
- Agnes, M.; Thanassoulas, A.; Stavropoulos, P.; Nounesis, G.; Miliotis, G.; Miriagou, V.; Athanasiou, E.; Benkovics, G.; Malanga, M.; Yannakopoulou, K. Designed positively charged cyclodextrin hosts with enhanced binding of penicillins as carriers for the delivery of antibiotics: The case of oxacillin. Int. J. Pharm. 2017, 531, 480–491. [Google Scholar] [CrossRef]
- Kfoury, M.; Landy, D.; Fourmentin, S. Characterization of Cyclodextrin/Volatile Inclusion Complexes: A Review. Molecules 2018, 23, 1204. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Saraswat, H.; Banoo, S.; Islam, M. Structural Determination of Cephalexin/β-Cyclodextrin Inclusion Complex and its Validation using Molecular Simulation Methods. Asian J. Chem. 2020, 32, 930–934. [Google Scholar] [CrossRef]
- Dubey, P.; Sharma, H.; Shah, S.; Tyagi, C.; Chandekar, A.; Jadon, R. Formulations and evaluation of Cyclodextrin complexed Ceadroxil loaded nanosponges. Int. J. Drug Deliv. 2017, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Park, E.; Kim, Y.; Lee, S.; Kwon, J.; Cho, H.; Lee, Y. A medusa-like β-cyclodextrin with 1-methyl-2-(2′-carboxyethyl) maleic anhydrides, a potential carrier for pH-sensitive drug delivery. J. Drug Target. 2014, 22, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, K.V.R.N.S.; Kumar, B.V.S.S.; Kumar, M. Preparation and evaluation of fast dissolving dosage forms of cefuroxime axetil. Int. J. Chem. Sci. 2012, 10, 2151–2164. [Google Scholar]
- Mizera, M.; Szymanowska, D.; Stasiłowicz, A.; Siąkowska, D.; Lewandowska, K.; Miklaszewski, A.; Plech, T.; Tykarska, E.; Cielecka-Piontek, J. Computer-Aided Design of Cefuroxime Axetil/Cyclodextrin System with Enhanced Solubility and Antimicrobial Activity. Biomolecules 2020, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieroba, B.; Kalisz, G.; Sroka-Bartnicka, A.; Płazińska, A.; Płaziński, W.; Starek, M.; Dąbrowska, M. Molecular Structure of Cefuroxime Axetil Complexes with α-, β-, γ-, and 2-Hydroxypropyl-β-Cyclodextrins: Molecular Simulations and Raman Spectroscopic and Imaging Studies. Int. J. Mol. Sci. 2021, 22, 5238. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Pore, Y.; Dhawale, S.; Burade, K.; Kuchekar, B. Physicochemical characterization of spray dried ternary micro-complexes of cefuroxime axetil with hydroxypropyl-β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2013, 76, 391–401. [Google Scholar] [CrossRef]
- Sapte, S.; Pore, Y. Inclusion complexes of cefuroxime axetil with β-cyclodextrin: Physicochemical characterization, molecular modeling and effect of l-arginine on complexation. J. Pharm. Anal. 2016, 6, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Verma, R.; Purohit, D.; Pandey, P.; Kumar, M.; Kumari, B.; Kaushik, D. Development of binary and ternary complex of cefuroxime axetil with cyclodextrin for improving pharmaceutical characteristics. Int. J. Appl. Pharm. 2020, 12, 107–117. [Google Scholar] [CrossRef]
- Dąbrowska, M.; Krzek, J.; Miękina, E. Stability analysis of cefaclor and its inclusion complexes of β-cyclodextrin by thin-layer chromatography and densitometry. JPC J. Planar Chromatogr. Mod. TLC 2012, 25, 127–132. [Google Scholar] [CrossRef]
- Misiuk, W. Investigation of inclusion complex of HP-γ-cyclodextrin with ceftazidime. J. Mol. Liq. 2016, 224, 387–392. [Google Scholar] [CrossRef]
- Kharwade, R.; More, S.; Khan, S.; Yeole, P.G. Stabilization of ceftazidime in liquid state using poloxamer and cyclodextrin. Int. J. Pharm. Pharm. Sci. 2010, 2, 156–161. [Google Scholar]
- Lim, H.; Jin, S.; Jeong, Y.; Kim, S.-B.; Jang, D.-J.; Kim, S.T. Preparation of hydroxypropyl-β-cyclodextrin-incorporated liposomes and evaluation of their rapid release property. J. Ind. Eng. Chem. 2021, 100, 59–62. [Google Scholar] [CrossRef]
- Bouattour, Y.; Neflot-Bissuel, F.; Traïkia, M.; Biesse-Martin, A.-S.; Frederic, R.; Yessaad, M.; Jouannet, M.; Wasiak, M.; Chennell, P.; Sautou, V. Cyclodextrins Allow the Combination of Incompatible Vancomycin and Ceftazidime into an Ophthalmic Formulation for the Treatment of Bacterial Keratitis. Int. J. Mol. Sci. 2021, 22, 10538. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Mondal, A.; Sannigrahi, S. Kinetic measurements of the hydrolytic degradation of cefixime: Effect of Captisol complexation and water-soluble polymers. J. Pharm. Pharmacol. 2008, 60, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Pamudji, J.S.; Mauludin, R.; Nurhabibah. Influence of β-cyclodextrin on Cefixime Stability in Liquid Suspension Dosage Form. Procedia Chem. 2014, 13, 119–127. [Google Scholar] [CrossRef]
- Mogal, P.V.; Derle, D.V. Cefixime, in General, Class 4 Drug but Individually Class 2 Drug. J. Med. Physiol. Ther. 2017, 1, 1000103. [Google Scholar]
- Mogal, P.; Derle, D. Functionality Advancement of Poorly Soluble Drug by Comparative Study of Solubilizing Techniques with Molecular Simulation to in vivo evaluation. Int. J. PharmTech Res. 2017, 10, 139–155. [Google Scholar] [CrossRef]
- Mehta, P.; More, A.S.; Kulkarni, A. Effect of hydrophilic polymers on cefixime complexation with β-cyclodextrin. Int. J. Curr. Pharm. Res. 2013, 5, 66–70. [Google Scholar]
- Jadhav, P.; Petkar, B.; Pore, Y.; Kulkarni, A.; Burade, K. Physicochemical and molecular modeling studies of cefixime–l-arginine–cyclodextrin ternary inclusion compounds. Carbohydr. Polym. 2013, 98, 1317–1325. [Google Scholar] [CrossRef]
- Cirri, M.; Mennini, N.; Nerli, G.; Rubia, J.; Casalone, E.; Melani, F.; Maestrelli, F.; Mura, P. Combined Use of Cyclodextrins and Amino Acids for the Development of Cefixime Oral Solutions for Pediatric Use. Pharmaceutics 2021, 13, 1923. [Google Scholar] [CrossRef]
- Agrawal, P.G.; Bhargava, S. Preparation & Characterization of Solid Inclusion Complex of Cefpodoxime Proxetil with β-Cyclodextrin. Curr. Drug Deliv. 2008, 5, 1–6. [Google Scholar] [CrossRef]
- Yurtdaş-Kırımlıoğlu, G. Spray dried nanospheres for inclusion complexes of cefpodoxime proxetil with β-cyclodextrin, 2-hydroxypopyl-β-cyclodextrin and methyl-β-cyclodextrin: Improved dissolution and enhanced antibacterial activity. Drug Dev. Ind. Pharm. 2021, 47, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Gündoğdu, E.; Başpınar, Y.; Köksal Karayıldırım, Ç.; Karasulu, E. Evaluation of cefpodoxime proxetil complex with hydroxypropyl-β-cyclodextrin in the presence of a water soluble polymer: Characterization and permeability studies. Fabad J. Pharm. Sci. 2011, 36, 137–148. [Google Scholar]
- Gundogdu, E.; Koksal, C.; Karasulu, E. Comparison of cefpodoxime proxetil release and antimicrobial activity from tablet formulations: Complexation with hydroxypropyl-β-cyclodextrin in the presence of water soluble polymer. Drug Dev. Ind. Pharm. 2012, 38, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Aleem, O.; Kuchekar, B.; Pore, Y.; Late, S. Effect of β-cyclodextrin and hydroxypropyl β-cyclodextrin complexation on physicochemical properties and antimicrobial activity of cefdinir. J. Pharm. Biomed. Anal. 2008, 47, 535–540. [Google Scholar] [CrossRef]
- Mohit, V.; Harshal, G.; Neha, D.; Vilasrao, K.; Rajashree, H. Effect of preparation method on complexation of Cefdinir with β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2010, 67, 39–47. [Google Scholar] [CrossRef]
- Mohit, V.; Harshal, G.; Neha, D.; Vilasrao, K.; Rajashree, H. A comparative study of complexation methods for cefdinir-hydroxypropyl-β-cyclodextrin system. J. Incl. Phenom. Macrocycl. Chem. 2011, 71, 57–66. [Google Scholar] [CrossRef]
- Guo, B.; Zhong, S.; Li, N.; Li, X.; Yi, J.; Jin, M. Dissolution enhancement of cefdinir with hydroxypropyl-β-cyclodextrin. Drug Dev. Ind. Pharm. 2013, 39, 1638–1643. [Google Scholar] [CrossRef]
- Morina, D.; Sessevmez, M.; Sinani, G.; Mülazımoğlu, L.; Cevher, E. Oral tablet formulations containing cyclodextrin complexes of poorly water soluble cefdinir to enhance its bioavailability. J. Drug Deliv. Sci. Technol. 2020, 57, 101742. [Google Scholar] [CrossRef]
- Liu, H.; Gao, C.; Qu, Z.; Liu, M.; Zhao, S. Preparation Method of Ceftiofur Acid Long-Acting Injection. Patent CN103230364A, 11 June 2014. [Google Scholar]
- Ruiz-Carretero, P.; Nacher, A.; Merino-Sanjuan, M.; Casabo, V.G. Pharmacokinetics and absolute bioavailability of oral cefuroxime axetil in the rat. Int. J. Pharm. 2000, 202, 89–96. [Google Scholar] [CrossRef]
- Paczkowska, M.; Mizera, M.; Szymanowska-Powałowska, D.; Lewandowska, K.; Błaszczak, W.; Gościańska, J.; Pietrzak, R.; Cielecka-Piontek, J. β-Cyclodextrin complexation as an effective drug delivery system for meropenem. Eur. J. Pharm. Biopharm. 2016, 99, 24–34. [Google Scholar] [CrossRef]
- Popielec, A.; Agnes, M.; Yannakopoulou, K.; Fenyvesi, É.; Loftsson, T. Self-assembled cyclodextrin-based nanoparticles for meropenem stabilization. J. Drug Deliv. Sci. Technol. 2018, 45, 20–27. [Google Scholar] [CrossRef]
- Raza, A.; Miles, J.A.; Sime, F.B.; Ross, B.P.; Roberts, J.A.; Popat, A.; Kumeria, T.; Falconer, J.R. PLGA encapsulated γ-cyclodextrin-meropenem inclusion complex formulation for oral delivery. Int. J. Pharm. 2021, 597, 120280. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-Y.; Yoon, H.-K.; Kim, W.-Y.; Shin, J.-T. Process for Preparing Ertapenem-Containing Lyophilized Formulation. Patent WO 2016/028002 A1, 25 January 2016. [Google Scholar]
- Paczkowska, M.; Szymanowska-Powałowska, D.; Mizera, M.; Siąkowska, D.; Błaszczak, W.; Piotrowska-Kempisty, H.; Cielecka-Piontek, J. Cyclodextrins as multifunctional excipients: Influence of inclusion into β-cyclodextrin on physicochemical and biological properties of tebipenem pivoxil. PLoS ONE 2019, 14, e0210694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentín, S.; Morales, A.; Sánchez, J.L.; Rivera, A. Safety and efficacy of doxycycline in the treatment of rosacea. Clin. Cosmet. Investig. Dermatol. 2009, 2, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Guimarães, P.P.G.; de Menezes, A.C.; Teixeira, K.I.R.; Denadai, Â.M.L.; Fills, R.A.; Cortés, M.E.; Sinisterra, R.D. Enhanced efficacy against bacterial biofilms via host:guest cyclodextrin-doxycycline inclusion complexes. J. Incl. Phenom. Macrocycl. Chem. 2021, 99, 197–207. [Google Scholar] [CrossRef]
- Kogawa, A.C.; Zoppi, A.; Quevedo, M.A.; Nunes Salgado, H.R.; Longhi, M.R. Increasing Doxycycline Hyclate Photostability by Complexation with β-Cyclodextrin. AAPS PharmSciTech 2014, 15, 1209–1217. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.; Rani, P.; Adhikari, L.; Gupta, M.; Semalty, A.; Semalty, M. Preparation and characterization of cyclodextrin complexes of doxycycline hyclate for improved photostability in aqueous solution. J. Incl. Phenom. Macrocycl. Chem. 2022, 102, 271–278. [Google Scholar] [CrossRef]
- Peraro, C.R.; Anconi, A.C.S.A.; Anconi, C.P.A. Formation of β-Cyclodextrin inclusion compound with doxycycline: A theoretical approach. Chem. Phys. Lett. 2018, 692, 140–145. [Google Scholar] [CrossRef]
- He, Z.-X.; Wang, Z.-H.; Zhang, H.-H.; Pan, X.; Su, W.-R.; Liang, D.; Wu, C.-B. Doxycycline and hydroxypropyl-β-cyclodextrin complex in poloxamer thermal sensitive hydrogel for ophthalmic delivery. Acta Pharm. Sin. B 2011, 1, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Chen, M.; He, Z.; Wang, Z.; Zhang, M.; He, Z.; Wan, Q.; Liang, D.; Repka, M.A.; Wu, C. Molecular Modeling-Based Inclusion Mechanism and Stability Studies of Doxycycline and Hydroxypropyl-β-Cyclodextrin Complex for Ophthalmic Delivery. AAPS PharmSciTech 2013, 14, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; He, Z.; Zhang, L.; Zhang, H.; Zhang, M.; Wen, X.; Quan, G.; Huang, X.; Pan, X.; Wu, C. Optimization of a doxycycline hydroxypropyl-β-cyclodextrin inclusion complex based on computational modeling. Acta Pharm. Sin. B 2013, 3, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Khodaverdi, E.; Kamali, H.; Hadizadeh, F.; Eisvand, F. Injectable In-Situ Forming Depot of Doxycycline Hyclate/α-Cyclodextrin Complex Using PLGA for Periodontitis Treatment: Preparation, Characterization, and In-Vitro Evaluation. Curr. Drug Deliv. 2020, 18, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, S.; Pan, Y.; Qu, W.; Tao, Y.; Chen, D.; Huang, L.; Liu, Z.; Wang, Y.; Yuan, Z. Preparation, characterization and pharmacokinetics of doxycycline hydrochloride and florfenicol polyvinylpyrroliddone microparticle entrapped with hydroxypropyl-β-cyclodextrin inclusion complexes suspension. Colloids Surf. B Biointerfaces 2016, 141, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A.S.; Anconi, C.P.A.; Dos Santos, H.F.; De Almeida, W.B.; Nascimento, C.S. Inclusion process of tetracycline in β and γ-cyclodextrins: A theoretical investigation. Chem. Phys. Lett. 2015, 626, 80–84. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Sandeep, S.; Divya, P.L. Synthesis and characterization of maleated cyclodextrin-grafted-silylated montmorillonite for the controlled release and colon specific delivery of tetracycline hydrochloride. RSC Adv. 2012, 2, 9555–9564. [Google Scholar] [CrossRef]
- Monteiro, A.P.F.; Rocha, C.M.S.L.; Oliveira, M.F.; Gontijo, S.M.L.; Agudelo, R.R.; Sinisterra, R.D.; Cortés, M.E. Nanofibers containing tetracycline/β-cyclodextrin: Physico-chemical characterization and antimicrobial evaluation. Carbohydr. Polym. 2017, 156, 417–426. [Google Scholar] [CrossRef]
- Hsiung, E.; Celebioglu, A.; Chowdhury, R.; Kilic, M.E.; Durgun, E.; Altier, C.; Uyar, T. Antibacterial nanofibers of pullulan/tetracycline-cyclodextrin inclusion complexes for Fast-Disintegrating oral drug delivery. J. Colloid Interface Sci. 2022, 610, 321–333. [Google Scholar] [CrossRef]
- Patel, M.N.; Dave, M.T.; Choksi, P.J. A Freeze Dried Parenteral Composition of Tigecycline and Process for Preparation Thereof. Patent WO 2017/118994 A1, 13 July 2017. [Google Scholar]
- Wiest, D.B.; Cochran, J.B.; Tecklenburg, F.W. Chloramphenicol Toxicity Revisited: A 12-Year-Old Patient with a Brain Abscess. J. Pediatr. Pharmacol. Ther. 2012, 17, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, F.; Usami, M.; Kimura, K.; Uekama, K. Crystallization and polymorphic transition behavior of chloramphenicol palmitate in 2-hydroxypropyl-β-cyclodextrin matrix. J. Incl. Phenom. Mol. Recognit. Chem. 1996, 25, 165–168. [Google Scholar] [CrossRef]
- Mashhood Ali, S.; Asmat, F.; Maheshwari, A. NMR spectroscopy of inclusion complex of d-(-)-chloramphenicol with β-cyclodextrin in aqueous solution. Il Farm. 2004, 59, 835–838. [Google Scholar] [CrossRef]
- Fatiha, M.; Leila, L.; Eddine, K.D.; Leila, N. Computational investigation of enol/keto chloramphenicol with β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2013, 77, 421–427. [Google Scholar] [CrossRef]
- Li, N.B.; Luo, H.Q.; Liu, S.P. Resonance Rayleigh scattering study of the inclusion complexation of chloramphenicol with β-cyclodextrin. Talanta 2005, 66, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Zuorro, A.; Fidaleo, M.; Lavecchia, R. Solubility Enhancement and Antibacterial Activity of Chloramphenicol Includedin Modified β-Cyclodextrins. Bull. Korean Chem. Soc. 2010, 31, 3460–3462. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-H.; Zhou, Y.-f. Inclusion interaction of chloramphenicol and heptakis (2,6-di-O-methyl)-β-cyclodextrin: Phase solubility and spectroscopic methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 83, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; Lopes, M.d.F.S.; Paz, F.A.A.; Braga, S.S. Chloramphenicol·cyclodextrin inclusion compounds: Co-dissolution and mechanochemical preparations and antibacterial action. CrystEngComm 2013, 15, 2822–2834. [Google Scholar] [CrossRef]
- Aiassa, V.; Zoppi, A.; Becerra, M.C.; Albesa, I.; Longhi, M.R. Enhanced inhibition of bacterial biofilm formation and reduced leukocyte toxicity by chloramphenicol:β-cyclodextrin:N-acetylcysteine complex. Carbohydr. Polym. 2016, 152, 672–678. [Google Scholar] [CrossRef]
- Cerutti, J.P.; Aiassa, V.; Fernández, M.A.; Longhi, M.R.; Quevedo, M.A.; Zoppi, A. Structural, physicochemical and biological characterization of chloramphenicol multicomponent complexes. J. Mol. Liq. 2021, 331, 115761. [Google Scholar] [CrossRef]
- Gannimani, R.; Ramesh, M.; Mtambo, S.; Pillay, K.; Soliman, M.E.; Govender, P. γ-Cyclodextrin capped silver nanoparticles for molecular recognition and enhancement of antibacterial activity of chloramphenicol. J. Inorg. Biochem. 2016, 157, 15–24. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, C.; Zhu, X.; Wang, X.; Wang, H.; Hu, G.; Fu, Q.; He, Z. Chloramphenicol/sulfobutyl ether-β-cyclodextrin complexes in an ophthalmic delivery system: Prolonged residence time and enhanced bioavailability in the conjunctival sac. Expert Opin. Drug Deliv. 2019, 16, 657–666. [Google Scholar] [CrossRef]
- Fan, G.; Zhang, L.; Shen, Y.; Shu, G.; Yuan, Z.; Lin, J.; Zhang, W.; Peng, G.; Zhong, Z.; Yin, L.; et al. Comparative muscle irritation and pharmacokinetics of florfenicol-hydroxypropyl-β-cyclodextrin inclusion complex freeze-dried powder injection and florfenicol commercial injection in beagle dogs. Sci. Rep. 2019, 9, 16739. [Google Scholar] [CrossRef]
- Tongiani, S.; Freehauf, K.A. Compounds and Methods for Enhancing Solubility of Florfenicol and Structurally-Related Antibiotics Using Cyclodextrins. Patent WO 2008/133901 A1, 11 June 2008. [Google Scholar]
- Rogel, C.; Mendoza, N.; Troncoso, J.; Gonzalez, J.; Von Plessing, C. Formulation and characterization of inclusion complexes using hydroxypropyl-β-cyclodextrin and florfenicol with chitosan microparticles. J. Chil. Chem. Soc. 2010, 56, 574–578. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, C.; Zhai, S.; Tan, M.; Zhao, J.; Zhu, X.; Wang, L.; Liu, Q.; Dai, T. Enrofloxacin/florfenicol loaded cyclodextrin metal-organic-framework for drug delivery and controlled release. Drug Deliv. 2021, 28, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.E.; Bajoria, R. The role of nitro-reduction and nitric oxide in the toxicity of chloramphenicol. Hum. Exp. Toxicol. 1999, 18, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Shehatta, I.S.; Ibrahim, M.S.; Sultan, M.R. The antimicrobial nalidixic acid as a probe for molecular recognition of α- and β-cyclodextrins. Can. J. Chem. 2002, 80, 1313–1320. [Google Scholar] [CrossRef]
- Mallick, S.; Sahu, A.; Pal, K. Dissolution behavior of nalidixic acid solid dispersions using water soluble dispersion carriers. Acta Pol. Pharm. 2004, 61, 21–30. [Google Scholar]
- Leyva, E.; Moctezuma, E.; Loredo-Carrillo, S.E.; Espinosa-González, C.G.; Cárdenas-Chaparro, A. Determination of the structure of quinolone-γ-cyclodextrin complexes and their binding constants by means of UV–Vis and 1H NMR. J. Incl. Phenom. Macrocycl. Chem. 2018, 91, 211–218. [Google Scholar] [CrossRef]
- Celebi, N.; Shirakura, O.; Machida, Y.; Nagai, T. The Inclusion Complex of Piromidic Acid with Dimethyl-ß-cyclodextrin in Aqueous Solution and in the Solid State. J. Incl. Phenom. 1987, 5, 407–413. [Google Scholar] [CrossRef]
- Celebi, N.; Nagai, T. Improvement of Dissolution Characteristics of Piromidic Acid by Dimethly–β–Cyclodextrin Complexation. Drug Dev. Ind. Pharm. 1988, 14, 63–75. [Google Scholar] [CrossRef]
- Iacovino, R.; Rapuano, F.; Caso, J.V.; Russo, A.; Lavorgna, M.; Russo, C.; Isidori, M.; Russo, L.; Malgieri, G.; Isernia, C. β-Cyclodextrin Inclusion Complex to Improve Physicochemical Properties of Pipemidic Acid: Characterization and Bioactivity Evaluation. Int. J. Mol. Sci. 2013, 14, 13022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfanou, F.; Michaleas, S.; Benaki, D.; Galanopoulou, O.; Voulgari, A.; Antoniadou-Vyza, E. Photostabilization of oxolinic acid in hydroxypropyl-β-cyclodextrins; implications for the effect of molecular self-assembly phenomena. J. Incl. Phenom. Macrocycl. Chem. 2009, 64, 289–297. [Google Scholar] [CrossRef]
- Koester, L.S.; Guterres, S.S.; Le Roch, M.; Eifler-Lima, V.L.; Zuanazzi, J.A.; Bassani, V.L. Ofloxacin/β-Cyclodextrin Complexation. Drug Dev. Ind. Pharm. 2001, 27, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X. Preparation and characterization of the inclusion complex of Ofloxacin with β-CD and HP-β-CD. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 173–179. [Google Scholar] [CrossRef]
- Amaro, B.; Souza Alves, C.C.; Ferreira, F.G.; Carvalho, E.P.; da Silva, J.; Souza, A.; Oswaldo, C., Jr.; Carli, P.A.S.; Machado, F.L.; Denadai, Â.; et al. Multifunctionality of βCD/Ofloxacin and HPβCD/Ofloxacin Complexes: Improvement of the Antimicrobial Activity and Apoptosis Induction on Lung Adenocarcinoma A549 Cells. J. Braz. Chem. Soc. 2020, 31, 2628–2637. [Google Scholar] [CrossRef]
- Misiuk, W.; Jozefowicz, M. Study on a host–guest interaction of hydroxypropyl-β-cyclodextrin with ofloxacin. J. Mol. Liq. 2015, 202, 101–106. [Google Scholar] [CrossRef]
- Rajendiran, N.; Mohandoss, T.; Thulasidhasan, J. Encapsulation of ciprofloxacin, sparfloxacin, and ofloxacin drugs with α- and β-cyclodextrins: Spectral and molecular modelling studies. Phys. Chem. Liq. 2016, 54, 193–212. [Google Scholar] [CrossRef]
- Padhan, P.; Sethy, A.; Behera, P.K. Host-guest interaction between Ofloxacin-β-Cyclodextrin complexes in acidic and neutral pH: A fluorescence quenching study. J. Photochem. Photobiol. A Chem. 2017, 337, 165–171. [Google Scholar] [CrossRef]
- Elbashir, A.A.; Dsugi, N.F.A.; Aboul-Enein, H.Y. Supramolecular Study on the Interaction between Ofloxacin and Methyl β-Cyclodextrin by Fluorescence Spectroscopy and its Analytical Application. J. Fluoresc. 2014, 24, 355–361. [Google Scholar] [CrossRef]
- Chao, J.; Liu, Y.; Zhang, Y.; Zhang, J.; Zhang, Y.; Guo, Z.; Wang, Y.; Qin, L.; Zhang, B. Investigation of the inclusion behavior of ofloxacin with methyl-β-cyclodextrin. J. Mol. Liq. 2014, 200, 404–409. [Google Scholar] [CrossRef]
- Mehrizi, M.; Amiri, S.; Hajir Bahrami, S.; Mirzaee, R. PVA nanofibers containing ofloxacin/α-cyclodextrin inclusion complexes: Improve ofloxacin water solubility. J. Text. Inst. 2020, 111, 669–681. [Google Scholar] [CrossRef]
- Chao, J.; Chen, L.; Xu, H.; Meng, D. Preparation and study on the solid inclusion complex of ciprofloxacin with β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2002, 58, 2809–2815. [Google Scholar] [CrossRef]
- Chao, J.; Meng, D.; Li, J.; Xu, H.; Huang, S. Preparation and study on the novel solid inclusion complex of ciprofloxacin with HP-β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 729–734. [Google Scholar] [CrossRef]
- Blanco-Fernandez, B.; Lopez-Viota, M.; Concheiro, A.; Alvarez-Lorenzo, C. Synergistic performance of cyclodextrin–agar hydrogels for ciprofloxacin delivery and antimicrobial effect. Carbohydr. Polym. 2011, 85, 765–774. [Google Scholar] [CrossRef]
- Singh, B.; Dhiman, A.; Rajneesh; Kumar, A. Slow release of ciprofloxacin from β- cyclodextrin containing drug delivery system through network formation and supramolecular interactions. Int. J. Biol. Macromol. 2016, 92, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Badr-Eldin, S.; Al, H.; Kotta, S.; Abdulhafiz, N. Self-Assembled Supramolecular Hydrogel Based on α-Cyclodextrin/Poloxamer Polypseudorotaxanes for Ocular Delivery of Ciprofloxacin. Int. J. Pharmacol. 2021, 17, 15–27. [Google Scholar] [CrossRef]
- Bozkir, A.; Denli, Z.; Kucukturkmen, B. Effect of hydroxypropyl-β-cyclodextrin on the solubility, stability and in-vitro release of ciprofloxacin for ocular drug delivery. Acta Pol. Pharm. 2012, 69, 719–724. [Google Scholar]
- Başaran, B.; Bozkir, A. Thermosensitive and pH induced in situ ophthalmic gelling system for ciprofloxacin hydrochloride: Hydroxypropyl-β-cyclodextrin complex. Acta Pol. Pharm. 2012, 69, 1137–1147. [Google Scholar]
- Macocinschi, D.; Filip, D.; Vlad, S.; Tuchilus, C.G.; Cristian, A.F.; Barboiu, M. Polyurethane/β-cyclodextrin/ciprofloxacin composite films for possible medical coatings with antibacterial properties. J. Mater. Chem. B 2014, 2, 681–690. [Google Scholar] [CrossRef]
- Moreira, M.P.; Andrade, G.R.S.; de Araujo, M.V.G.; Kubota, T.; Gimenez, I.F. Ternary cyclodextrin polyurethanes containing phosphate groups: Synthesis and complexation of ciprofloxacin. Carbohydr. Polym. 2016, 151, 557–564. [Google Scholar] [CrossRef]
- Masoumi, S.; Amiri, S.; Bahrami, S.H. PCL-based nanofibers loaded with ciprofloxacin/cyclodextrin containers. J. Text. Inst. 2018, 109, 1044–1053. [Google Scholar] [CrossRef]
- Aytac, Z.; Ipek, S.; Erol, I.; Durgun, E.; Uyar, T. Fast-dissolving electrospun gelatin nanofibers encapsulating ciprofloxacin/cyclodextrin inclusion complex. Colloids Surf. B Biointerfaces 2019, 178, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Villegas, G.; Martínez-Hernández, R.; Morales, J.; Olayo, R. Incorporation of ciprofloxacin/beta cyclodextrin complex to polylactic acid electrospun fibers and modelling the release behavior. Rev. Mex. De Ing. Química 2019, 18, 737–747. [Google Scholar] [CrossRef]
- Sun, L.; Yang, S.; Qian, X.; An, X. Cyclodextrin and cellulose combination product developed by click chemistry: Fascinating design for inclusion of ciprofloxacin. Cellulose 2020, 27, 5955–5970. [Google Scholar] [CrossRef]
- Dhevaraj, J.; Vembu, S.; Maqbool, I.; Prasad, N.R.; Pazhamalai, S.; Gopalakrishnan, M. Synthesis, characterization, sustain delivery studies and anti-bacterial evaluation of β and γ -cyclodextrin core ciprofloxacin. Drug Invent. Today 2019, 11, 642–651. [Google Scholar]
- Dhiman, P.; Bhatia, M. Microwave assisted quaternized cyclodextrin grafted chitosan (QCD-g-CH) nanoparticles entrapping ciprofloxacin. J. Polym. Res. 2021, 28, 176. [Google Scholar] [CrossRef]
- Garcia-Fernandez, M.J.; Maton, M.; Benzine, Y.; Tabary, N.; Baptiste, E.J.; Gargouri, M.; Bria, M.; Blanchemain, N.; Karrout, Y. Ciprofloxacin loaded vascular prostheses functionalized with poly-methylbeta- cyclodextrin: The importance of in vitro release conditions. J. Drug Deliv. Sci. Technol. 2019, 53, 101166. [Google Scholar] [CrossRef]
- Blanchemain, N.; Laurent, T.; Haulon, S.; Traisnel, M.; Neut, C.; Kirkpatrick, J.; Morcellet, M.; Hildebrand, H.F.; Martel, B. In vitro study of a HPγ-cyclodextrin grafted PET vascular prosthesis for application as anti-infectious drug delivery system. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 675–681. [Google Scholar] [CrossRef]
- Chao, J.-B.; Tong, H.-B.; Liu, D.-S.; Huang, S.-P. Preparation and characterization of inclusion complexes of pefloxacin mesylate with three kinds of cyclodextrins. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 64, 166–170. [Google Scholar] [CrossRef]
- Nanjwade, V.; Manvi, F.V.; Nanjwade, B. Formulation and Evaluation of Dispersible Tablets of Lomefloxacin HCl. Int. J. Drug Dev. Res. 2013, 5, 103–113. [Google Scholar]
- Cappello, B.; Iervolino, M.; Miro, A.; Chetoni, P.; Burgalassi, S.; Saettone, M.F. Formulation and Preliminary in vivo Testing of Rufloxacin-Cyclodextrin Ophthalmic Solutions. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Chattah, A.K.; Mroue, K.H.; Pfund, L.Y.; Ramamoorthy, A.; Longhi, M.R.; Garnero, C. Insights into Novel Supramolecular Complexes of Two Solid Forms of Norfloxacin and β-Cyclodextrin. J. Pharm. Sci. 2013, 102, 3717–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chierentin, L.; Garnero, C.; Chattah, A.K.; Delvadia, P.; Karnes, T.; Longhi, M.R.; Salgado, H.R.N. Influence of β-cyclodextrin on the Properties of Norfloxacin Form A. AAPS PharmSciTech 2015, 16, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.S.; Chierentin, L.; Bongioanni, A.; Salgado, H.R.N.; Longhi, M.R.; Garnero, C. β-cyclodextrin complexation as an approach to enhance the biopharmaceutical properties of Norfloxacin B Hydrate. Carbohydr. Res. 2019, 485, 107818. [Google Scholar] [CrossRef] [PubMed]
- Garnero, C.; Chattah, A.K.; Aloisio, C.; Fabietti, L.; Longhi, M. Improving the Stability and the Pharmaceutical Properties of Norfloxacin Form C through Binary Complexes with β-Cyclodextrin. AAPS PharmSciTech 2018, 19, 2255–2263. [Google Scholar] [CrossRef]
- Guyot, M.; Fawaz, F.; Bildet, J.; Bonini, F.; Lagueny, A.M. Physicochemical characterization and dissolution of norfloxacin/cyclodextrin inclusion compounds and PEG solid dispersions. Int. J. Pharm. 1995, 123, 53–63. [Google Scholar] [CrossRef]
- Loh, G.O.K.; Tan, Y.T.F.; Peh, K.-K. Enhancement of norfloxacin solubility via inclusion complexation with β-cyclodextrin and its derivative hydroxypropyl-β-cyclodextrin. Asian J. Pharm. Sci. 2016, 11, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Maia, P.P.; de Sousa, S.M.R.; De Almeida, W.B.; Guimarães, L.; Nascimento, C.S. Computational investigation on the host–guest inclusion process of norfloxacin into β-cyclodextrin. J. Mol. Modeling 2016, 22, 220. [Google Scholar] [CrossRef]
- Li, J.; Zhao, C.; Chao, J. Investigation on the inclusion behavior of Norfloxacin with 2-methyl-β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 325–331. [Google Scholar] [CrossRef]
- Mendes, C.; Wiemes, B.P.; Buttchevitz, A.; Christ, A.P.; Ribas, K.G.; Adams, A.I.H.; Silva, M.A.S.; Oliveira, P.R. Investigation of β-cyclodextrin–norfloxacin inclusion complexes. Part 1. Preparation, physicochemical and microbiological characterization. Expert Rev. Anti-Infect. Ther. 2015, 13, 119–129. [Google Scholar] [CrossRef]
- Mendes, C.; Buttchevitz, A.; Barison, A.; Ocampos, F.M.M.; Bernardi, L.S.; Oliveira, P.R.; Silva, M.A.S. Investigation of β-cyclodextrin–norfloxacin inclusion complexes. Part 2. Inclusion mode and stability studies. Expert Rev. Anti-Infect. Ther. 2015, 13, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Loh, G.O.K.; Tan, Y.T.F.; Peh, K.K. Effect of HPMC concentration on β-cyclodextrin solubilization of norfloxacin. Carbohydr. Polym. 2014, 101, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Dua, K.; Mv Ramana, D.; Sara, U.; Malipeddi, H.; Agrawal, A.; Garg, V.; Pabreja, K. Investigation of enhancement of solubility of norfloxacin beta-cyclodextrin in presence of acidic solubilizing additives. Curr. Drug Deliv. 2007, 4, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Ponte, M.; Aloisio, C.; Romero-Guerra, D.M.; Gracia-Vásquez, S.; Garnero, C.; Longhi, M. Binary and ternary complexes of norfloxacin to improve the solubility of the active pharmaceutical ingredient. Ther. Deliv. 2018, 9, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, X. Preparation and characterization of a ternary inclusion complex comprising the norfloxacin/β-cyclodextrin complex incorporated in a liposome. J. Incl. Phenom. Macrocycl. Chem. 2015, 82, 311–321. [Google Scholar] [CrossRef]
- Mendes, C.; Meirelles, G.C.; Barp, C.G.; Assreuy, J.; Silva, M.A.S.; Ponchel, G. Cyclodextrin based nanosponge of norfloxacin: Intestinal permeation enhancement and improved antibacterial activity. Carbohydr. Polym. 2018, 195, 586–592. [Google Scholar] [CrossRef]
- Sharma, R.; Bathe, R.S. Complexation of poorly water soluble drug norfloxacin with cyclodextrin. Int. J. Biomed. Adv. Res. 2014, 5, 631–640. [Google Scholar] [CrossRef]
- Calsavara, L.P.V.; Zanin, G.M.; de Moraes, F.F. Enrofloxacin inclusion complexes with cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2012, 73, 219–224. [Google Scholar] [CrossRef]
- Ding, Y.; Pang, Y.; Vara Prasad, C.V.N.S.; Wang, B. Formation of inclusion complex of enrofloxacin with 2-hydroxypropyl-β-cyclodextrin. Drug Deliv. 2020, 27, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, C.; Zhao, J.; Tan, M.; Zhai, S.; Wei, Y.; Wang, L.; Dai, T. Electrospun Fibrous Membrane Containing a Cyclodextrin Covalent Organic Framework with Antibacterial Properties for Accelerating Wound Healing. ACS Biomater. Sci. Eng. 2021, 7, 3898–3907. [Google Scholar] [CrossRef]
- Mourya, V.K.; Saini, T.R.; Balasubramaniyam, V.; Shete, J.S.; Jadhav, G.S. Molecular Inclusion of Sparfloxacin with Hydroxypropyl Beta Cyclodextrin. Braz. J. Pharm. Sci. 2002, 64, 568–572. [Google Scholar]
- Chao, J.-B.; Tong, H.-B.; Huang, S.-P.; Liu, D.-S. Preparation and study on the solid inclusion complex of sparfloxacin with β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2004, 60, 161–166. [Google Scholar] [CrossRef]
- Baby, B.; Harsha, N.S.; Jayaveera, K.N.; Abraham, A. Formulation and Evaluation of a Robust Drug Delivery System by Using Levofloxacin-Chitosan/Hydroxypropyl β-Cyclodextrin Nanoparticles. Drug Deliv. Lett. 2013, 3, 12–17. [Google Scholar] [CrossRef]
- De Gaetano, F.; Marino, A.; Marchetta, A.; Bongiorno, C.; Zagami, R.; Cristiano, M.C.; Paolino, D.; Pistarà, V.; Ventura, C.A. Development of Chitosan/Cyclodextrin Nanospheres for Levofloxacin Ocular Delivery. Pharmaceutics 2021, 13, 1293. [Google Scholar] [CrossRef] [PubMed]
- Yunus Basha, R.; Sampath Kumar, T.S.; Doble, M. Dual delivery of tuberculosis drugs via cyclodextrin conjugated curdlan nanoparticles to infected macrophages. Carbohydr. Polym. 2019, 218, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kang, Q. Synthesis of conjugates of β-cyclodextrin with polyamidoamine dendrimers and their molecular inclusion interaction with levofloxacin lactate. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 55–61. [Google Scholar] [CrossRef]
- Sanbhal, N.; Saitaer, X.; Li, Y.; Mao, Y.; Zou, T.; Sun, G.; Wang, L. Controlled Levofloxacin Release and Antibacterial Properties of β-Cyclodextrins-Grafted Polypropylene Mesh Devices for Hernia Repair. Polymers 2018, 10, 493. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.L.; Sun, J.; Zhang, Y.; Zhu, N.; Han, L.-M.; Suo, Q.-L. Preparation, Characterization and in vitro Evaluation of Tosufloxacin Tosylate and Hydroxypropyl-beta-cyclodextrin Inclusion Complex. Indian J. Pharm. Sci. 2019, 81, 249–258. [Google Scholar] [CrossRef]
- Sun, J.; Hong, H.; Zhu, N.; Han, L.; Suo, Q. Response surface methodology to optimize the preparation of tosufloxacin tosylate/hydroxypropyl-β-cyclodextrin inclusion complex by supercritical antisolvent process. J. Mol. Struct. 2019, 1198, 126939. [Google Scholar] [CrossRef]
- Sun, J.; Hong, H.; Zhu, N.; Han, L.; Suo, Q. Spectroscopic Analysis and Dissolution Properties Study of Tosufloxacin Tosylate/Hydroxypropyl-β-Cyclodextrin Inclusion Complex Prepared by Solution-Enhanced Dispersion with Supercritical CO2. J. Pharm. Innov. 2020, 15, 603–616. [Google Scholar] [CrossRef]
- Dsugi, N.F.A.; Elbashir, A.A. Supramolecular interaction of Moxifloxacin and β-cyclodextrin spectroscopic characterization and analytical application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 137, 804–809. [Google Scholar] [CrossRef]
- Szabó, Z.-I.; Deme, R.; Mucsi, Z.; Rusu, A.; Mare, A.D.; Fiser, B.; Toma, F.; Sipos, E.; Tóth, G. Equilibrium, structural and antibacterial characterization of moxifloxacin-β-cyclodextrin complex. J. Mol. Struct. 2018, 1166, 228–236. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Le-Deygen, I.M.; Uporov, I.V.; Kudryashova, E.V. A study of the physicochemical properties and structure of moxifloxacin complex with methyl-β-cyclodextrin. Colloid J. 2017, 79, 668–676. [Google Scholar] [CrossRef]
- Sukhoverkov, K.V.; Le-Deygen, I.M.; Egorov, A.M.; Kudryashova, E.V. Physicochemical Properties of the Inclusion Complex of Moxifloxacin with Hydroxypropyl-β-Cyclodextrin Synthesized by RESS. Russ. J. Phys. Chem. B 2018, 12, 1193–1204. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Le-Deygen, I.M.; Kudryashova, E.V. The Effect of Molecular Architecture of Sulfobutyl Ether β-Cyclodextrin Nanoparticles on Physicochemical Properties of Complexes with Moxifloxacin. Colloid J. 2018, 80, 312–319. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Le-Deygen, I.M.; Belogurova, N.G.; Kudryashova, E.V. Effect of cross-linking on the inclusion complex formation of derivatized β-cyclodextrins with small-molecule drug moxifloxacin. Carbohydr. Res. 2020, 498, 108183. [Google Scholar] [CrossRef] [PubMed]
- Skuredina, A.A.; Tychinina, A.S.; Le-Deygen, I.M.; Golyshev, S.A.; Belogurova, N.G.; Kudryashova, E.V. The formation of quasi-regular polymeric network of cross-linked sulfobutyl ether derivative of β-cyclodextrin synthesized with moxifloxacin as a template. React. Funct. Polym. 2021, 159, 104811. [Google Scholar] [CrossRef]
- Dsugi, N.F.A.; Elbashir, A.A.; Suliman, F.E.O. Supramolecular interaction of gemifloxacin and hydroxyl propyl β-cyclodextrin spectroscopic characterization, molecular modeling and analytical application. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 151, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Kłosińska-Szmurło, E.; Grudzień, M.; Betlejewska-Kielak, K.; Pluciński, F.A.; Biernacka, J.; Mazurek, A.P. Physico-chemical properties of lomefloxacin, levofloxacin and moxifloxacin relevant to Biopharmaceutics Classification System. Acta Chim. Slov. 2014, 61, 827–834. [Google Scholar] [PubMed]
- Kłosińska-Szmurło, E.; Pluciński, F.A.; Grudzień, M.; Betlejewska-Kielak, K.; Biernacka, J.; Mazurek, A.P. Experimental and theoretical studies on the molecular properties of ciprofloxacin, norfloxacin, pefloxacin, sparfloxacin, and gatifloxacin in determining bioavailability. J. Biol. Phys. 2014, 40, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Kłosińska-Szmurło, E.; Paweł, M.; Monika, G.; Betlejewska-Kielak, K. A New Computational Approach to the Classification of Fluoroquinolones According to the Biopharmaceutical Classification System. Curr. Comput.-Aided Drug Des. 2017, 13, 60–74. [Google Scholar] [CrossRef]
- Pierfitte, C.; Royer, R.J.; Moore, N.; Bégaud, B. The link between sunshine and phototoxicity of sparfloxacin. Br. J. Clin. Pharmacol. 2000, 49, 609–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Yu, X.; Wang, S.; Blasier, R.; Markel, D.; Mao, G.; Shi, T.; Ren, W. Cyclodextrin-erythromycin complexes as a drug delivery device for orthopedic application. Int. J. Nanomed. 2011, 6, 3173–3186. [Google Scholar] [CrossRef] [Green Version]
- Marian, E.; Tünde, J.; Vicas, L.; Kacso, I.; Miclaus, M.O.; Bratu, I. Inclusion Compounds of Erythromycin with β-cyclodextrin. Rev. De Chim. 2011, 62, 1065–1068. [Google Scholar]
- Marian, E.; Mureşan, M.; Tünde, J.; Vicas, L. Evaluation of antimicrobial activity of some types of inclusion complexes of erythromycin with β-cyclodextrin on Staphylococcus aureus. Farmacia 2013, 61, 518–525. [Google Scholar]
- Masood, F.; Yasin, T.; Bukhari, H.; Mujahid, M. Characterization and application of roxithromycin loaded cyclodextrin based nanoparticles for treatment of multidrug resistant bacteria. Mater. Sci. Eng. C 2016, 61, 1–7. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.-J.; Jeong, J.-H.; Kim, Y.; Park, J.; Jeong, D.I.; Kim, H.J.; Hwang, C.; Ahn, S.-H.; Ko, H.-J.; et al. Fast dissolving nanofiber mat for the local antimicrobial application of roxithromycin in oral cavity. Mater. Sci. Eng. C 2021, 131, 112537. [Google Scholar] [CrossRef]
- Salem, I.I.; Düzgünes, N. Efficacies of cyclodextrin-complexed and liposome-encapsulated clarithromycin against Mycobacterium avium complex infection in human macrophages. Int. J. Pharm. 2003, 250, 403–414. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhong, D.; Chen, Y.; Li, S. Investigation and Physicochemical Characterization of Clarithromycin–Citric Acid–Cyclodextrins Ternary Complexes. Drug Dev. Ind. Pharm. 2007, 33, 163–171. [Google Scholar] [CrossRef]
- Zhang, X.; Zou, M.; Li, S.; Chen, X.; Zhong, D. Bioavailability of Clarithromycin Cyclodextrin Ternary Complexes Upon Oral Administration to Healthy Beagle Dogs. Drug Dev. Ind. Pharm. 2008, 34, 1048–1053. [Google Scholar] [CrossRef]
- Mulimani, U.; Goswami, B.; Rughwani, B. Dissolution Enhancement of Clarithromycin Using Ternary Cyclodextrin Complexation. Int. J. Pharm. Res. Sch. (IJPRS) 2018, 7, 18–29. [Google Scholar] [CrossRef]
- Shete, N.A.; Swami, V.M.; Chaudhari, A.; Khabiya, P. A Design of Experiment Approach for Optimization and Characterization of Clarithromycin Ternary System Using Spray Drying. J. Drug Deliv. Ther. 2020, 10, 211–220. [Google Scholar] [CrossRef]
- El Harti, J.; Cherrah, Y.; Bouklouze, A. Improvement of Water Solubility of Josamycin by Inclusion Complex with γ-Cyclodextrin. ISRN Anal. Chem. 2012, 2012, 673564. [Google Scholar] [CrossRef] [Green Version]
- Calcagnile, M.; Bettini, S.; Damiano, F.; Talà, A.; Tredici, S.M.; Pagano, R.; Di Salvo, M.; Siculella, L.; Fico, D.; De Benedetto, G.E.; et al. Stimulatory Effects of Methyl-β-cyclodextrin on Spiramycin Production and Physical–Chemical Characterization of Nonhost@Guest Complexes. ACS Omega 2018, 3, 2470–2478. [Google Scholar] [CrossRef] [Green Version]
- Saita, M.G.; Aleo, D.; Melilli, B.; Patti, A. Effect of cyclodextrin additives on azithromycin in aqueous solution and insight into the stabilization mechanism by sulfobutyl ether-β-cyclodextrin. Int. J. Pharm. 2019, 566, 674–679. [Google Scholar] [CrossRef]
- Maddukuri, S.; Radha, G. Formulation and characterisation of azithromycin dihydrate inclusion complexes using derivatives of β-cyclodextrins as complexing agents. Int. Res. J. Pharm. 2019, 10, 77–85. [Google Scholar] [CrossRef]
- Zhao, M.-R.; Wang, L.-S.; Liu, H.-W.; Wang, Y.-J.; Yang, H. Preparation, physicochemical characterization and in vitro dissolution studies of azithromycin-cyclodextrin inclusion complexes. J. Incl. Phenom. Macrocycl. Chem. 2016, 85, 137–149. [Google Scholar] [CrossRef]
- Raval, M.; Bagada, H. Formulation and Evaluation of Cyclodextrin-Based Thermosensitive In Situ Gel of Azithromycin for Periodontal Delivery. J. Pharm. Innov. 2021, 16, 67–84. [Google Scholar] [CrossRef]
- DiSanto, A.R.; Tserng, K.Y.; Chodos, D.J.; DeSante, K.A.; Albert, K.S.; Wagner, J.G. Comparative Bioavailability Evaluation of Erythromycin Base and Its Salts and Esters. I. Erythromycin Estolate Capsules Versus Enteric-Coated Erythromycin Base Tablets. J. Clin. Pharmacol. 1980, 20, 437–443. [Google Scholar] [CrossRef]
- Krause, K.; Serio, A.; Kane, T.; Connolly, L. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [Green Version]
- Pryka, R.D.; Rodvold, K.A.; Rotschafer, J.C. Teicoplanin: An investigational glycopeptide antibiotic. Clin. Pharm. 1988, 7, 647–658. [Google Scholar] [PubMed]
- Sagitha, P.; Reshmi, C.R.; Sundaran, S.P.; Binoy, A.; Mishra, N.; Sujith, A. β-Cyclodextrin functionalized polyurethane nano fibrous membranes for drug delivery. J. Drug Deliv. Sci. Technol. 2021, 65, 102759. [Google Scholar] [CrossRef]
- Skiba, M.; Bounoure, F.; Barbot, C.; Arnaud, P.; Skiba, M. Development of cyclodextrin microspheres for pulmonary drug delivery. J. Pharm. Pharm. Sci. 2005, 8, 409–418. [Google Scholar] [PubMed]
- Parvez, S.; Yadagiri, G.; Gedda, M.R.; Singh, A.; Singh, O.P.; Verma, A.; Sundar, S.; Mudavath, S.L. Modified solid lipid nanoparticles encapsulated with Amphotericin B and Paromomycin: An effective oral combination against experimental murine visceral leishmaniasis. Sci. Rep. 2020, 10, 12243. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Sorrenti, M.; Rossi, S.; Catenacci, L.; Sandri, G.; Bonferoni, M.C.; Caramella, C.; Bettinetti, G. Vancomycin–Triacetyl Cyclodextrin Interaction Products for Prolonged Drug Delivery. Pharm. Dev. Technol. 2008, 13, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Zarif, M.S.; Afidah, A.R.; Abdullah, J.M.; Razak, S. Physicochemical characterization of vancomycin and its complexes with β-cyclodextrin. Biomed. Res. 2012, 23, 513–520. [Google Scholar]
- Zarif, M.S.; Abdullah, J.M.; Afidah, A.R.; Shafee, S.S.; Mustafa, Z.; Razak, S.A. Cytotoxicity evaluation of vancomycin and its complex with beta-cyclodextrin on human glial cell line. Pak. J. Pharm. Sci. 2012, 25, 831–837. [Google Scholar]
- Thatiparti, T.R.; von Recum, H.A. Cyclodextrin Complexation for Affinity-Based Antibiotic Delivery. Macromol. Biosci. 2010, 10, 82–90. [Google Scholar] [CrossRef]
- Thatiparti, T.R.; Juric, D.; von Recum, H.A. Pseudopolyrotaxane Formation in the Synthesis of Cyclodextrin Polymers: Effects on Drug Delivery, Mechanics, and Cell Compatibility. Bioconjugate Chem. 2017, 28, 1048–1058. [Google Scholar] [CrossRef]
- Salih, M.; Omolo, C.A.; Agrawal, N.; Walvekar, P.; Waddad, A.Y.; Mocktar, C.; Ramdhin, C.; Govender, T. Supramolecular amphiphiles of Beta-cyclodextrin and Oleylamine for enhancement of vancomycin delivery. Int. J. Pharm. 2020, 574, 118881. [Google Scholar] [CrossRef]
- Simões, S.M.N.; Veiga, F.; Torres-Labandeira, J.J.; Ribeiro, A.C.F.; Sandez-Macho, M.I.; Concheiro, A.; Alvarez-Lorenzo, C. Syringeable Pluronic–α-cyclodextrin supramolecular gels for sustained delivery of vancomycin. Eur. J. Pharm. Biopharm. 2012, 80, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Bettinetti, G.; Sorrenti, M.; Catenacci, L.; Ferrari, F.; Rossi, S.; Salvadeo, I.; Carraro, P. Solid-state interactions and drug release of teicoplanin in binary combinations with peracetylated α-, β-, and γ-cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 329–332. [Google Scholar] [CrossRef]
- Angelova, A.; Ringard-Lefebvre, C.; Baszkin, A. Drug–Cyclodextrin Association Constants Determined by Surface Tension and Surface Pressure Measurements: I. Host–Guest Complexation of Water Soluble Drugs by Cyclodextrins: Polymyxin B–β Cyclodextrin System. J. Colloid Interface Sci. 1999, 212, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Andersen, F.M.; Bundgaard, H. Inclusion complexation of metronidazole benzoate with β-cyclodextrin and its depression of anhydrate-hydrate transition in aqueous suspensions. Int. J. Pharm. 1984, 19, 189–197. [Google Scholar] [CrossRef]
- Huang, Q.; Li, B.; Yang, S.; Ma, P.; Wang, Z. Preparation and Cyclodextrin Solubilization of the Antibacterial Agent Benzoyl Metronidazole. Sci. World J. 2013, 2013, 306476. [Google Scholar] [CrossRef] [PubMed]
- Malli, S.; Bories, C.; Ponchel, G.; Loiseau, P.M.; Bouchemal, K. Phase solubility studies and anti-Trichomonas vaginalis activity evaluations of metronidazole and methylated β-cyclodextrin complexes: Comparison of CRYSMEB and RAMEB. Exp. Parasitol. 2018, 189, 72–75. [Google Scholar] [CrossRef]
- Celebioglu, A.; Uyar, T. Metronidazole/Hydroxypropyl-β-Cyclodextrin inclusion complex nanofibrous webs as fast-dissolving oral drug delivery system. Int. J. Pharm. 2019, 572, 118828. [Google Scholar] [CrossRef]
- Bensouiki, S.; Belaib, F.; Sindt, M.; Rup-Jacques, S.; Magri, P.; Ikhlef, A.; Meniai, A.-H. Synthesis of cyclodextrins-metronidazole inclusion complexes and incorporation of metronidazole—2-hydroxypropyl-β-cyclodextrin inclusion complex in chitosan nanoparticles. J. Mol. Struct. 2022, 1247, 131298. [Google Scholar] [CrossRef]
- Lahiani-Skiba, M.; Bounoure, F.; Shawky-Tous, S.; Arnaud, P.; Skiba, M. Optimization of entrapment of metronidazole in amphiphilic β-cyclodextrin nanospheres. J. Pharm. Biomed. Anal. 2006, 41, 1017–1021. [Google Scholar] [CrossRef]
- Chadha, R.; Jain, D.V.S.; Aggarwal, A.; Singh, S.; Thakur, D. Binding constants of inclusion complexes of nitroimidazoles with β-cyclodextrins in the absence and presence of PVP. Thermochim. Acta 2007, 459, 111–115. [Google Scholar] [CrossRef]
- Jagdale, S.; Kulkarni, A.; Chabukswar, A.; Kuchekar, B. Design and Evaluation of Microwave Induced Solid Dispersion of Tinidazole and Molecular Modelling with β-cyclodextrin. Lett. Drug Des. Discov. 2016, 13, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, P.; Pu, H.; Qian, H.; Sun, Q.; Zhao, L.; Li, M.; Li, H. Study on the synthesis and drug-loading optimization of beta-cyclodextrin polymer microspheres containing ornidazole. J. Drug Deliv. Sci. Technol. 2020, 58, 101836. [Google Scholar] [CrossRef]
- Natesan, S.; Sowrirajan, C.; Yousuf, S.; Enoch, I.V.M.V. Mode of encapsulation of Linezolid by β-Cyclodextrin and its role in bovine serum albumin binding. Carbohydr. Polym. 2015, 115, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.; Bose, A.; Bindhani, S.; Kar, R.K.; Pani, N.R.; Nayak, A.K. Effect of hydrophilic polymer on solubility and taste masking of linezolid in multi-component cyclodextrin inclusion complex: Physicochemical characterization and molecular docking. J. Drug Deliv. Sci. Technol. 2021, 66, 102876. [Google Scholar] [CrossRef]
- Bednarek, E.; Bocian, W.; Michalska, K. Nuclear magnetic resonance spectroscopic study of the inclusion complex of (R)-tedizolid with HDAS-β-CD, β-CD, and γ-cyclodextrin in aqueous solution. J. Pharm. Biomed. Anal. 2019, 169, 170–180. [Google Scholar] [CrossRef]
- Paczkowska-Walendowska, M.; Rosiak, N.; Tykarska, E.; Michalska, K.; Płazińska, A.; Płaziński, W.; Szymanowska, D.; Cielecka-Piontek, J. Tedizolid-Cyclodextrin System as Delayed-Release Drug Delivery with Antibacterial Activity. Int. J. Mol. Sci. 2021, 22, 115. [Google Scholar] [CrossRef]
- Jover, A.; Budal, R.M.; Al-Soufi, W.; Meijide, F.; Vázquez Tato, J.; Yunes, R.A. Spectra and structure of complexes formed by sodium fusidate and potassium helvolate with β- and γ-cyclodextrin. Steroids 2003, 68, 55–64. [Google Scholar] [CrossRef]
- Marian, E.; Tita, B.; Duteanu, N.; Vicas, L.; Ciocan, S.; Jurca, T.; Antal, L.; Tica, O.; Mureşan, M.; Pallag, A.; et al. Antimicrobial activity of fusidic acid inclusion complexes. Int. J. Infect. Dis. 2020, 101, 65–73. [Google Scholar] [CrossRef]
- Carvalho, S.G.; Siqueira, L.A.; Zanini, M.S.; dos Santos Matos, A.P.; Quaresma, C.H.; da Silva, L.M.; de Andrade, S.F.; Severi, J.A.; Villanova, J.C.O. Physicochemical and in vitro biological evaluations of furazolidone-based β-cyclodextrin complexes in Leishmania amazonensis. Res. Vet. Sci. 2018, 119, 143–153. [Google Scholar] [CrossRef]
- Carvalho, S.G.; Cipriano, D.F.; de Freitas, J.C.C.; Junior, M.Â.S.; Ocaris, E.R.Y.; Teles, C.B.G.; de Jesus Gouveia, A.; Rodrigues, R.P.; Zanini, M.S.; Villanova, J.C.O. Physicochemical characterization and in vitro biological evaluation of solid compounds from furazolidone-based cyclodextrins for use as leishmanicidal agents. Drug Deliv. Transl. Res. 2020, 10, 1788–1809. [Google Scholar] [CrossRef]
- Gupta, S.; Govil, D.; Kakar, P.N.; Prakash, O.; Arora, D.; Das, S.; Govil, P.; Malhotra, A. Colistin and polymyxin B: A re-emergence. Indian J. Crit. Care Med. 2009, 13, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Valetti, S.; Mura, S.; Stella, B.; Couvreur, P. Rational design for multifunctional non-liposomal lipid-based nanocarriers for cancer management: Theory to practice. J. Nanobiotechnol. 2013, 11, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| BP | HP-β-CD | n/a | binary | dihydropyridine | in solution | HPLC (stability study) | 727–937 (pH-dependent) | n/a | [38] |
| HP-β-CD | n/a | binary | — | in solution | HPLC (stability study) | temperature-dependent | n/a | [39] | |
| α-, β-, γ-CD | n/a | binary | — | in solution | HPLC (stability study) | dependent on the type of CD | n/a | [40] | |
| HP-β-CD | 1:1 | binary | — | in solution | NMR, IMC, MM | n/a | n/a | [41] | |
| 13 different CDs | 1:1 | binary | — | in solution | HPLC (stability study) | dependent on the type of CD | n/a | [42] | |
| β-CD, RM-β-CD, γ-CD, RM-γ-CD, Dimeb, Trimeb, Trimeg | 1:1 | binary | — | freeze-drying | HPLC (stability study), DSC, FT-IR, NMR | n/a | n/a | [43] | |
| α-, β-, γ-CD | 1:1 | binary | — | in solution | ITC | 1025 (α-CD), 1001 (β-CD), 1297 (γ-CD) | n/a | [44] | |
| PMP | β-CD | n/a | binary | — | saturated aqueous solution method | UV-VIS, DSC, XRPD | n/a | n/a | [45] |
| ampicillin | β-CD | 1:1 | n/a | n/a | n/a | fast atom bombardment MS, circular dichroism spectroscopy | n/a | n/a | [46] |
| β-CD | 2:1 | n/a | n/a | n/a | UV-VIS, vapour pressure osmometric methods | n/a | n/a | [47] | |
| β-CD, HP-β-CD | 1:1 | binary | — | in solution | microcalorimetry, NMR, MM | dependent on the type of CD, stoichiometry, and pH | n/a | [48] | |
| β-CD, γ-CD, heptakis [6-(2-carboxyethyl)thio-6-deoxy]-β-CD, octakis [6-(2-carboxyethyl)thio-6-deoxy]-γ-CD, octakis (6-amino-6-deoxy)-β-CD | usually 1:1 | binary | — | in solution | NMR, MS | 19 (γ-CD) 17 (octakis [6-(2-carboxyethyl)thio-6-deoxy]-γ-CD) | n/a | [34] | |
| β-CD | 1:1 | binary | — | in solution | NMR | n/a | n/a | [49] | |
| β-CD, telechelic polymer of β-CD-PEG-β-CD | dependent on the system | prodrug | PEG | as described 4 | FT-IR, NMR, UV-VIS, DSC | n/a | dependent on the system | [50] | |
| ampicillin | β- and γ-CD connected with D-mannose and D-N-acetylglucosamine | n/a | binary | — | in solution | NMR, white light reflectance spectroscopy | dependent on the type of CD | n/a | [51] |
| α-, β- and γ-CD | 1:1 | binary | — | in solution | ITC | 1400 (α-CD), 1047 (β-CD), 1023 (γ-CD) | n/a | [44] | |
| β-CD | n/a | NPs | ZnO | as described 4 | UV-VIS, FT-IR, AFM, particle size analysis, zeta potential | n/a | n/a | [52] | |
| pivampicillin | α-, β-, γ-, HP-α-, HP-β-, HP-γ-, M-β-CD | 1:1 | binary | — | co-precipitation | DSC, FT-IR, MM, machine learning | n/a | n/a | [53] |
| amoxicillin | β-CD | n/a | binary | — | in solution | PSS, NMR, HPLC | 10.72 | 5.88 → 7.09 mM (in 20 mM β-CD) | [54] |
| β-CD | 1:1 | binary | — | freeze-drying | FT-IR, NMR, TGA, DSC, HPLC | 1878 | n/a | [55] | |
| HP-β-CD | 1:1, 1:2 | binary | — | in solution | IMC, MM, HPLC (stability) | dependent on the mode of complex formation | n/a | [56] | |
| α-, β-, γ-CD | n/a | binary | — | in solution | IMC, petri-dish bioassay method | n/a | n/a | [57] | |
| β-CD, telechelic polymer of β-CD-PEG-β-CD | dependent on the system | prodrug | PEG | as described 4 | FT-IR, NMR, UV-VIS, DSC | n/a | dependent on the system | [50] | |
| β-CD | 1:1 | NPs | chitosan/sodium alginate/ graphene oxide | kneading, microwave-assisted, co-precipitation | PSS, UV-VIS, NMR, XRPD, DSC, SEM | 219.5 | PSS 6 | [58] | |
| dicloxacillin | β-CD, γ-CD, heptakis [6-(2-carboxyethyl)thio-6-deoxy]-β-CD, octakis [6-(2-carboxyethyl)thio-6-deoxy]-γ-CD, octakis (6-amino-6-deoxy)-β-CD | usually 1:1 | binary | — | in solution | NMR, MS | 622 (octakis [6-(2-carboxyethyl)thio-6-deoxy]-γ-CD) | n/a | [34] |
| α-, β-, γ-, and HP-β-CD | 1:1 | binary | — | in solution | PSS, NMR, MM, UV-VIS (stability) | dependent on stoichiometry and pH | PSS 6 | [59] | |
| cloxacillin sodium | β-CD | 1:1 | binary | — | co-precipitation | NMR, FL, FT-IR | 1.73 × 103 | n/a | [60] |
| methicillin | per-6-(4-methoxylbenzyl)-amino-6-deoxy-β-CD | 1:1 | binary | — | in solution | NMR, antibacterial activity | n/a | n/a | [61] |
| oxacillin | anionic and cationic thioether-substituted β- and γ-CD derivatives | 1:1 | binary | — | in solution | NMR, ITC, biological experiments | up to 1700 (with octakis (6-(2-aminoethylthio)-6- deoxy)-γ-CD) | n/a | [62] |
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| cefalexin | β-CD | 1:1 | binary | — | in solution | FL, UV-VIS | 533 (pH 5.0) | n/a | [64] |
| β-CD, telechelic polymer of β-CD-PEG-β-CD | dependent on the system | prodrug | PEG | as described 4 | FT-IR, NMR, UV-VIS, DSC | n/a | dependent on the system | [50] | |
| cefadroxil | β-CD | 1:4 by weight | nanosponges | diphenylcarbonate | as described 4 | FT-IR, DSC, SEM, size, polydispersity index, zeta potential, optical microscopy, in vivo studies | n/a | n/a | [65] |
| cefradine | β-CD | as described 4 | a medusa-like drug | 1-methyl-2-(2′-carboxyethyl) maleic anhydride, carboxylate dimethyl maleic anhydride | as described 4 | HPLC, NMR, fluorescence correlation spectroscopy, cytotoxicity | n/a | n/a | [66] |
| cefuroxime axetil | β-CD | 1:1 | binary | — | kneading | PSS, UV-VIS, XRPD, FT-IR, dissolution | 3037 | PSS 6 | [67] |
| α-, β-, γ-, HP-α-, HP-β-, HP-γ-, M-β-CD | 1:1 | binary | — | co-precipitation | DSC, FT-IR, MM, machine learning | n/a | n/a | [53] | |
| HP-β-CD | 1:1 | binary | — | co-precipitation | DSC, FT-IR, XRPD, MM, dissolution, antibacterial activity | n/a | PSS 6 | [68] | |
| α-, β-, γ-, HP-β-CD | 1:2 by weight | binary | — | grinding + tablet press | Raman spectroscopy and imaging, MM | n/a | n/a | [69] | |
| HP-β-CD | 1:1 | ternary | PVP K30, HPMC, poloxamer 188, PEG 4000, Aerosil®®® 200 | spray-drying | PSS, UV-VIS, FT-IR, XRPD, DSC, SEM, particle size analysis, dissolution | 382.7 | PSS 6 | [70] | |
| β-CD | 1:1 | ternary | L-arginine | spray-drying | PSS, saturation solubility study, dissolution, UV-VIS, XRPD, DSC, SEM, MM | 339.7 (binary), 491.0 (ternary) | 24.4 (binary), 47.5 (ternary) | [71] | |
| β-CD | n/a | ternary | PVP K-30 | kneading | PSS, UV-VIS, FT-IR, DSC, XRPD, dissolution, taste evaluation | n/a | PSS 6 | [72] | |
| cefaclor | β-CD | 1:2 | binary | — | grinding + tablet press | TLC-densitometry, NMR | n/a | n/a | [73] |
| ceftazidime | HP-γ-CD | 1:1 | binary | — | freeze-drying | UV-VIS, FT-IR, NMR, MM | 1500 | n/a | [74] |
| β-CD, HP-β-CD | n/a | formulation | poloxamer 127, cremophore RH | solvent evaporation, kneading, micromicellisation | HPLC, UV-VIS, FT-IR, XRPD, NMR | n/a | n/a | [75] | |
| HP-β-CD | n/a | liposome | phosphatidylcholine | as described 4 | FT-IR, NMR | n/a | n/a | [76] | |
| ceftazidime + vancomycin | HP-β-CD, HP-γ-CD | 1:1 | drugs + CD | — | in solution | HPLC, NMR, UV-VIS, solubility, turbidimetry, microbiological studies | n/a | PSS 6 | [77] |
| ceftriaxone | β-CD | n/a | NPs | ZnO | as described 4 | UV-VIS, FT-IR, AFM, particle size analysis, zeta potential | n/a | n/a | [52] |
| cefixime | Captisol (SBE-β-CD) | 1:1 | ternary | hypromellose E5, povidone K30, macrogol 4000 (PEG 4000) | kneading | PSS, HPLC, FT-IR | 11.5 | PSS 6 | [78] |
| β-CD | 1:2 | binary | — | freeze-drying, kneading | FT-IR, XRPD, SEM, HPLC (stability) | n/a | n/a | [79] | |
| β-CD | n/a | binary | — | kneading, solvent evaporation, freeze-drying, microwave irradiation, spray-drying | saturation solubility studies, HPLC, DSC, XRPD, SEM, dissolution, ex vivo permeability, in vivo study | n/a | up to 6.5-fold | [80] | |
| HP-β-CD | 1:1 | binary | — | kneading, solvent evaporation, spray-drying, freeze-drying, solvent evaporation, microwave irradiation | PSS, HPLC, ATR, NMR, XRPD, SEM, DSC, dissolution, antimicrobial studies, MM | 73.9 | up to 8.2-fold (spray-drying) | [81] | |
| β-CD | 1:1 | ternary | PVP K-30, HPMC K4 | freeze-drying | PSS, UV-VIS, DSC, XRPD, FT-IR, dissolution | 138.9 (binary), 305.2 (with PVP), 460.0 (with HPMC) | PSS 6 | [82] | |
| β-CD, HP-β-CD | 1:1 | ternary | L-arginine | spray-drying | PSS, UV-VIS, FT-IR, XRPD, DSC, SEM, particle size analysis, dissolution, MM | β-CD: 300 (binary), 2835 (ternary), HP-β-CD: 440 (binary), 1257 (ternary) | PSS 6 | [83] | |
| HP-β-CD, SBE-β-CD (Captisol), γ-CD | 1:1, 1:2 | ternary | different amino acids | in solution | PSS, UV-VIS, NMR, MM, microbiological studies | 20 (with SBE-β-CD) | up to 1.5 (SBE-β-CD) | [84] | |
| cefetamet pivoxil | α-, β-, γ-, HP-α-, HP-β-, HP-γ-, M-β-CD | 1:1 | binary | — | co-precipitation | DSC, FT-IR, MM, machine learning | n/a | n/a | [53] |
| cefpodoxime proxetil | β-CD | 1:2 | binary | — | solvent evaporation, freeze-drying | PSS, UV-VIS, FT-IR, DSC, XRPD, SEM, dissolution, antimicrobial studies, HPLC | n/a | PSS 6 | [85] |
| β-CD, HP-β-CD, M-β-CD | 1:1 | binary | — | spray-drying | PSS, saturated aqueous solubility, particle size analysis, zeta potential, HPLC, SEM, DSC, FT-IR, NMR, antibacterial activity | 134.9 (β-CD), 267.9 (HP-β-CD), 267.3 (M-β-CD) | 0.241 → 0.637 (β-CD), 1.201 (HP-β-CD), 1.340 (M-β-CD) mg/mL | [86] | |
| HP-β-CD | 1:1 | ternary | CMC sodium | kneading | PSS, FT-IR, DSC, SEM, HPLC, permeability studies (Caco-2 cells) | 430.8 (binary), 730.4 (ternary) | PSS 6 | [87] | |
| HP-β-CD | 1:1 | ternary | CMC sodium | kneading | PSS, FT-IR, DSC, SEM, HPLC, antimicrobial activity | 430.8 (binary), 730.4 (ternary) | PSS 6 | [88] | |
| cefdinir | β-CD, HP-β-CD | 1:1 | binary | — | kneading | PSS, UV-VIS, XRPD, FT-IR, dissolution, antimicrobial studies | 120.4 (β-CD), 58.6 (HP-β-CD) | by 101% (β-CD), by 23.4% (HP-β-CD) | [89] |
| β-CD | 1:1 | binary | — | kneading, co-evaporation, spray-drying, microwave irradiation | PSS, UV-VIS, DSC, FT-IR, SEM, NMR, dissolution | 119.8 | PSS 6 | [90] | |
| HP-β-CD | 1:1 | binary | — | kneading, co-evaporation, spray-drying, microwave irradiation | PSS, UV-VIS, DSC, FT-IR, SEM, NMR, XRPD, dissolution | 53.7 | PSS 6 | [91] | |
| HP-β-CD | 1:1 | binary | — | freeze-drying | PSS, saturation solubility study, SEM, XRPD, DSC, FT-IR, dissolution | 65.3 | 2.36-fold | [92] | |
| β-CD, γ-CD, HP-β-CD, SBE7-β-CD | 1:1 | binary | — | freeze-drying | PSS, HPLC, DSC, FT-IR, XRPD, SEM, antimicrobial activity | 100.3 (β-CD), 186.6 (γ-CD), 114.1 (HP-β-CD), 986.5 (SBE7-β-CD) | PSS 6 | [93] | |
| ceftiofur | HP-β-CD | n/a | formulation | sodium alginate poloxamer 407, 188 | as described in the patent | HPLC | n/a | 72.7 times (0.03 → 2.18 mg/mL) | [94] |
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| meropenem | β-CD | 1:1 | binary | — | kneading | PSS, UV-VIS, FT-IR, DSC, HPLC (stability), dissolution, MM, antimicrobial activity | n/a | ~15% | [96] |
| (2-hydroxy-3-N,N,N-trimethylamino)propyl- β-CD | 1:1 | NPs | CMC | as described 4 | HPLC, UV-VIS, NMR, particle size measurements, zeta potential, permeation studies | n/a | n/a | [97] | |
| γ-CD | 1:1 | NPs | PLGA | liquid CO2 method | UV-VIS, TGA, DSC, XRPD, FT-IR, SEM, NMR, MM, HPLC, in vitro release, permeability (Caco-2 cells), laser scanning confocal microscopy, antibacterial assay | n/a | n/a | [98] | |
| ertapenem | HP-β-CD | n/a | binary | — | freeze-drying | HPLC | n/a | n/a | [99] |
| tebipenem pivoxil | β-CD | 1:1 | binary | — | grinding | PSS, HPLC, FT-IR, DSC, Raman, dissolution, MM, antibacterial activity, permeability (Caco-2 cells) | 0.1539 | n/a | [100] |
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| doxycycline | HP-β-CD | 1:2 | binary | — | freeze-drying | FT-IR, NMR, IMC, SEM, DLS, zeta potential, microbiological studies | n/a | n/a | [102] |
| β-CD | 1:1 | binary | — | freeze-drying | TGA, DTA, FT-IR, NMR, ITC, DLS, zeta potential, antimicrobial tests, cytotoxicity assay | 503 | n/a | [32] | |
| β-CD | 1:1 | binary | — | freeze-drying | NMR, MM, FT-IR, DSC, TGA, photostability (HPLC), microbiological studies | n/a | n/a | [103] | |
| α-, γ-, HP-β-CD, RM-β-CD | n/a | binary | — | freeze-drying | UV-VIS, SEM, XRPD, photostability (UV-VIS), permeation studies | n/a | n/a | [104] | |
| β-CD | 1:1 | binary | — | in silico only | DFT | n/a | n/a | [105] | |
| HP-β-CD | 1:1 | hydrogel | MgCl2, poloxamer P407 and P188 | as described 4 | PSS, TGA, XRPD, FT-IR, measurement of gelation temperature, in vitro release, stability (HPLC) | 31.2 (in water) 120.8 (in MgCl2) | PSS 6 | [106] | |
| HP-β-CD | n/a | ternary | MgCl2 | freeze-drying | SEM, UV-VIS, NMR, MM, stability (HPLC), antibacterial activity | n/a | n/a | [107] | |
| HP-β-CD | 1:4 | ternary | MgCl2 | freeze-drying | HPLC (assay, stability studies), optical microscopy, NMR | n/a | n/a | [108] | |
| α-CD | 1:1 | in situ forming implant | PLGA | freeze-drying | FT-IR, XRPD, DSC, stability (UV-VIS), in vitro release (HPLC), SEM, MM, cytotoxicity | n/a | n/a | [109] | |
| HP-β-CD | n/a | microparticles | PVP, HPMC | vacuum drying | SEM, UV-VIS, HPLC, particle size analysis, stability, in vitro release, pharmacokinetic study, antibacterial activity | n/a | n/a | [110] | |
| tetracycline | β-, γ-CD | 1:1 | binary | — | in silico | semi-empirical and DFT calculations | n/a | n/a | [111] |
| maleated β-CD | n/a | hydrogel | 3-methylacryloxy- propyltrimethoxy silylated montmorillonite | as described 4 | SEM, FT-IR, XRPD, elemental analysis, UV-VIS, in vitro release, in vitro biocompatibility test | n/a | n/a | [112] | |
| β-CD | 1:1 | NFs | PCL | freeze-drying + electrospinning | thermal analysis, XRPD, FT-IR, SEM, electrical conductivity, viscosity, in vitro release, microbiological test | n/a | n/a | [113] | |
| HP-β-CD | 1:2 | NFs | pullulan | electrospinning | PSS, UV-VIS, conductivity and viscosity measurements, SEM, FT-IR, XRPD, TGA, dissolution, in vitro release, disintegration, antibacterial test, DFT | 21 | ~2.1-fold | [114] | |
| tigecycline | SBE-β-CD | n/a | formulation | HCl | freeze-drying | n/a | n/a | n/a | [115] |
| API | CD | API:CD Ratio | System | Addition | Preparation | Characterisation | Stability Constant Kc (M−1) | Solubility Improvement | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| erythromycin | β-CD | 1:3.3 (w/w) | binary | — | solvent evaporation, kneading | UV-VIS, NMR, XRPD, SEM, stability, antibacterial activity, in vivo studies, cytotoxicity | n/a | n/a | [212] |
| β-CD | 1:1 | binary | — | kneading, co-precipitation, freeze-drying | FT-IR, XRPD, DSC | n/a | n/a | [213] | |
| β-CD | n/a | binary | — | kneading, co-precipitation, freeze-drying | antimicrobial activity | n/a | n/a | [214] | |
| roxithromycin | β-CD, HP-β-CD | 1:1 | NPs | PLGA | solvent evaporation | SEM, FT-IR, XRPD, UV-VIS, antimicrobial studies | n/a | n/a | [215] |
| HP-β-CD | 1:1 | NFs | PVA, D-α-tocopheryl PEG succinate | electrospinning | PSS, HPLC, FT-IR, DSC, XRPD, XPS, wetting test, disintegration, in vitro release, cytotoxicity, antimicrobial activity, toxicity, pharmacokinetic study | 36.1 | PSS 6 | [216] | |
| clarithromycin | β-CD | 1:1 | binary | — | solvent evaporation | HPLC, antimicrobial activity | n/a | 700-fold (pH 7.4) | [217] |
| β-CD | 1:1 | ternary | citric acid | co-evaporation, freeze-drying | PSS, HPLC, IR, DSC, SEM, XRPD | 102.4 (in water) 161.2 (in 6 mM CA) | PSS 6 | [218] | |
| β-CD | 1:1 | ternary | citric acid | freeze-drying | dissolution, pharmacokinetic studies | n/a | n/a | [219] | |
| β-CD | n/a | ternary | Soluplus | slurry evaporation, kneading | PSS, HPLC, DSC, MM, in vitro dissolution | 1702 | 6-fold | [220] | |
| HP-β-CD | n/a | ternary | PVP K30 | kneading, spray-drying | DSC, XRPD, FT-IR, particle size analysis, in vitro release | n/a | n/a | [221] | |
| josamycin propionate | γ-CD | 1:2 | binary | — | co-precipitation | PSS, UV-VIS, XRPD, FT-IR, NMR | 3060 | 0.35 → 4.60 mg/mL (in 250 mg/mL γ-CD) | [222] |
| spiramycin | α-, β-, γ-, M-β-CD | 1:3 | binary | — | in solution | UV-VIS, ATR-FT-IR, MM | n/a | n/a | [223] |
| azithromycin | SBE-β-CD | n/a | binary | — | in solution | HPLC, NMR | n/a | n/a | [224] |
| β-CD, epichlorohydrin-β-CD, SBE-β-CD | 1:1, 1:2 | binary | — | kneading, freeze-drying | PSS, UV-VIS, FT-IR, DSC, XRPD, SEM, in vitro dissolution | PSS 6 | PSS 6 | [225] | |
| HP-β-CD and its polymer | 1:1 | polymer | epichloro- hydrin | saturated solution method | UV-VIS, FT-IR, XRPD, SEM, solubility, dissolution | n/a | 34.2 → 311.3 (HP-β-CD), 628.5 mg/L (polymer) | [226] | |
| HP-β-CD | 1:1 | gel formulation | poloxamer 407, Carbopol 934P | kneading | PSS, UV-VIS, XRPD, SEM, DSC, physicochemical properties of gel | 6.04 × 104 | 2.3 → 49.8 mg/L | [227] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczar, D.; Michalska, K. Cyclodextrin Inclusion Complexes with Antibiotics and Antibacterial Agents as Drug-Delivery Systems—A Pharmaceutical Perspective. Pharmaceutics 2022, 14, 1389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14071389
Boczar D, Michalska K. Cyclodextrin Inclusion Complexes with Antibiotics and Antibacterial Agents as Drug-Delivery Systems—A Pharmaceutical Perspective. Pharmaceutics. 2022; 14(7):1389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14071389
Chicago/Turabian StyleBoczar, Dariusz, and Katarzyna Michalska. 2022. "Cyclodextrin Inclusion Complexes with Antibiotics and Antibacterial Agents as Drug-Delivery Systems—A Pharmaceutical Perspective" Pharmaceutics 14, no. 7: 1389. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14071389