2.1. Classically Used Transfection Efficacy Assays Fail to Predict Protein Production in Industrial Settings
Polyplex (PEI) and lipoplex-based approaches are the most frequently used transient transfection methods [
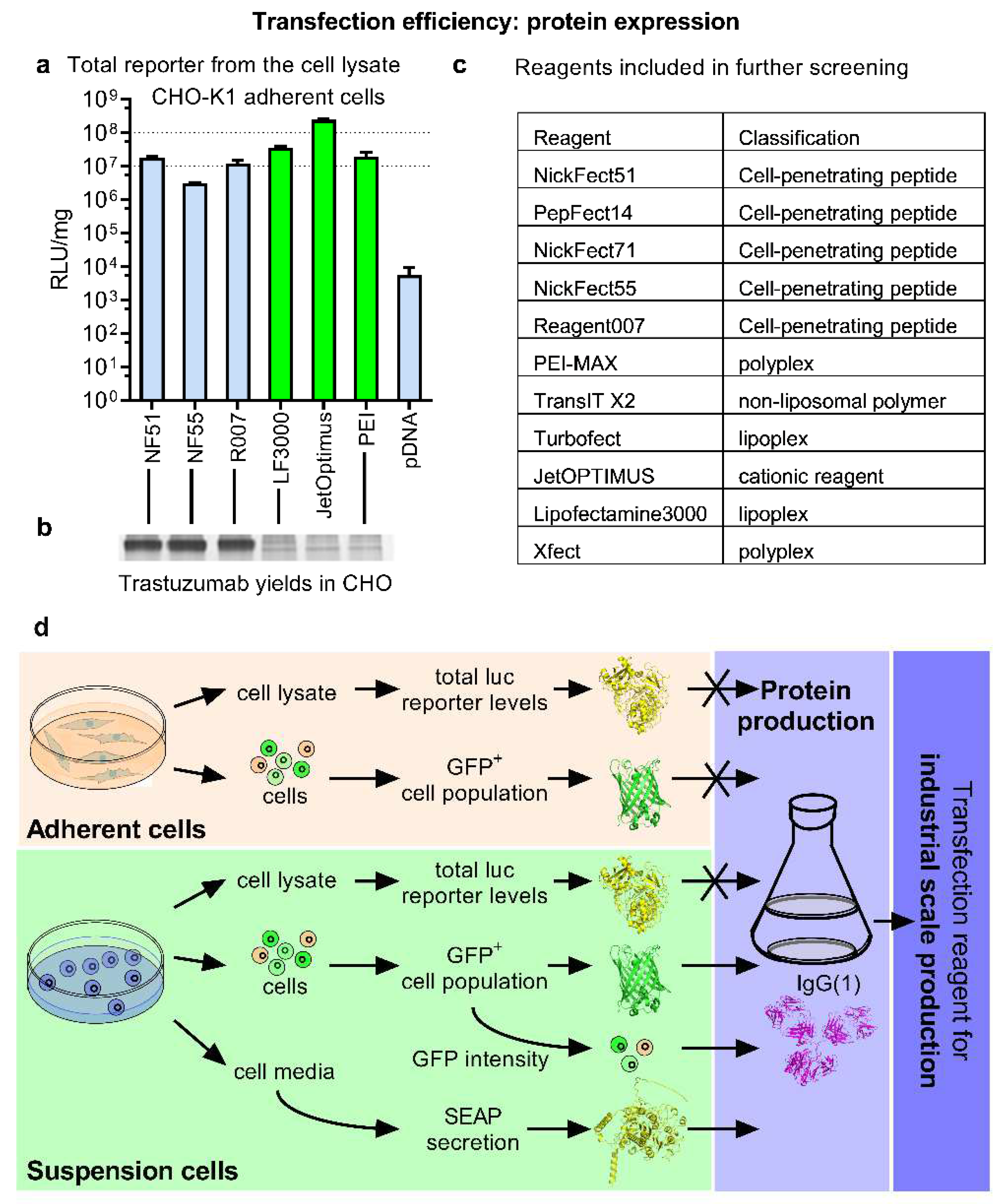
4]. The transfection efficacy can be assessed by the quantitation of the expressed reporter. The total luciferase amount is often measured from the cell lysate in an adherent cell culture between 24 h and 48 h post-transfection. Luciferase reporter assays are sensitive, fast, require simple equipment, and are widely available. Since the desired outcome in the industry is maximizing the yields of protein-of-interest, it is therefore reasonable to focus on the quantitation of total reporter protein.
The pDNA transfection efficacy (
Figure 1a,b) of a small set of well-known lipoplex, polyplex, as well as cell-penetrating peptide (CPP)-based transfection methods were compared (
Figure 1c) in CHO cells, which is the primary expression platform [
3,
11]. We normalized the relative luminescence units (RLU) with total protein, as this does not change the tendencies between the reagents (
Figure S1), but allows the comparison of RLUs from different experiments. Since each transfection reagent has its own instructions for use, leaving room for optimization in many cases, assay conditions were selected by optimizing the reagent amount (
Figure S2a), N/P ratio (
Figure S2b), charge ratio (
Figure S2c), media change and pDNA dose (
Figure S3a–c) to maximize the transfection output. Generally, a high-protein expression was observed with all the methods, although lipoplex and polyplex proved to be the most effective (
Figure 2a). CPP-mediated transfection, albeit efficient, did not quite reach the levels shown by jetOptimus or Lipofectamine. When taking into account the mean values, CPPs also performed slightly less efficiently than PEI-Max.
As a next step we investigated how these reporter performances translated into the industrial protein expression settings. The production of an example protein, therapeutic mAb Trastuzumab, was used to achieve this with the QMCF protein expression technology [
12] in CHO suspension cells over a one-week production cycle. The QMCF includes Epstein-Barr Virus nuclear antigen 1 (EBNA-1) and mouse polyomavirus Large T antigen (PyLT) elements in the producer cells and plasmid expression vectors with the aim of enhancing the retention and replication of the protein expression vector in the cells and allowing significant extension of the “transient” period in the protein production. Other protein expression methods with similar mechanisms are also available [
3]. We hypothesized that all the transfection methods were able to produce reasonable yields of mAb, especially the top performers from
Figure 1a as these mediated high reporter protein expression from the same cell line in a short time frame. Unexpectedly, significant differences from the assumed output were observed—the CPP methods NF55 [
13], NF51 [
14], as well as the Reagent007 all significantly outperformed both the lipoplex and polyplex approaches in produced protein levels (
Figure 2b). Considering this, we conclude that the fast and easy luciferase reporter lab assay fails to predict industrial protein production performance (
Figure 2d).
Considering that clinically relevant recombinant protein production in mammalian cells is, by its nature, technically challenging, the whole field benefits from being able to predict large-scale performance without weeks of production in cell cultures [
15]. The availability of fast lab-scale screening methods would contribute to the development of improved transfection methods for protein-based drug production, and protein production in general.
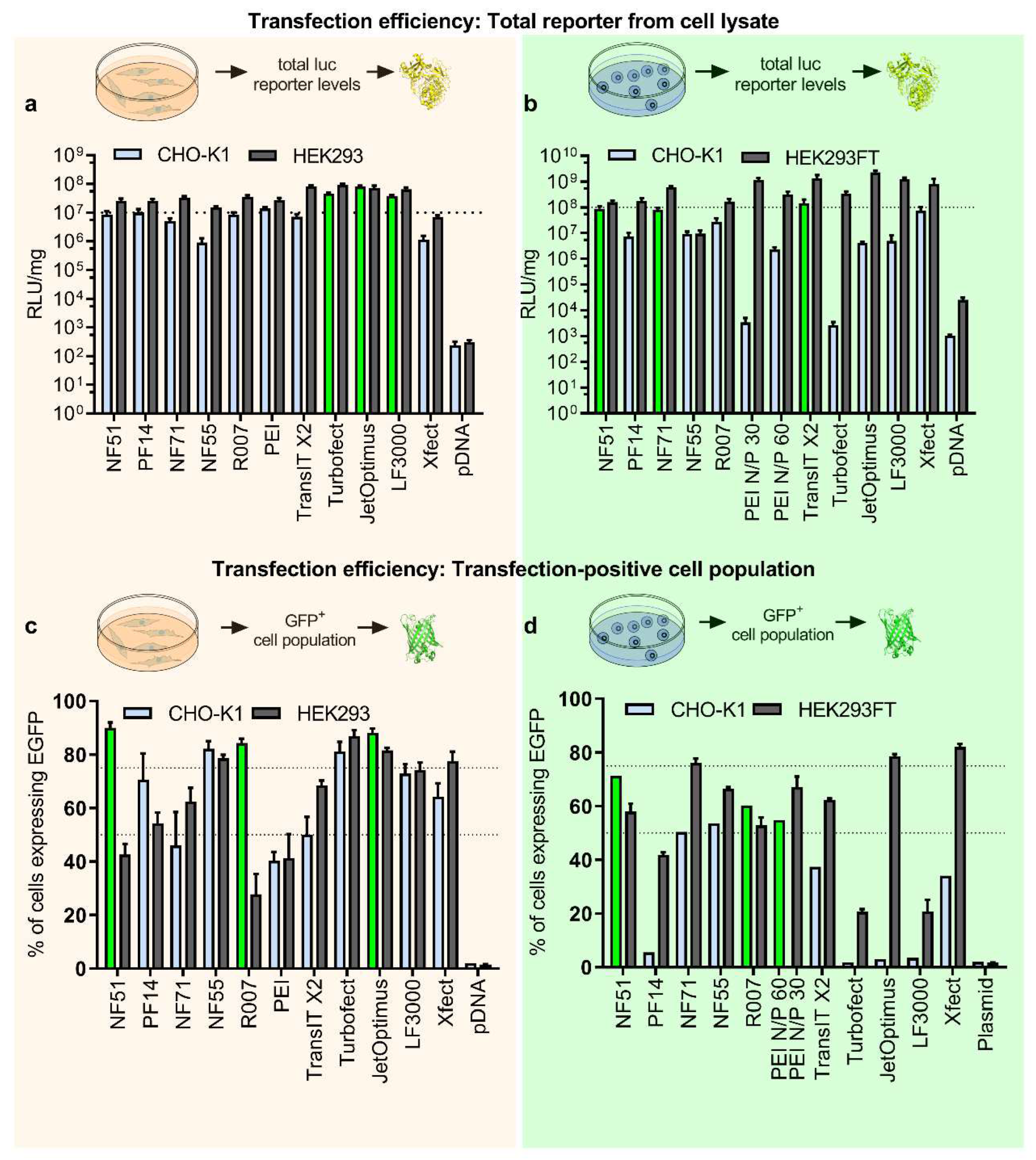
To investigate the transfection efficacies and possibilities for protein production further, we expanded the selection of transfection methods and included CPPs NF71 [
16], PF14 [
17], and other known examples of polymers and liposomes (
Figure 1c). Furthermore, to explore if the abovementioned discrepancy was specific to CHO, we included another cell line, HEK293. HEK293 is a well-established producer cell line because of its ease of use for both cell growth and transfection, as well as its excellent protein yields. HEK293 is frequently utilized as the expression system to produce recombinant proteins and viruses for gene therapies [
4]. Again, to be able to compare different reagents, optimal conditions for reagents in HEK cells were determined to maximize the transfection result (
Figures S2a,b and S3b,c). We completed luciferase expression with the extended set of transfection methods and observed that the output efficacies were in line with those observed with the initial smaller set in
Figure 1a. The top performers were liposomal and polymeric approaches with statistically significantly higher values (
Figure 2a, light blue bars, top three are highlighted in green). For the HEK293 cell line, the differences between the methods were smaller, but liposomal and PEI-based approaches were still the top performers (
Figure 2a).
An important difference between small-scale and the industrial cell factories is the use of suspension cultures. With the use of suspension cells, much higher cell densities can be achieved than possible with adherent cell cultures. Therefore, as a next step, we assessed the transfection efficacies in suspension culture by using a total luciferase quantitation assay. The transfection time and volume, presence of serum in the media, transfection reagent dose, pDNA dose, and N/P ratio were optimized to maximize the expressed protein levels (
Figures S4a,b and S5a–d).
In the suspension culture, the contrast between high and low performers was further pronounced (
Figure 2a vs.
Figure 2b). Notably, the top performers in CHO suspension culture were markedly different from what were observed in adherent conditions, and included two peptide-based methods (NF51 and NF71) and one polyplex-based (TransIT) method (
Figure 2b, highlighted in green). Importantly, when comparing the high and low performers in the suspension culture to the initial set of methods included in mAb production (
Figure 1b), the predictive value of the suspension culture transfection was higher than that observed in adherent cell culture. One of the most efficient protein producers—NF51—was now among the top three transfection methods, and none of the nonperformers in mAb production were among the best transfection methods in CHO suspension culture (
Figure 2b vs.
Figure 1b).
2.2. The Transfected Cell Population Is an Important Protein Expression Efficacy Predictor
Although the amount of the total expressed protein (
Figure 2a,b) could be an intuitive way for estimating transfection performance, long-term protein production is also dependent on another important aspect—a successful division of transfected cells. Each transfection-positive cell clone gives rise to approximately 100 to 500 protein-producing offspring, depending on the cell line and cultivation conditions [
18]. For example, a cell that is successfully transfected (estimated at an early time point, such as in
Figure 2), but is unable to undergo cell division and produce a high number of daughter cells (which will only become apparent after several days post-transfection, such as in
Figure 1b) is probably not contributing to high levels of expressed protein yields. To assess the effect of the transfection-positive cell population, two separate lines of reasoning were utilized. First, to estimate the fraction of cells that contribute to transfection, we determined the proportion of cells that were successfully transfected. Second, to observe the cells that fail to contribute to protein expression, we determined the number of live/dead cells post-transfection.
The transfection-positive cell population was estimated via EGFP reporter and by determining fluorescent-positive cells post-transfection. This assessment was first performed in adherent cells, and it was observed that the predictability for protein yields had further increased: now two top protein producers in CHO—NF51 and Reagent007—were among the top transfectors (
Figure 1b vs.
Figure 2c). However, some of the nonperforming protein producers were also flagged among the efficient transfection methods (such as jetOptimus in CHO in
Figure 2c). Further analysis by dividing transfected cells into subpopulations of low, medium, and high EGFP+ did not offer additional insight (
Figure S6).
Next, we investigated whether the transfection-positive cell count improves protein production prediction with the use of suspension culture —similar to what was observed in the case of total luciferase quantitation. The regular time period for protein production is approximately a week, therefore we wanted to confirm that the chosen pGFP reporter (which did not include any plasmid retention and replication enhancers) was expressed over several days. In both of the suspension cell lines the EGFP signals were present beyond five days (
Figure S7a,b). Additionally, it was concluded that a 48 h end-point was the optimal timeframe for the assay in suspension cells (
Figure S7c), as there was no significant increase in transfection-positive cell population after the 48 h mark. From transfection-positive cell count profiles (
Figure 2d) it was observed that the highest transfection-positive percentages were achieved with CPP-based methods and PEI-Max in CHO cells, whereas in HEK cells the CPP NF71, Xfect, and jetOptimus were the top performers (
Figure 2d).
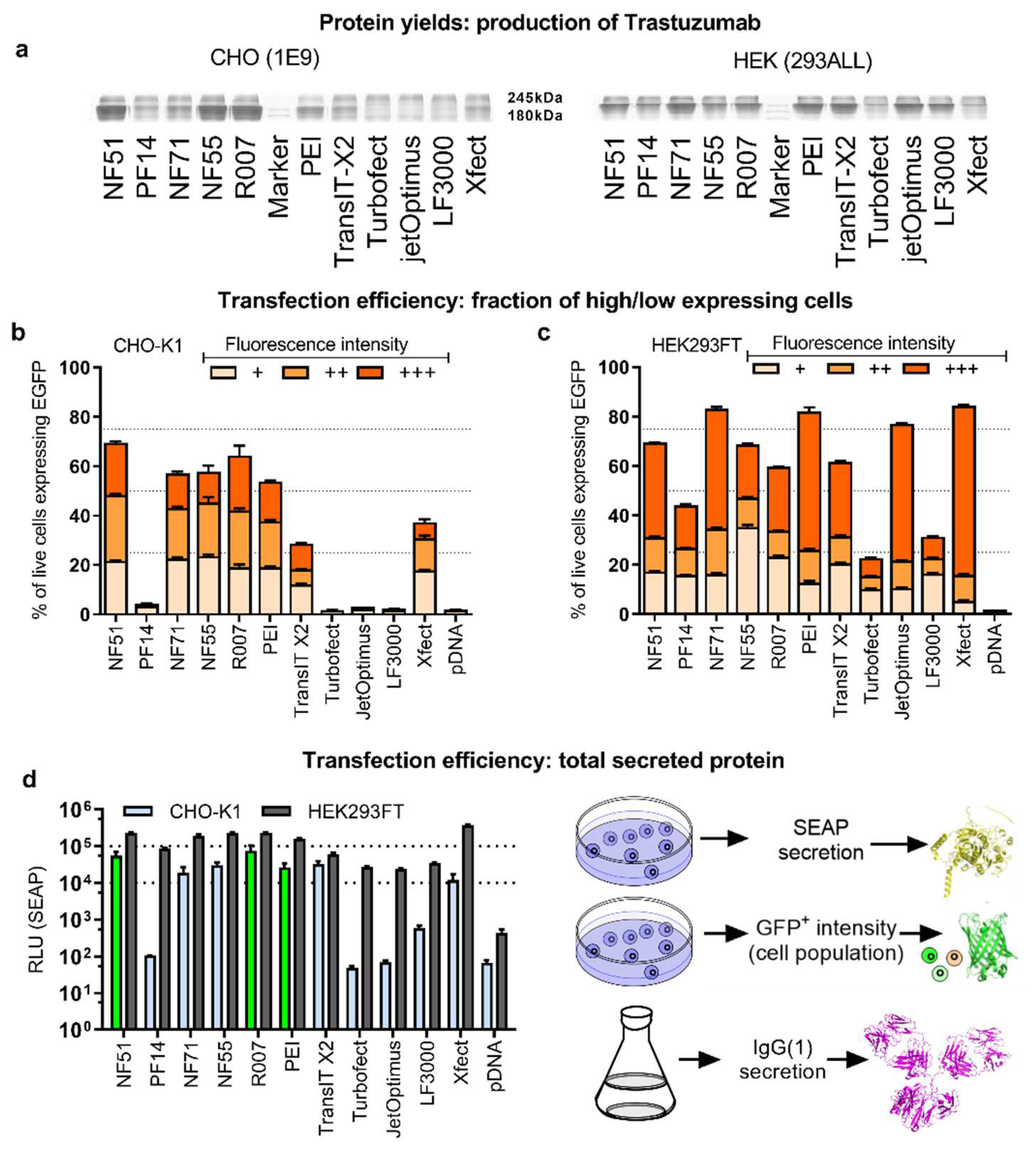
To analyze the impact of the efficacy assays (
Figure 2) on the prediction of protein production, a second round of mAb production was performed to compare all the transfection methods. Moreover, it was done in CHO as well as HEK293 producer cells over a 1-week production cycle (
Figure 3a). The superiority of the CPP transfection over polymeric and liposomal methods was observed in CHO cells (
Figure 3a). The noted CPPs NF51, NF55 and R007 achieved a considerably higher level of the expressed protein, compared to all the other methods. The CPPs NF51 and NF55 were developed previously, and although their high efficacy for pDNA transfection has been shown [
13,
14], their utility in protein production settings as well as their superiority over polyplex and lipoplex approaches had not been explored before.
In order to estimate the protein production predictive power, transfection-positive cell counts were compared to protein production efficacies (
Figure 2d vs.
Figure 3a, statistical analysis for all groups throughout the work are shown in
Supplementary Table S2, and plots in
Supplementary Table S3). It was observed that transfection-positive cell counts (
Figure 2d) successfully flagged both the performers (highlighted in green) and non-performers in mAb production: NF51, NF55, Reagent007 and PEI-Max were among the best transfectors as well as the highest protein-producing groups (although PEI-Max cannot be considered on par with the CPP methods). Conversely, the rest of the methods lacked in transfection and mAb production.
In HEK293, the protein was produced most effectively when transfected with the CPP NF71, followed by polyplex methods PEI-Max, TransIT, and jetOptimus (
Figure 3a). The CPP NF71 was developed recently [
16] and has been shown as an efficient siRNA and miRNA transfection reagent [
16,
19], but it has not been demonstrated in any of the pDNA-based applications before. However, in HEK cells neither the reporter assays (total protein or transfection-positive cell population) nor the culturing condition (adherent or suspension) were able to predict performance in mAb production, thus further investigation is needed for this cell line.
2.3. Expressed Protein Yields Are Predicted by the Total Secreted Protein Reporter
In order to establish whether specific subpopulations of cells with high- or low-expressed protein levels determine the outcome of the ultimate protein yields, the GFP+ cell populations were further investigated. The transfection-positive suspension cell subpopulations were divided into three subcategories: cells with low, medium, and high expressed protein levels (
Figure 3b–c,
Figures S8 and S9). However, none of these sub-fractions improved prediction precision, as the subpopulation proportions correlated primarily with the total GFP+ cells.
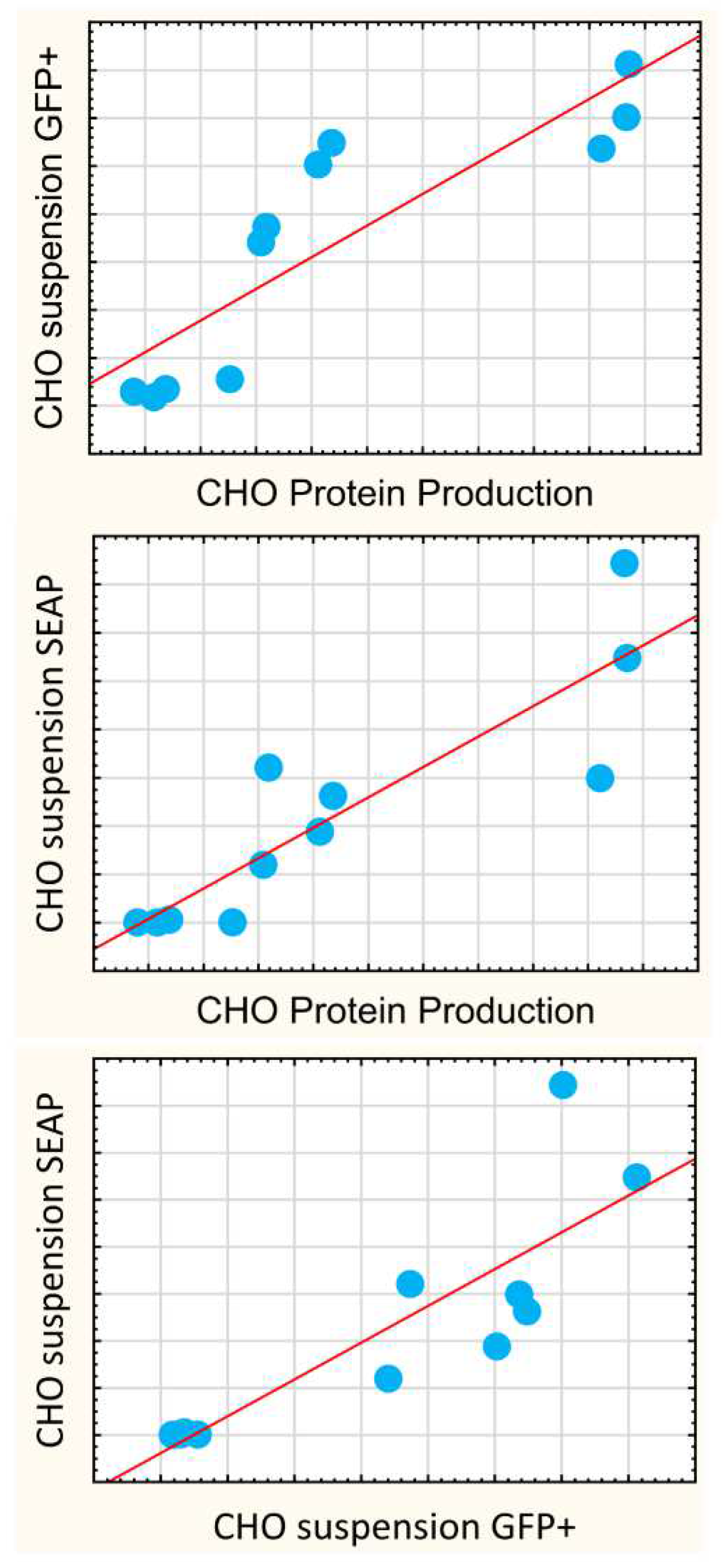
Considering that the ultimate goal lies in the effective production of large mammalian proteins that depend on processing in ER and Golgi, and are often secreted, a reporter protein that is also critically dependent on the ER and Golgi processing, and is secreted out of the cell—the secreted alkaline phosphatase (SEAP)—was included. The profiles of pSEAP transfection (
Figure 3d) indicated that the SEAP expression in the CHO suspension culture correlated strongly with the mAb protein yields (r = 0.88,
p < 0.05). In the HEK293 suspension culture, SEAP expression correlated with the number of GFP+ cells, but, unlike in CHO, neither of these efficacy assays correlated with mAb production.
2.4. High Viability of Cells Does Not Correlate with High Protein Expression
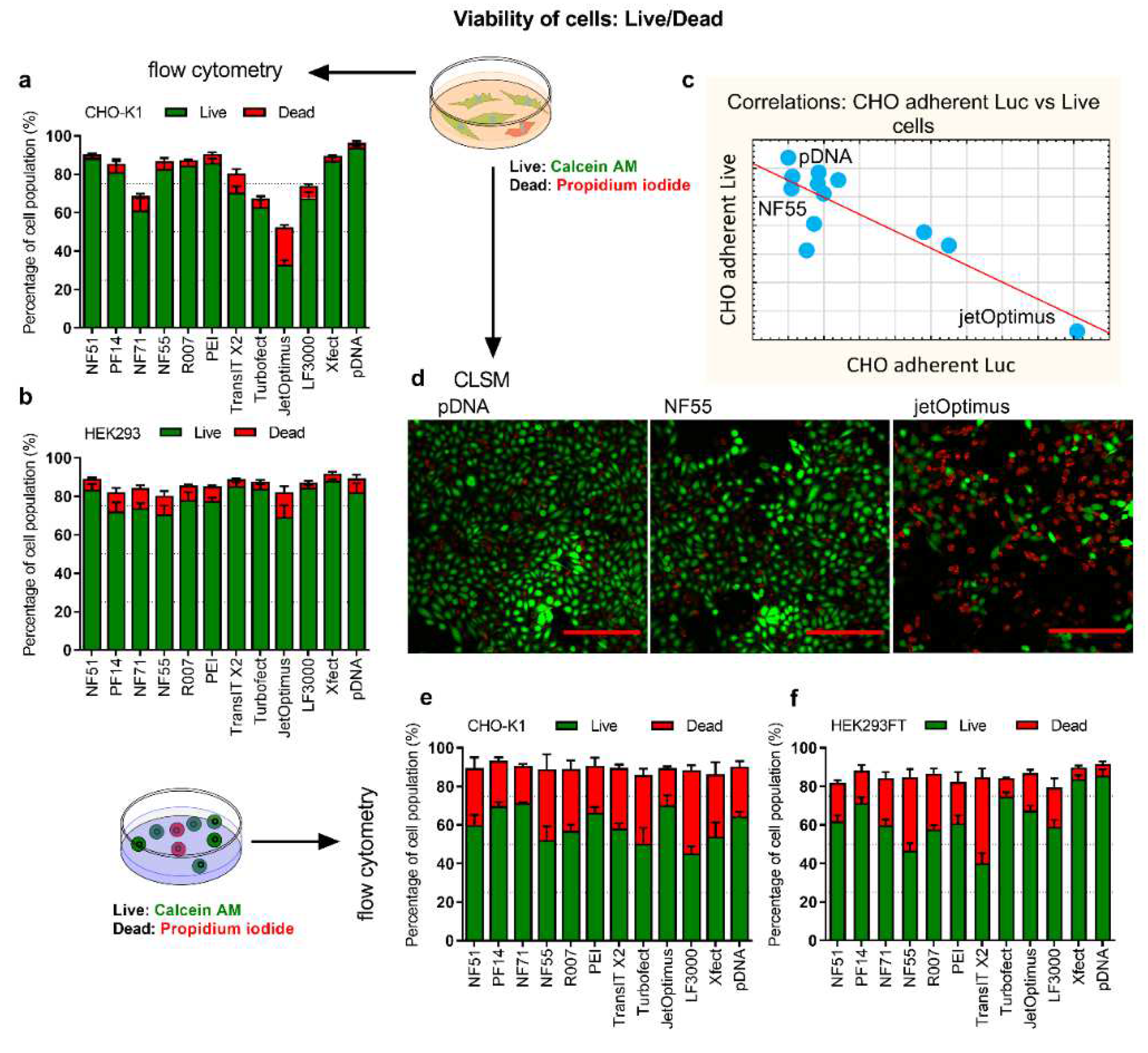
The successful expression of proteins is dependent on the number of viable cells. In order to characterize the cells that fail to contribute to transfection, the numbers of live and dead cells were analyzed. Although BrdU (
Figure S10a) and MTS assays (
Figure S10b) were considered, the live/dead assay was selected due to its high optimization range, compatibility with different detection methods, and because it reflected both live cells and cells with compromised cell membranes in a cell population (
Figure 4a,b,d–f,
Figures S11–S13). The aim was to analyze whether the proportion of (living) cells correlated with the expressed protein amount and if there was a higher number of live cells in the groups that performed well in protein production.
We observed that in the CHO-adherent cells, some of the transfection methods were accompanied by an apparent reduction of viable cells. Controversially, the most “toxic” methods were concurrently very efficient transfection mediators in the adherent culture: the Pearson coefficient between adherent CHO total luciferase expression and the number of live cells was r = −0.85 (
p < 0.05). Two different cases from this correlation were visualized with the CLSM: jetOptimus as the highest total luciferase transfection, and the least number of live cells. NF55 was one of the least performing in total luciferase expression in adherent CHO, but among the group with the highest number of viable cells (
Figure 4c,d). In suspension cells, there was no strong correlation between cell viability (
Figure 4e,f) and other assays.
On the one hand, we can conclude that reduced cell viability post-transfection may not mean much in adherent conditions, i.e., it is not necessarily a negative occurrence by itself. However, in the context of protein production in defined media, the total luciferase expression in adherent cell culture is not correlated with the protein yields. If anything, the correlation is negative (r = −0.59, but statistically not significant).
2.5. Correlations between Protein Production and Screening Methods
We were most interested in finding connections that predict long-term and large-scale protein production efficacies from fast- and small-scale assays. To achieve this, we screened a number of methods that reflected different aspects of transfection efficacies and toxicities in two producer cell lines. We constructed a correlation matrix between the numerical outputs of all the assays for both the cell lines. The correlation matrix is presented in
Supplementary Table S3.
The most significant conclusions from the above are as follows. First, in CHO, the protein production yields correlated with both the suspension expressions of SEAP (r = 0.88,
p < 0.05) and GFP+ population (
Figure 5, r = 0.86,
p < 0.05), but did not correlate with luciferase quantitation in neither adherent nor suspension media (
Supplementary Table S3, Figure S15). This forced us to conclude a surprising implication: luciferase reporter quantitation assay, despite its excellent technical aspects, is not a good screening assay for predicting efficacies in protein expression. Conversely, the application of SEAP and GFP reporters should be encouraged as these create valuable data that can be used for protein production.
The second implication is, as clearly illustrated in the correlation analysis (
Supplementary Table S3), that the adherent culture methods generally fail to predict the effects of transfection in suspension conditions. Neither transfection efficacy assays, nor viability/toxicity assays correlated with protein production efficacies in CHO and HEK293. Hence, when working with the applications of protein expression in mammalian cells, suspension culturing is a must, even in research laboratory settings.
Third, although the current report exemplifies several useful predictors for the CHO producer cell line in terms of efficacy assays and transfection methods, the utility and implications for the HEK293 are, unfortunately, less significant. Although we had demonstrated that a CPP-based transfection with NF71 significantly outperformed polyplex and lipoplex methods in protein production in HEK293, predictive transfection assays for this cell line should be explored in the future.
Finally, in the current report, we presented cell-penetrating peptide-based transient transfection that significantly outperformed the most widely used method in protein production industrial settings, the PEI. We showed that NF55 and NF51 were efficient for CHO-based production, and NF71 is an excellent performer in HEK293 cells. These methods have the potential to replace PEI in industrial settings and offer higher yields for therapeutic protein production. Considering that the current report only intended to find correlative connections between various efficacy assays and transfection reagents, it offers little proof that would explain the mechanisms behind the observations. For example, it would be useful to mechanistically explain why the CPPs NF55, NF51 and R007 are significantly more efficient in protein expression than PEI or LF3000. With that knowledge, the process can be further improved. The mechanism for their success may be either increased efficacy in transfection, or less negative effects in cellular growth processes. More specifically, the process of association with the nucleic acid, interactions with the cell membrane, endosomal escape, long-term effects of cellular accumulation on cell viability are among the questions that should be answered in a follow-up study.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




