
Stabilization of Graphene Oxide Dispersion in Plasma-like Isotonic Solution Containing Aggregating Concentrations of Bivalent Cations
 , , , and
, , , and
Abstract
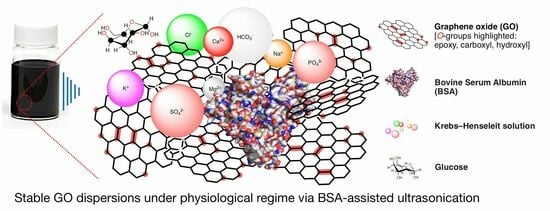
:
1. Introduction
2. Materials and Methods
2.1. Analyzed Materials
2.2. Graphene Oxide Sonication
2.3. Graphene Oxide Dilution with Bovine Serum Albumin (BSA)
2.4. Graphene Oxide–Bovine Serum Albumin Dilution with Krebs–Henseleit Solution
2.5. Graphene Oxide Particle Size Analysis
2.6. pH Measurement
2.7. Transmission Electron Microscopy
2.8. Cryogenic Transmission Electron Microscopy
2.9. Atomic Force Microscope Analysis
2.10. Functional Groups Determination
2.11. Statistics and Data Analysis
3. Results
3.1. Graphene Oxide Water Dispersions Stock Characteristics
3.2. Graphene Oxide Dilution
3.3. Graphene Oxide Sonication
3.4. Graphene Oxide Dilution with Bovine Serum Albumin
3.5. Graphene Oxide Dispersions with Bovine Serum Albumin and Krebs–Henseleit Solution
3.6. Graphene Oxide as Analyzed by Transmission Electron Microscopy and Cryo-Transmission Electron Microscopy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuila, T.; Bose, S.; Kumar, A.; Khanra, P. Progress in Materials Science Chemical functionalization of graphene and its applications. Prog. Mater. Sci. 2012, 57, 1061–1105. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Eda, G.; Chhowalla, M. Graphene-based Composite Thin Films for Electronics. Nano Lett. 2009, 9, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Costinas, C.; Salagean, C.A.; Cotet, L.C.; Baia, M.; Todea, M.; Magyari, K.; Baia, L. Insights into the Stability of Graphene Oxide Aqueous Dispersions. Nanomaterials 2022, 12, 4489. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.M.; Gonçalves, C.; Sousa, D.M.; Ferreira, A.R.; Magalh, D.; Goncąlves, C.; Sousa, D.M.; Ferreira, A.R.; Moreira, J.A.; Goncąlves, I.C.; et al. Smaller particle size and higher oxidation improves biocompatibility of graphene-based materials. Carbon N. Y. 2016, 99, 318–329. [Google Scholar] [CrossRef]
- Khoei, A.R.; Khorrami, M.S. Mechanical properties of graphene oxide: A molecular dynamics study. Fuller. Nanotub. Carbon Nanostructures 2016, 24, 594–603. [Google Scholar] [CrossRef]
- Tripathi, D.; Ray, P.; Singh, A.V.; Kishore, V.; Singh, S.L. Durability of Slippery Liquid-Infused Surfaces: Challenges and Advances. Coatings 2023, 13, 1095. [Google Scholar] [CrossRef]
- Yang, K.; Chen, B.; Zhu, X.; Xing, B. Aggregation, Adsorption, and Morphological Transformation of Graphene Oxide in Aqueous Solutions Containing Different Metal Cations. Environ. Sci. Technol. 2016, 50, 11066–11075. [Google Scholar] [CrossRef]
- Bai, R.G.; Muthoosamy, K.; Manickam, S.; Hilal-Alnaqbi, A. Graphene-based 3D scaffolds in tissue engineering: Fabrication, applications, and future scope in liver tissue engineering. Int. J. Nanomed. 2019, 14, 5753–5783. [Google Scholar] [CrossRef]
- Chen, S.L.; Chen, C.Y.; Hsieh, J.C.H.; Yu, Z.Y.; Cheng, S.J.; Hsieh, K.Y.; Yang, J.W.; Kumar, P.V.; Lin, S.F.; Chen, G.Y. Graphene oxide-based biosensors for liquid biopsies in cancer diagnosis. Nanomaterials 2019, 9, 1725. [Google Scholar] [CrossRef]
- Sharma, H.; Mondal, S. Functionalized graphene oxide for chemotherapeutic drug delivery and cancer treatment: A promising material in nanomedicine. Int. J. Mol. Sci. 2020, 21, 6280. [Google Scholar] [CrossRef]
- Alemi, F.; Maleki, M.; Mir, M.; Ebrahimi-kalan, A.; Zarei, M.; Yousefi, B.; Rashtchizadeh, N.; Laboratories, C.; Disorders, M.; Medicine, M.; et al. Synthesis, characterization, and evaluation of pH-sensitive doxorubicin-loaded functionalized graphene oxide in osteosarcoma cells. BioImpacts 2023, 13, 207–218. [Google Scholar] [CrossRef]
- Zhao, M.; Shi, j.; Cai, W.; Liu, K.; Shen, K.; Li, Z.; Wang, Y.; Hu, D. Advances on Graphene-Based Nanomaterials and Mesenchymal Stem Cell-Derived Exosomes Applied in Cutaneous Wound Healing. Int. J. Nanomed. 2021, 16, 2647–2665. [Google Scholar] [CrossRef]
- Bao, D.; Sun, J.; Gong, M.; Shi, J.; Qin, B.; Deng, K.; Liu, G.; Zeng, S.; Xiang, Z.; Fu, S. Combination of graphene oxide and platelet-rich plasma improves tendon–bone healing in a rabbit model of supraspinatus tendon reconstruction. Regen. Biomater. 2021, 8, rbab045. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zhou, M.; Chen, L.; Fang, H.; Liu, S.; Zhou, C.; Sun, J.; Wang, Z. Alendronate loaded graphene oxide functionalized collagen sponge for the dual effects of osteogenesis and anti-osteoclastogenesis in osteoporotic rats. Bioact. Mater. 2020, 5, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.S.; An, S.S.A.; Yi, D.K. Oxidative stress and antibacterial properties of a graphene oxide-cystamine nanohybrid. Int. J. Nanomed. 2015, 10, 549–556. [Google Scholar] [CrossRef]
- Gomes, R.N.; Borges, I.; Pereira, A.T.; Maia, A.F.; Pestana, M.; Magalhães, F.D.; Pinto, A.M.; Gonçalves, I.C. Antimicrobial graphene nanoplatelets coatings for silicone catheters. Carbon N. Y. 2018, 139, 635–647. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Mukherjee, S.P.; Gallud, A.; Burkert, S.C.; Bistarelli, S.; Bellucci, S.; Bottini, M.; Star, A.; Fadeel, B. Biological interactions of carbon-based nanomaterials: From coronation to degradation. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Di Santo, R.; Digiacomo, L.; Quagliarini, E.; Capriotti, A.L.; Laganà, A.; Chiozzi, R.Z.; Caputo, D.; Cascone, C.; Coppola, R.; Pozzi, D.; et al. Personalized graphene oxide-protein corona in the human plasma of pancreatic cancer patients. Front. Bioeng. Biotechnol. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Hu, W.; Peng, C.; Lv, M.; Li, X.; Zhang, Y.; Chen, N.; Fan, C.; Huang, Q. Protein corona-mediated mitigation of cytotoxicity of graphene oxide. ACS Nano 2011, 5, 3693–3700. [Google Scholar] [CrossRef]
- Duan, G.; Kang, S.; Tian, X.; Garate, J.A.; Zhao, L.; Ge, C.; Zhou, R. Protein corona mitigates the cytotoxicity of graphene oxide by reducing its physical interaction with cell membrane. Nanoscale 2015, 7, 15214–15224. [Google Scholar] [CrossRef]
- Huang, P.-J.J.; Pautler, R.; Shanmugaraj, J.; Labbé, G.; Liu, J. Inhibiting the VIM-2 Metallo-β-Lactamase by Graphene Oxide and Carbon Nanotubes. ACS Appl. Mater. Interfaces 2015, 7, 9898–9903. [Google Scholar] [CrossRef] [PubMed]
- Criado, M.P.; Fraschini, C.; Salmieri, S.; Becher, D.; Sáfrány, Á.; Lacroix, M. Evaluation of Antioxidant Cellulose Nanocrystals and Applications in Gellan Gum Films. Ind. Biotechnol. 2015, 11, 59–68. [Google Scholar] [CrossRef]
- Beatson, R.P. Determination of Sulfonate Groups and Total Sulfur. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 473–484. ISBN 978-3-642-74065-7. [Google Scholar]
- Salgin, S.; Salgin, U.; Bahadir, S. Zeta potentials and isoelectric points of biomolecules: The effects of ion types and ionic strengths. Int. J. Electrochem. Sci. 2012, 7, 12404–12414. [Google Scholar] [CrossRef]
- Wang, C.G.; Lan, L.; Liu, Y.P.; Tan, H.F. Defect-guided wrinkling in graphene. Comput. Mater. Sci. 2013, 77, 250–253. [Google Scholar] [CrossRef]
- Shen, X.; Lin, X.; Yousefi, N.; Jia, J.; Kim, J.-K. Wrinkling in graphene sheets and graphene oxide papers. Carbon N. Y. 2014, 66, 84–92. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Gliga, A.R.; Lazzaretto, B.; Brandner, B.; Fielden, M.; Vogt, C.; Newman, L.; Rodrigues, A.F.; Shao, W.; Fournier, P.M.; et al. Graphene oxide is degraded by neutrophils and the degradation products are non-genotoxic. Nanoscale 2018, 10, 1180–1188. [Google Scholar] [CrossRef]
- Rai, M.; Paudel, N.; Sakhrie, M.; Gemmati, D.; Khan, I.A.; Tisato, V.; Kanase, A.; Schulz, A.; Singh, A.V. Perspective on Quantitative Structure–Toxicity Relationship (QSTR) Models to Predict Hepatic Biotransformation of Xenobiotics. Livers 2023, 3, 448–462. [Google Scholar] [CrossRef]
- Kurapati, R.; Russier, J.; Squillaci, M.A.; Treossi, E.; Ménard-Moyon, C.; Del Rio-Castillo, A.E.; Vazquez, E.; Samorì, P.; Palermo, V.; Bianco, A. Dispersibility-Dependent Biodegradation of Graphene Oxide by Myeloperoxidase. Small 2015, 11, 3985–3994. [Google Scholar] [CrossRef] [PubMed]
- Bolibok, P.; Koter, S.; Kaczmarek-Kędziera, A.; Kowalczyk, P.; Łukomska, B.; Łukomska, O.; Boncel, S.; Wiśniewski, M.; Kaneko, K.; Terzyk, A.P. Liquid phase adsorption induced nanosizing of graphene oxide. Carbon N. Y. 2021, 183, 948–957. [Google Scholar] [CrossRef]
- Kuziel, A.W.; Milowska, K.Z.; Chau, P.L.; Boncel, S.; Koziol, K.K.; Yahya, N.; Payne, M.C. The True Amphipathic Nature of Graphene Flakes: A Versatile 2D Stabilizer. Adv. Mater. 2020, 32, 2000608. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Dayem, A.A.; Kwon, D.-N.; Kim, J.-H. Biocompatibility effects of biologically synthesized graphene in primary mouse embryonic fibroblast cells. Nanoscale Res. Lett. 2013, 8, 393. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, J.; Hao, X.; Zhang, C.; Wei, N.; Zhang, C. Wetting properties of defective graphene oxide: A molecular simulation study. Molecules 2018, 23, 1439. [Google Scholar] [CrossRef]
- Peng, E.; Todorova, N.; Yarovsky, I. Effects of Size and Functionalization on the Structure and Properties of Graphene Oxide Nanoflakes: An in Silico Investigation. ACS Omega 2018, 3, 11497–11503. [Google Scholar] [CrossRef]
- Lin, L.; Zhuang, X.; Huang, R.; Song, S.; Wang, Z.; Wang, S.; Cheng, L.; Zhu, R. Size-dependent effects of suspended graphene oxide nanoparticles on the cellular fate of mouse neural stem cells. Int. J. Nanomed. 2020, 15, 1421–1435. [Google Scholar] [CrossRef]
- Mendes, R.G.; Koch, B.; Bachmatiuk, A.; Ma, X.; Sanchez, S.; Damm, C.; Schmidt, O.G.; Gemming, T.; Eckert, J.; Rümmeli, M.H. A size dependent evaluation of the cytotoxicity and uptake of nanographene oxide. J. Mater. Chem. B 2015, 3, 2522–2529. [Google Scholar] [CrossRef]
- Sattari, S.; Adeli, M.; Beyranvand, S.; Nemati, M. Functionalized graphene platforms for anticancer drug delivery. Int. J. Nanomed. 2021, 16, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.C.; Pak, P.J.; Joo, Y.H.; Lee, H.S.; Chung, N. In vitro and in vivo comparison of the immunotoxicity of single- and multi-layered graphene oxides with or without pluronic F-127. Sci. Rep. 2016, 6, 38884. [Google Scholar] [CrossRef]
- Wang, A.; Pu, K.; Dong, B.; Liu, Y.; Zhang, L.; Zhang, Z.; Duan, W.; Zhu, Y. Role of surface charge and oxidative stress in cytotoxicity and genotoxicity of graphene oxide towards human lung fibroblast cells. J. Appl. Toxicol. 2013, 33, 1156–1164. [Google Scholar] [CrossRef]
- Cohignac, V.; Landry, M.J.; Ridoux, A.; Pinault, M.; Annangi, B.; Gerdil, A.; Herlin-boime, N.; Haruta, M.; Codogno, P.; Boczkowski, J.; et al. Carbon nanotubes, but not spherical nanoparticles, block autophagy by a shape-related targeting of lysosomes in murine macrophages. Autophagy 2018, 14, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Herrera, C.E.; Singh, B.; Singh, S.; Singh, R.K.; Kumar, R. Graphene oxide: An efficient material and recent approach for biotechnological and biomedical applications. Mater. Sci. Eng. C 2018, 86, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Jin, X.; Wang, F.; You, Y.; Chu, D.; Zetterlund, P.B.; Joshi, R.K. Cation-induced coagulation in graphene oxide suspensions. Mater. Today Chem. 2019, 13, 139–146. [Google Scholar] [CrossRef]
- Wu, L.; Liu, L.; Gao, B.; Muñoz-Carpena, R.; Zhang, M.; Chen, H.; Zhou, Z.; Wang, H. Aggregation kinetics of graphene oxides in aqueous solutions: Experiments, mechanisms, and modeling. Langmuir 2013, 29, 15174–15181. [Google Scholar] [CrossRef] [PubMed]
- Onsager, L.; Onsager, L. The effect of shape on the interaction of colloidal particles. Ann. N. Y. Acad. Sci. 2006, 51, 627–659. [Google Scholar] [CrossRef]
- Li, D.; Müller, M.B.; Gilje, S.; Kaner, R.B.; Wallace, G.G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 2008, 3, 101–105. [Google Scholar] [CrossRef]
- Suter, J.L.; Coveney, P.V. Principles governing control of aggregation and dispersion of aqueous graphene oxide. Sci. Rep. 2021, 11, 22460. [Google Scholar] [CrossRef]
- Amirov, R.R.; Shayimova, J.; Nasirova, Z.; Dimiev, A.M. Chemistry of graphene oxide. Reactions with transition metal cations. Carbon N. Y. 2017, 116, 356–365. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Gao, Y.; Zhao, Y. Control of number of graphene layers using ultrasound in supercritical CO2 and their application in lithium-ion batteries. J. Supercrit. Fluids 2014, 85, 95–101. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Rosillo-Lopez, M.; Lee, T.J.; Bella, M.; Hart, M.; Salzmann, C.G. Formation and chemistry of carboxylic anhydrides at the graphene edge. RSC Adv. 2015, 5, 104198–104202. [Google Scholar] [CrossRef]
- Araújo, M.P.; Soares, O.S.G.P.; Fernandes, A.J.S.; Pereira, M.F.R.; Freire, C. Tuning the surface chemistry of graphene flakes: New strategies for selective oxidation. RSC Adv. 2017, 7, 14290–14301. [Google Scholar] [CrossRef]
- Iwakura, Y.; Uno, K.; Oya, M. Polymerization of DL-alanine NCA and L-alanine NCA. J. Polym. Sci. Part A-1 Polym. Chem. 1967, 5, 2867–2874. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.L. New insight on an old reaction—The aqueous hydrolysis of acetic anhydride. J. Phys. Org. Chem. 2012, 25, 1105–1111. [Google Scholar] [CrossRef]
- Guthrie, J.P. Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a ran. CJC West. Univ. Virtual Compil. 2014, 1, 2342–2354. [Google Scholar] [CrossRef]
- Tao, L.; Han, J.; Tao, F.-M. Correlations and Predictions of Carboxylic Acid pKa Values Using Intermolecular Structure and Properties of Hydrogen-Bonded Complexes. J. Phys. Chem. A 2008, 112, 775–782. [Google Scholar] [CrossRef]
- Keshavan, S.; Calligari, P.; Stella, L.; Fusco, L.; Delogu, L.G.; Fadeel, B. Nano-bio interactions: A neutrophil-centric view. Cell Death Dis. 2019, 10, 569. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Tokarczyk, K.; Jachimska, B.; Mulheran, P.A. Bovine Serum Albumin Adsorption at a Silica Surface Explored by Simulation and Experiment. J. Phys. Chem. B 2017, 121, 3975–3986. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, Y.; Chen, W.; Wang, K.; Zhu, L. Concentration Dependent Effects of Bovine Serum Albumin on Graphene Oxide Colloidal Stability in Aquatic Environment. Environ. Sci. Technol. 2018, 52, 7212–7219. [Google Scholar] [CrossRef]
- Kubiak-Ossowska, K.; Jachimska, B.; Mulheran, P.A. How Negatively Charged Proteins Adsorb to Negatively Charged Surfaces: A Molecular Dynamics Study of BSA Adsorption on Silica. J. Phys. Chem. B 2016, 120, 10463–10468. [Google Scholar] [CrossRef]
- Li, R.; Wu, Z.; Wangb, Y.; Ding, L.; Wang, Y. Role of pH-induced structural change in protein aggregation in foam fractionation of bovine serum albumin. Biotechnol. Rep. 2016, 9, 46–52. [Google Scholar] [CrossRef]
- Šimšíková, M. Interaction of graphene oxide with albumins: Effect of size, pH, and temperature. Arch. Biochem. Biophys. 2016, 593, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, B.; Tang, D.; Yu, C. Concurrent aggregation and transport of graphene oxide in saturated porous media: Roles of temperature, cation type, and electrolyte concentration. Environ. Pollut. 2018, 235, 350–357. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name, Stage and Number (Averaged Data) | Z-Ave (d.nm) | PDI | Averaged Int 1 (d.nm) | Averaged Int 2 (d.nm) | Averaged Int 3 (d.nm) | Area Int 1 (%) | Area Int 2 (%) | Area Int 3 (%) |
|---|---|---|---|---|---|---|---|---|
| GO Stock | ||||||||
| GO1 pu | 3557.0 | 0.916 | 740.1 | 185.1 | 97.6 | 95.0 | 3.0 | 1.3 |
| GO2 pu | 4363.0 | 0.668 | 1481.0 | 188.3 | 5119.0 | 33.7 | 27.3 | 21.1 |
| GO3 pu | 1077.0 | 0.717 | 1049.0 | 4642.0 | 267.2 | 60.8 | 20.5 | 18.7 |
| GO4 pu | 3386.0 | 0.844 | 657.3 | 360.4 | 266.9 | 56.7 | 18.7 | 12.4 |
| GO Diluted | ||||||||
| GO1 140 µg/mL | 1344.0 | 0.412 | 913.9 | 391.4 | 4526.0 | 63.1 | 21.8 | 9.0 |
| GO2 140 µg/mL | 1759.0 | 0.612 | 830.0 | 2066.0 | 5207.0 | 73.6 | 23.0 | 3.1 |
| GO3 140 µg/mL | 768.8 | 0.559 | 1163.0 | 265.2 | 4906.0 | 66.5 | 25.7 | 7.9 |
| GO4 140 µg/mL (42) | 26300 | 0.758 | 2347.0 | 1734.0 | 4557.0 | 21.7 | 20.7 | 20.0 |
| GO Sonicated | ||||||||
| GO1 140 µg/mL 6 × 50 W | 628.0 | 0.518 | 382.9 | 92.8 | 4417.0 | 92.1 | 5.2 | 2.6 |
| GO2 140 µg/mL 6 × 50 W | 654.2 | 0.552 | 197.3 | 508.8 | 54.1 | 79.9 | 19.9 | 0.2 |
| GO3 140 µg/mL 6 × 50 W | 532.1 | 0.472 | 767.9 | 4419.0 | 0 | 89.7 | 10.3 | 0 |
| GO4 140 µg/mL 6 × 50 W (42) | 536.1 | 0.489 | 751.1 | 4628.0 | 0 | 93.8 | 6.2 | 0 |
| GO With BSA | ||||||||
| GO1 50 µg/mL BSA 400 mg/L | 357.9 | 0.38 | 285.7 | 3981.0 | 0 | 96.2 | 3.8 | 0 |
| GO2 50 µg/mL BSA 400 mg/L | 408.2 | 0.418 | 408.6 | 4405.0 | 0 | 95.8 | 4.2 | 0 |
| GO3 50 µg/mL BSA 400 mg/L | 509.7 | 0.391 | 716.7 | 4657.0 | 0 | 94.6 | 5.4 | 0 |
| GO4 50 µg/mL BSA 400 mg/L | 441.8 | 0.448 | 486.4 | 4486.0 | 0 | 96.0 | 4.0 | 0 |
| GO with BSA and Krebs–Henseleit | ||||||||
| GO1 10 µg/mL BSA 80 mg/L KH | 421.1 | 0.385 | 193.6 | 0 | 0 | 100 | 0 | 0 |
| GO2 10 µg/mL BSA 80 mg/L KH | 382.6 | 0.363 | 253.9 | 66.9 | 0 | 99.6 | 0.4 | 0 |
| GO3 10 µg/mL BSA 80 mg/L KH | 440.2 | 0.406 | 294.4 | 1839.0 | 0 | 92.5 | 7.5 | 0 |
| GO4 10 µg/mL BSA 80 mg/L KH (42) | 490.1 | 0.435 | 234.0 | 0 | 0 | 100 | 0 | 0 |
| GO Functional Groups by Titration | COOH mmol/g | SO3H mmol/g | NH2 mmol/g | Phenol OH mmol/g |
|---|---|---|---|---|
| GO1 | ||||
| before sonication | 1.13 | 1.17 | nt * | nt |
| post-sonication 6 × 50 W 1 | 3.5 | 0.1 | nt | <0.1 |
| post-sonication 6 × 50W 2 | 4 | 0.15 | nt | <0.1 |
| GO2 | ||||
| before sonication | 1.35 | 1.57 | nt | nt |
| post-sonication 6 × 50W 1 | 3.7 | 0.1 | nt | <0.1 |
| post-sonication 6 × 50W 2 | 3.7 | 0.1 | nt | <0.1 |
| GO3 | ||||
| before sonication | 2.52 | nd | nt | nt |
| post-sonication 6 × 50W 1 | 4.5 | 0.2 | nt | nt |
| post-sonication 6 × 50W 2 | 4.3 | 0.3 | nt | nt |
| GO4 | ||||
| before sonication | 2.49 | nd | nt | nt |
| post-sonication 6 × 50W 1 | 4 | 0.1 | 1.2 | nt |
| post-sonication 6 × 50W 2 | 3.9 | 0.2 | 1 | nt |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasoń, M.Z.; Paradowska, A.; Fronczek, M.; Lejawa, M.; Kamieńska, N.; Krejca, M.; Kolanowska, A.; Boncel, S.; Radomski, M.W. Stabilization of Graphene Oxide Dispersion in Plasma-like Isotonic Solution Containing Aggregating Concentrations of Bivalent Cations. Pharmaceutics 2023, 15, 2495. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15102495
Krasoń MZ, Paradowska A, Fronczek M, Lejawa M, Kamieńska N, Krejca M, Kolanowska A, Boncel S, Radomski MW. Stabilization of Graphene Oxide Dispersion in Plasma-like Isotonic Solution Containing Aggregating Concentrations of Bivalent Cations. Pharmaceutics. 2023; 15(10):2495. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15102495
Chicago/Turabian StyleKrasoń, Marcin Z., Anna Paradowska, Martyna Fronczek, Mateusz Lejawa, Natalia Kamieńska, Michał Krejca, Anna Kolanowska, Sławomir Boncel, and Marek W. Radomski. 2023. "Stabilization of Graphene Oxide Dispersion in Plasma-like Isotonic Solution Containing Aggregating Concentrations of Bivalent Cations" Pharmaceutics 15, no. 10: 2495. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15102495