Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration

Abstract
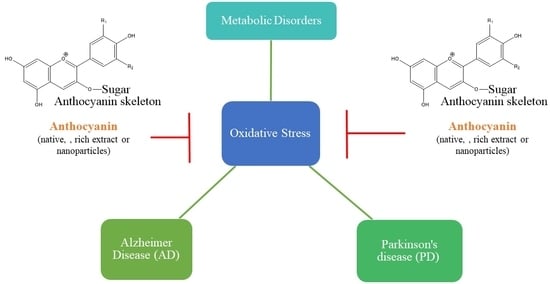
:
1. Introduction
1.1. Metabolic Syndrome and Oxidative Stress
1.2. Reactive Oxygen Species (ROS): Sources and Free Radical-Scavenging Mechanisms
2. Oxidative Stress and Human Disorders
3. Oxidative Stress and Brain
3.1. Oxidative Stress and Alzheimer’s disease (AD)
3.1.1. Sources of Oxidative Stress in AD
Mitochondrial Abnormalities
Glial Cell Activation
Advanced Glycation End Products
Redox-Active Metals: Iron, Copper, and Zinc
Amyloid beta (Aβ) Deposition
Alterations in Cell Signaling Pathways
3.2. Oxidative Stress and Parkinson’s Disease
3.2.1. Sources of Oxidative Stress in PD
Dopamine Metabolism
Mitochondrial Dysfunction
Neuroinflammation
4. Medications for AD and PD
4.1. Synthetic Approaches (Drugs)
4.2. Natural Approaches (Antioxidants)
5. Anthocyanins: A Natural Antioxidant
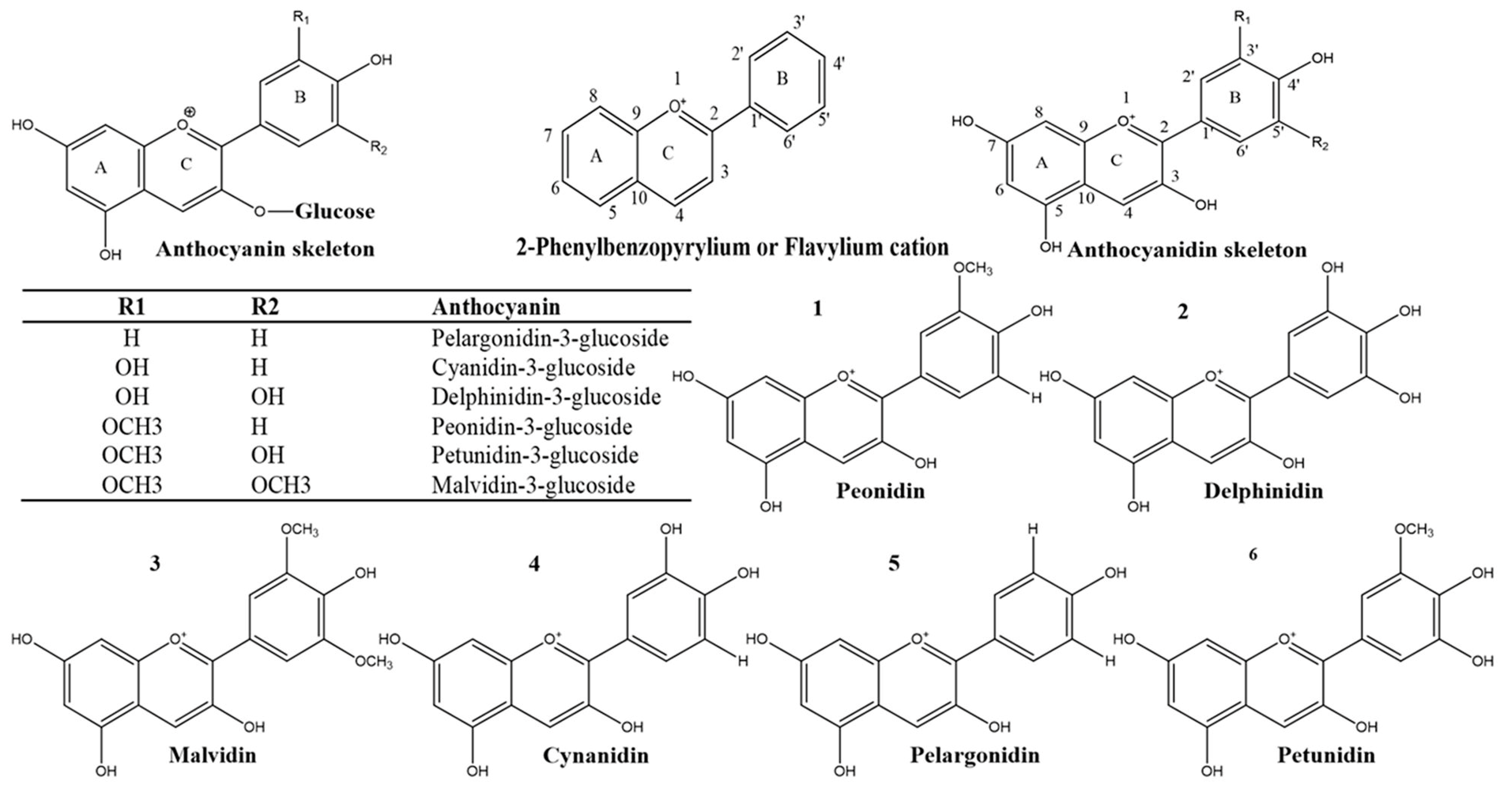
5.1. Chemistry, Structure-Activity Relationship and Mode of Action
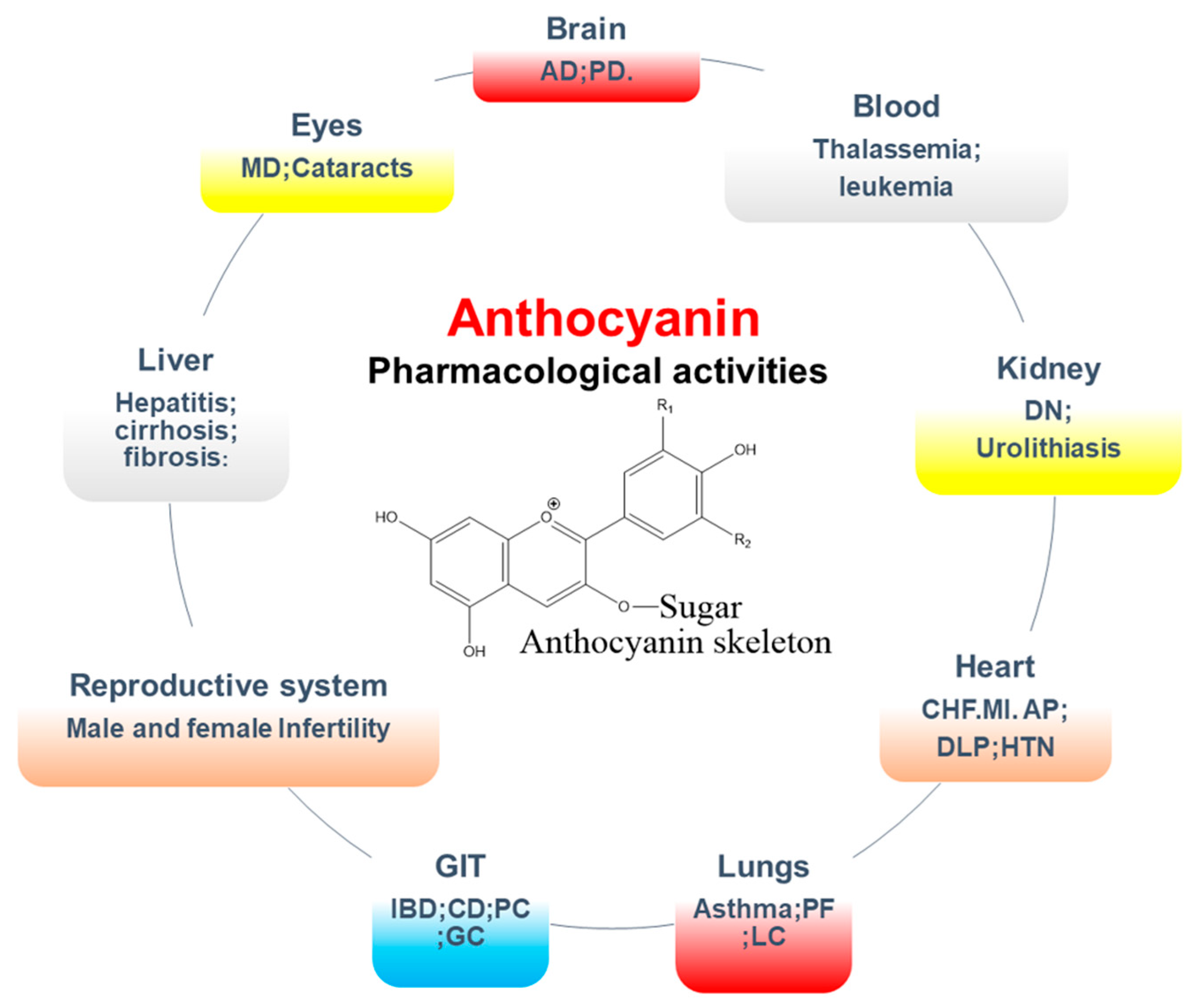
5.2. Pharmacological Activities of Anthocyanins
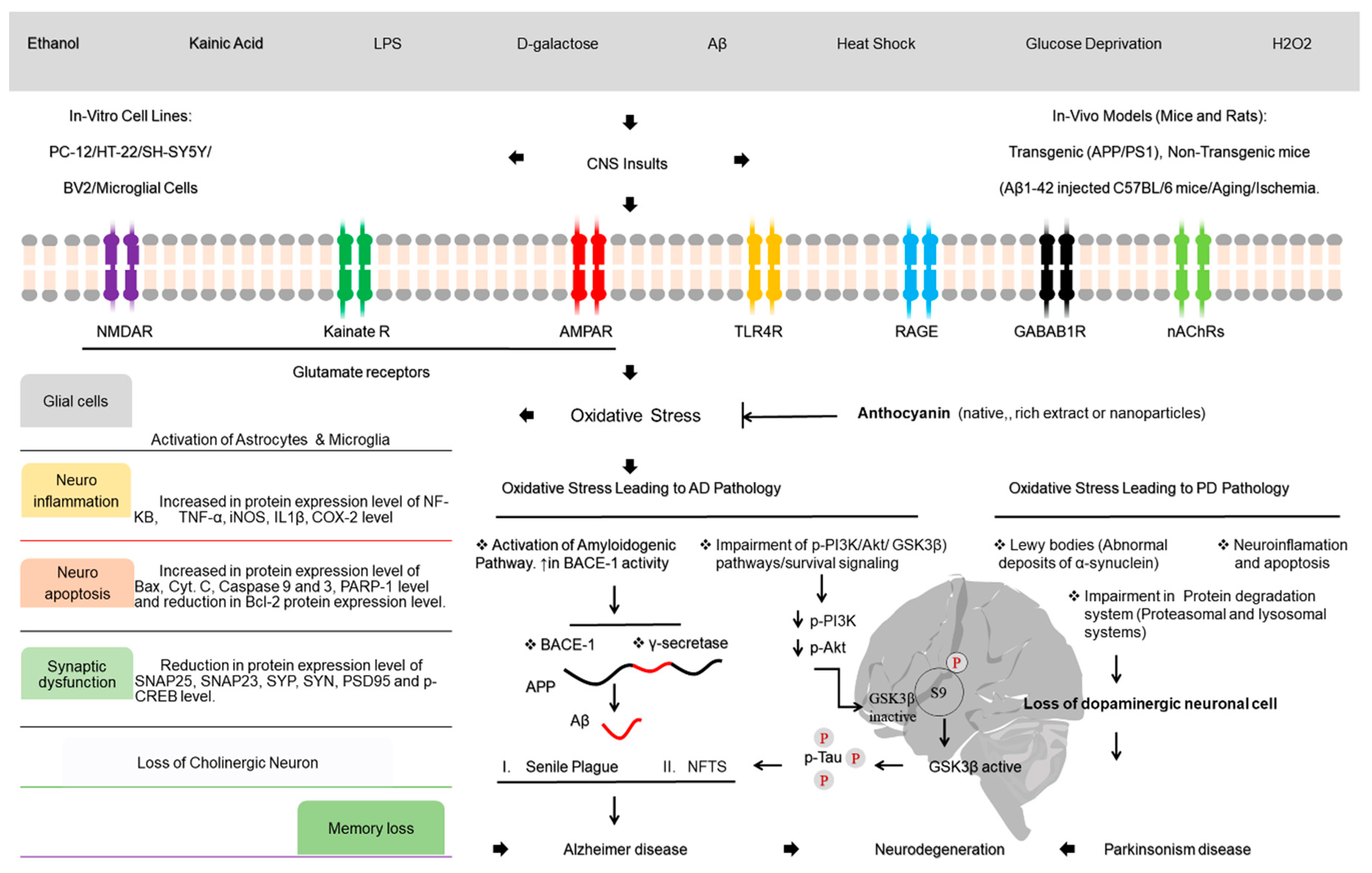
6. Anthocyanins: A Neuroprotective Agent
Nanoparticle Approach of Anthocyanins in Neuroprotection
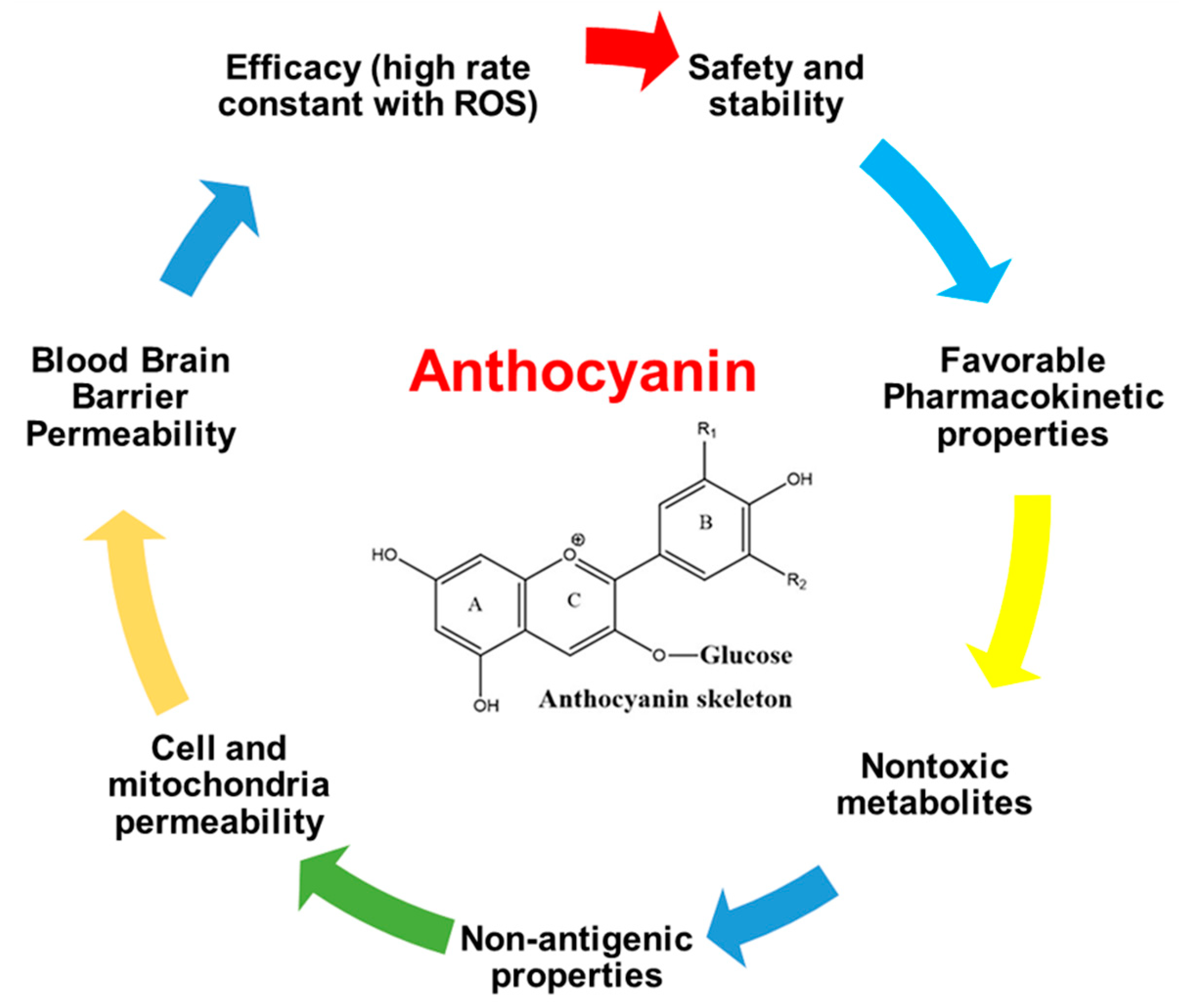
7. Safety and Toxicological Aspects of Anthocyanins
8. Anthocyanins as Mitochondrial Drug-Like Antioxidants and Their Possible Combinations
9. Antioxidants Under Clinical Trials
10. Conclusion and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yubero-Serrano, E.M.; Delgado-Lista, J.; Pena-Orihuela, P.; Perez-Martinez, P.; Fuentes, F.; Marin, C.; Tunez, I.; Tinahones, F.J.; Perez-Jimenez, F.; Roche, H.M.; et al. Oxidative stress is associated with the number of components of metabolic syndrome: LIPGENE study. Exp. Mol. Med. 2013, 45, e28. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Expert Panel on Detection, Evaluation; Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; The IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef]
- Yadav, U.C.; Rani, V.; Deep, G.; Singh, R.K.; Palle, K. Oxidative Stress in Metabolic Disorders: Pathogenesis, Prevention, and Therapeutics. Oxid. Med. Cell. Longev. 2016, 2016, 9137629. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Agarwal, A.; Hamada, A.; Esteves, S.C. Insight into oxidative stress in varicocele-associated male infertility: Part 1. Nat. Rev. Urol. 2012, 9, 678–690. [Google Scholar] [CrossRef]
- Poyton, R.O.; Castello, P.R.; Ball, K.A.; Woo, D.K.; Pan, N. Mitochondria and hypoxic signaling: A new view. Ann. N. Y. Acad. Sci. 2009, 1177, 48–56. [Google Scholar] [CrossRef]
- Bolton, J.L.; Trush, M.A.; Penning, T.M.; Dryhurst, G.; Monks, T.J. Role of quinones in toxicology. Chem. Res. Toxicol. 2000, 13, 135–160. [Google Scholar] [CrossRef]
- Yildirim, A.; Mavi, A.; Oktay, M.; Kara, A.A.; Algur, O.F.; Bilaloglu, V. Comparison of antioxidant and antimicrobial activities of tilia (Tilia argentea Desf ex DC), sage (Salvia triloba L.), and black tea (Camellia sinensis) extracts. J. Agric. Food Chem. 2000, 48, 5030–5034. [Google Scholar] [CrossRef]
- Yu, B.P. Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 1994, 74, 139–162. [Google Scholar] [CrossRef]
- Chopra, S.; Wallace, H.M. Induction of spermidine/spermine N1-acetyltransferase in human cancer cells in response to increased production of reactive oxygen species. Biochem. Pharmacol. 1998, 55, 1119–1123. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. The definition and measurement of antioxidants in biological systems. Free Radic. Biol. Med. 1995, 18, 125–126. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Lehoux, S. Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 2006, 71, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [Green Version]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Joint Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef]
- Babizhayev, M.A.; Yegorov, Y.E. Reactive Oxygen Species and the Aging Eye: Specific Role of Metabolically Active Mitochondria in Maintaining Lens Function and in the Initiation of the Oxidation-Induced Maturity Onset Cataract--A Novel Platform of Mitochondria-Targeted Antioxidants With Broad Therapeutic Potential for Redox Regulation and Detoxification of Oxidants in Eye Diseases. Am. J. Ther. 2016, 23, e98–e117. [Google Scholar] [CrossRef]
- Rosanna, D.P.; Salvatore, C. Reactive oxygen species, inflammation, and lung diseases. Curr. Pharm. Des. 2012, 18, 3889–3900. [Google Scholar] [CrossRef]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Leung, P.S.; Chan, Y.C. Role of oxidative stress in pancreatic inflammation. Antioxid. Redox Signal. 2009, 11, 135–165. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Gupta, S.; Sharma, R.K. Role of oxidative stress in female reproduction. Reprod. Biol. Endocrinol. 2005, 3, 28. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 2005, 75, 207–246. [Google Scholar] [CrossRef]
- Kell, D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 2010, 84, 825–889. [Google Scholar] [CrossRef]
- Moreira, P.I.; Smith, M.A.; Zhu, X.; Nunomura, A.; Castellani, R.J.; Perry, G. Oxidative stress and neurodegeneration. Ann. N. Y. Acad. Sci. 2005, 1043, 545–552. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef]
- Benzi, G.; Moretti, A. Are reactive oxygen species involved in Alzheimer’s disease? Neurobiol. Aging 1995, 16, 661–674. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox. Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [Green Version]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53 (Suppl. 1), S96–S102. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. (Vienna) 1998, 105, 855–870. [Google Scholar] [CrossRef]
- Onyango, I.G.; Khan, S.M. Oxidative stress, mitochondrial dysfunction, and stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2006, 3, 339–349. [Google Scholar] [CrossRef]
- De la Torre, J.C. Cerebromicrovascular pathology in Alzheimer’s disease compared to normal aging. Gerontology 1997, 43, 26–43. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Streck, E.L.; Matte, C.; Vieira, P.S.; Calcagnotto, T.; Wannmacher, C.M.; Wajner, M.; Wyse, A.T. Impairment of energy metabolism in hippocampus of rats subjected to chemically-induced hyperhomocysteinemia. Biochim. Biophys. Acta 2003, 1637, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; ran Ma, T.; Miranda, R.D.; Balestra, M.E.; Mahley, R.W.; Huang, Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18694–18699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikelenboom, P.; Veerhuis, R. The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 1996, 17, 673–680. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Van Muiswinkel, F.L.; Veerhuis, R.; Eikelenboom, P. Amyloid beta protein primes cultured rat microglial cells for an enhanced phorbol 12-myristate 13-acetate-induced respiratory burst activity. J. Neurochem. 1996, 66, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; McGeer, P.L. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J. Neurosci. Res. 1997, 49, 229–235. [Google Scholar] [CrossRef]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Luth, H.J.; Munch, G.; Arendt, T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002, 953, 135–143. [Google Scholar] [CrossRef]
- Luth, H.J.; Holzer, M.; Gartner, U.; Staufenbiel, M.; Arendt, T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res. 2001, 913, 57–67. [Google Scholar] [CrossRef]
- Reynolds, W.F.; Rhees, J.; Maciejewski, D.; Paladino, T.; Sieburg, H.; Maki, R.A.; Masliah, E. Myeloperoxidase polymorphism is associated with gender specific risk for Alzheimer’s disease. Exp. Neurol. 1999, 155, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Schmitt, D.; Hoff, H.F.; Hazen, S.L. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J. Clin. Investig. 1999, 103, 1547–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.M.; Requena, J.R.; Crowley, J.R.; Thorpe, S.R.; Heinecke, J.W. The myeloperoxidase system of human phagocytes generates Nepsilon-(carboxymethyl)lysine on proteins: A mechanism for producing advanced glycation end products at sites of inflammation. J. Clin. Investig. 1999, 104, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Munch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A.; et al. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Hartshorn, M.A.; Multhaup, G.; Goldstein, L.E.; Scarpa, R.C.; Cuajungco, M.P.; Gray, D.N.; Lim, J.; Moir, R.D.; et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 1999, 38, 7609–7616. [Google Scholar] [CrossRef]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar] [CrossRef] [PubMed]
- Su, X.Y.; Wu, W.H.; Huang, Z.P.; Hu, J.; Lei, P.; Yu, C.H.; Zhao, Y.F.; Li, Y.M. Hydrogen peroxide can be generated by tau in the presence of Cu(II). Biochem. Biophys. Res. Commun. 2007, 358, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.J.; Bush, A.I.; Masters, C.L.; Cappai, R.; Li, Q.X. Metals and amyloid-beta in Alzheimer’s disease. Int. J. Exp. Pathol. 2005, 86, 147–159. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H., Jr.; de Paradis, M.; Tanzi, R.E.; Wasco, W. The amyloid beta-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J. Biol. Chem. 1994, 269, 26618–26621. [Google Scholar] [PubMed]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009, 284, 34648–34657. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin rescue oxidative stress-mediated neuroinflammation/neurodegeneration and memory impairment in scopolamine-induced amnesia mice model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: Implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic. Res. 2002, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar] [CrossRef]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef] [PubMed]
- Carriedo, S.G.; Sensi, S.L.; Yin, H.Z.; Weiss, J.H. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 240–250. [Google Scholar] [CrossRef]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflamm. 2016, 13, 286. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.L.; Ferreira, A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J. Biol. Chem. 2006, 281, 28079–28089. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Bozyczko-Coyne, D.; O’Kane, T.M.; Wu, Z.L.; Dobrzanski, P.; Murthy, S.; Vaught, J.L.; Scott, R.W. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J. Neurochem. 2001, 77, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [CrossRef] [PubMed]
- Troy, C.M.; Rabacchi, S.A.; Xu, Z.; Maroney, A.C.; Connors, T.J.; Shelanski, M.L.; Greene, L.A. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001, 77, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.; Wiggert, B.; Petersen, R.B.; Perry, G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1994, 145, 42–47. [Google Scholar] [PubMed]
- Pappolla, M.A.; Omar, R.A.; Kim, K.S.; Robakis, N.K. Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer’s disease. Am. J. Pathol. 1992, 140, 621–628. [Google Scholar] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural Transm. (Vienna) 2005, 112, 455–469. [Google Scholar] [CrossRef]
- Tamagno, E.; Parola, M.; Bardini, P.; Piccini, A.; Borghi, R.; Guglielmotto, M.; Santoro, G.; Davit, A.; Danni, O.; Smith, M.A.; et al. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J. Neurochem. 2005, 92, 628–636. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J. Neural Transm. (Vienna) 2001, 108, 1397–1415. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; Bisaglia, M.; Lazzarini, E.; Tabares, L.C.; Beltramini, M.; Bubacco, L. Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: Possible implications for Parkinson’s disease. PLoS ONE 2012, 7, e38026. [Google Scholar] [CrossRef] [PubMed]
- Girotto, S.; Sturlese, M.; Bellanda, M.; Tessari, I.; Cappellini, R.; Bisaglia, M.; Bubacco, L.; Mammi, S. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J. Biol. Chem. 2012, 287, 18738–18749. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.M.; Arthur, R.E., Jr.; Thomas, D.M.; Elferink, L.A. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: Possible relevance to Parkinson’s disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Gluck, M.R.; Zeevalk, G.D. Inhibition of brain mitochondrial respiration by dopamine and its metabolites: Implications for Parkinson’s disease and catecholamine-associated diseases. J. Neurochem. 2004, 91, 788–795. [Google Scholar] [CrossRef]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Shamoto-Nagai, M.; Maruyama, W.; Yi, H.; Akao, Y.; Tribl, F.; Gerlach, M.; Osawa, T.; Riederer, P.; Naoi, M. Neuromelanin induces oxidative stress in mitochondria through release of iron: Mechanism behind the inhibition of 26S proteasome. J. Neural Transm. (Vienna) 2006, 113, 633–644. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Cannon, J.R.; Drolet, R.; Mastroberardino, P.G. Lessons from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 2010, 31, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Sone, N.; Saitoh, T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 1987, 48, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.L.; Hao, R.; Chen, L.F.; Lai, C.H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and alpha-Synuclein on Stress-induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef] [PubMed]
- Van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin Mitigates MPTP-Induced Loss of Nigrostriatal Dopaminergic Neurons and Accumulation of alpha-Synuclein via the Nrf2/HO-1 Pathway. Mol. Neurobiol. 2019, 56, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Fernandez Barreiro, A.; Poza, M.; Herrero, M.T. Parkinson’s disease and inflammatory changes. Neurotox. Res. 2003, 5, 411–418. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2019, 56, 2774–2790. [Google Scholar] [CrossRef]
- Beach, T.G.; Sue, L.I.; Walker, D.G.; Lue, L.F.; Connor, D.J.; Caviness, J.N.; Sabbagh, M.N.; Adler, C.H. Marked microglial reaction in normal aging human substantia nigra: Correlation with extraneuronal neuromelanin pigment deposits. Acta Neuropathol. 2007, 114, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Meek, P.D.; McKeithan, K.; Schumock, G.T. Economic considerations in Alzheimer’s disease. Pharmacotherapy 1998, 18, 68–73; discussion 79–82. [Google Scholar] [PubMed]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; Giusti, M.M. Anthocyanins in Health and Disease; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Upadhyay, P.; Panjwani, D.; Yadav, A.K. Neuropathology staging and treatment strategies of Alzheimer’s disease: An update. Int. J. Nutr. Pharmacol. Neurol. Dis. 2014, 4, 28. [Google Scholar] [CrossRef]
- DeLoach, T.; Beall, J. Diuretics: A possible keystone in upholding cognitive health. Ment. Health Clin. 2018, 8, 33–40. [Google Scholar] [CrossRef]
- Yasar, S.; Xia, J.; Yao, W.; Furberg, C.D.; Xue, Q.L.; Mercado, C.I.; Fitzpatrick, A.L.; Fried, L.P.; Kawas, C.H.; Sink, K.M.; et al. Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 2013, 81, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, J.M. Evidence to Consider Angiotensin II Receptor Blockers for the Treatment of Early Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 259–279. [Google Scholar] [CrossRef]
- Kehoe, P.G. The Coming of Age of the Angiotensin Hypothesis in Alzheimer’s Disease: Progress Toward Disease Prevention and Treatment? J. Alzheimers Dis. 2018, 62, 1443–1466. [Google Scholar] [CrossRef]
- Goodison, W.V.; Frisardi, V.; Kehoe, P.G. Calcium channel blockers and Alzheimer’s disease: Potential relevance in treatment strategies of metabolic syndrome. J. Alzheimers Dis. 2012, 30 (Suppl. 2), S269–S282. [Google Scholar] [CrossRef]
- Casey, J. Potassium-sparing diuretics might reduce risk of Alzheimer’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 293. [Google Scholar] [CrossRef]
- Nivsarkar, M.; Banerjee, A.; Padh, H. Cyclooxygenase inhibitors: A novel direction for Alzheimer’s management. Pharmacol. Rep. 2008, 60, 692–698. [Google Scholar]
- Fleisher, A.S.; Raman, R.; Siemers, E.R.; Becerra, L.; Clark, C.M.; Dean, R.A.; Farlow, M.R.; Galvin, J.E.; Peskind, E.R.; Quinn, J.F.; et al. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch. Neurol. 2008, 65, 1031–1038. [Google Scholar] [CrossRef]
- Vassar, R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 89. [Google Scholar] [CrossRef]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 2013, 154, 375–387. [Google Scholar] [CrossRef]
- Donner, T. Insulin—Pharmacology, Therapeutic Regimens and Principles of Intensive Insulin Therapy. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: Bristol, MA, USA, 2000. [Google Scholar]
- Tobinick, E.L.; Gross, H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J. Neuroinflamm. 2008, 5, 2. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Yan, H.; Tang, X.C. Non-cholinergic effects of huperzine A: Beyond inhibition of acetylcholinesterase. Cell. Mol. Neurobiol. 2008, 28, 173–183. [Google Scholar] [CrossRef]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef]
- Kulkarni, S.K.; Dhir, A. Withania somnifera: An Indian ginseng. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Smith, J.V.; Paramasivam, V.; Burdick, A.; Curry, K.J.; Buford, J.P.; Khan, I.; Netzer, W.J.; Xu, H.; Butko, P. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc. Natl. Acad. Sci. USA 2002, 99, 12197–12202. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Eckman, E.A.; Eckman, C.B. Reductions in levels of the Alzheimer’s amyloid beta peptide after oral administration of ginsenosides. FASEB J. 2006, 20, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Pandi-Perumal, S.R.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin in Alzheimer’s disease and other neurodegenerative disorders. Behav. Brain Funct. 2006, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Janicki, S.C.; Schupf, N. Hormonal influences on cognition and risk for Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Vrieling, A.; Buck, K.; Kaaks, R.; Chang-Claude, J. Adult weight gain in relation to breast cancer risk by estrogen and progesterone receptor status: A meta-analysis. Breast Cancer Res. Treat. 2010, 123, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, J.G.; Quitkin, F.M.; McGrath, P.; Harrison, W.; Tricamo, E. Adverse reactions to monoamine oxidase inhibitors. Part II. Treatment correlates and clinical management. J. Clin. Psychopharmacol. 1985, 5, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Pahan, K. Lipid-lowering drugs. Cell Mol. Life Sci. 2006, 63, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Maji, D.; Shaikh, S.; Solanki, D.; Gaurav, K. Safety of statins. Indian J. Endocrinol. Metab. 2013, 17, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J. Dopamine agonists: Their role in the treatment of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2000, 68, 685–689. [Google Scholar] [CrossRef]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef]
- Bonifacio, M.J.; Palma, P.N.; Almeida, L.; Soares-da-Silva, P. Catechol-O-methyltransferase and its inhibitors in Parkinson’s disease. CNS Drug Rev. 2007, 13, 352–379. [Google Scholar] [CrossRef]
- Olanow, C.W. Tolcapone and hepatotoxic effects. Tasmar Advisory Panel. Arch. Neurol 2000, 57, 263–267. [Google Scholar] [CrossRef]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural insight into Parkinson’s disease treatment from drug-inhibited DOPA decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S287–S296. [Google Scholar] [CrossRef]
- Brocks, D.R. Anticholinergic drugs used in Parkinson’s disease: An overlooked class of drugs from a pharmacokinetic perspective. J. Pharm. Pharm. Sci. 1999, 2, 39–46. [Google Scholar]
- Mizoguchi, K.; Yokoo, H.; Yoshida, M.; Tanaka, T.; Tanaka, M. Amantadine increases the extracellular dopamine levels in the striatum by re-uptake inhibition and by N-methyl-D-aspartate antagonism. Brain Res. 1994, 662, 255–258. [Google Scholar] [CrossRef]
- Ratheesh, G.; Tian, L.; Venugopal, J.R.; Ezhilarasu, H.; Sadiq, A.; Fan, T.P.; Ramakrishna, S. Role of medicinal plants in neurodegenerative diseases. Biomanuf. Rev. 2017, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Obrenovich, M.E.; Nair, N.G.; Beyaz, A.; Aliev, G.; Reddy, V.P. The role of polyphenolic antioxidants in health, disease, and aging. Rejuvenat. Res. 2010, 13, 631–643. [Google Scholar] [CrossRef]
- Darvesh, A.S.; Carroll, R.T.; Bishayee, A.; Geldenhuys, W.J.; Van der Schyf, C.J. Oxidative stress and Alzheimer’s disease: Dietary polyphenols as potential therapeutic agents. Expert Rev. Neurother. 2010, 10, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Smeriglio, A.; Barreca, D.; Bellocco, E.; Trombetta, D. Chemistry, Pharmacology and Health Benefits of Anthocyanins. Phytother. Res. 2016, 30, 1265–1286. [Google Scholar] [CrossRef] [PubMed]
- Mazza, G.J. Anthocyanins and heart health. Annali-Istituto Superiore Di Sanita 2007, 43, 369–374. [Google Scholar]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Zia-Ul-Haq, M.; Saad, B. Anthocyanins and Human Health: Biomolecular and Therapeutic Aspects, Introduction to Anthocyanins; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Miguel, M.G. Anthocyanins: Antioxidant and/or anti-inflammatory activities. JAPS 2011, 1, 7–15. [Google Scholar]
- Zhu, F.; Cai, Y.Z.; Yang, X.; Ke, J.; Corke, H. Anthocyanins, hydroxycinnamic acid derivatives, and antioxidant activity in roots of different chinese purple-fleshed sweetpotato genotypes. J. Agric. Food Chem. 2010, 58, 7588–7596. [Google Scholar] [CrossRef]
- Zafra-Stone, S.; Yasmin, T.; Bagchi, M.; Chatterjee, A.; Vinson, J.A.; Bagchi, D. Berry anthocyanins as novel antioxidants in human health and disease prevention. Mol. Nutr. Food Res. 2007, 51, 675–683. [Google Scholar] [CrossRef]
- Jeong, C.-H.; Jang, C.-W.; Kum, D.-C.; Yul Lee, K.; Yuan Lee, S.; Jin Hur, S.; Lee, S.-J. Protective effects of berry extracts on hydrogen peroxide-induced rat brain neuronal cell damage in vitro. J. Food Nutr. Res. 2014, 2, 277–280. [Google Scholar] [CrossRef]
- Hwang, J.-W.; Kim, E.-K.; Lee, S.-J.; Kim, Y.-S.; Moon, S.-H.; Jeon, B.-T.; Sung, S.-H.; Kim, E.-T.; Park, P.-J. Antioxidant activity and protective effect of anthocyanin oligomers on H2O2-triggered G2/M arrest in retinal cells. J. Agric. Food chem. 2012, 60, 4282–4288. [Google Scholar] [CrossRef]
- Kelsey, N.; Hulick, W.; Winter, A.; Ross, E.; Linseman, D. Neuroprotective effects of anthocyanins on apoptosis induced by mitochondrial oxidative stress. Nutr. Neurosci. 2011, 14, 249–259. [Google Scholar] [CrossRef]
- Lu, J.; Wu, D.M.; Zheng, Y.L.; Hu, B.; Zhang, Z.F. Purple sweet potato color alleviates d-galactose-induced brain aging in old mice by promoting survival of neurons via PI3K pathway and inhibiting cytochrome c-mediated apoptosis. Brain Pathol. 2010, 20, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Toufektsian, M.-C.; De Lorgeril, M.; Nagy, N.; Salen, P.; Donati, M.B.; Giordano, L.; Mock, H.-P.; Peterek, S.; Matros, A.; Petroni, K. Chronic dietary intake of plant-derived anthocyanins protects the rat heart against ischemia-reperfusion injury. J. Nutr. 2008, 138, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.H.; Yeh, C.T.; Yen, G.C. Anthocyanins induce the activation of phase II enzymes through the antioxidant response element pathway against oxidative stress-induced apoptosis. J. Agric. Food Chem. 2007, 55, 9427–9435. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Choi, J.H.; Yun, H.J.; Han, E.H.; Kim, H.G.; Kim, J.Y.; Park, B.H.; Khanal, T.; Choi, J.M.; Chung, Y.C.; et al. Anthocyanins from purple sweet potato attenuate dimethylnitrosamine-induced liver injury in rats by inducing Nrf2-mediated antioxidant enzymes and reducing COX-2 and iNOS expression. Food Chem. Toxicol. 2011, 49, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Steffen, Y.; Gruber, C.; Schewe, T.; Sies, H. Mono-O-methylated flavanols and other flavonoids as inhibitors of endothelial NADPH oxidase. Arch. Biochem. Biophys. 2008, 469, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P. Factors influencing the measurement of bioavailability, taking calcium as a model. J. Nutr. 2001, 131, 1344s–1348s. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.D.; Pereira-Caro, G.; Ludwig, I.A.; Clifford, M.N.; Crozier, A. Anthocyanins and Flavanones Are More Bioavailable than Previously Perceived: A Review of Recent Evidence. Ann. Rev. Food Sci. Technol. 2017, 8, 155–180. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Ge, J.; Yue, P.; Yue, X.; Fu, R.; Liang, J.; Gao, X. Loading of anthocyanins on chitosan nanoparticles influences anthocyanin degradation in gastrointestinal fluids and stability in a beverage. Food Chem. 2017, 221, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.U.; Shah, S.A.; Badshah, H.; Khan, M.; Kim, M.O. Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Abeta1-42-induced oxidative stress. J. Nanobiotechnol. 2017, 15, 12. [Google Scholar] [CrossRef]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Abeta1-42-induced neuroinflammation and neurodegeneration via the NF-KB /JNK/GSK3beta signaling pathway. Nanomedicine 2017, 13, 2533–2544. [Google Scholar] [CrossRef]
- He, J.; Giusti, M.M. Anthocyanins: Natural colorants with health-promoting properties. Ann. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; Tan, C.; Selig, M.J. Anthocyanidins and Anthocyanins. Food Sci. 2019, 218–223. [Google Scholar] [CrossRef]
- Bartikova, H.; Skalova, L.; Drsata, J.; Bousova, I. Interaction of anthocyanins with drug-metabolizing and antioxidant enzymes. Curr. Med. Chem. 2013, 20, 4665–4679. [Google Scholar] [CrossRef] [PubMed]
- Shukitt-Hale, B.; Kalt, W.; Carey, A.N.; Vinqvist-Tymchuk, M.; McDonald, J.; Joseph, J.A. Plum juice, but not dried plum powder, is effective in mitigating cognitive deficits in aged rats. Nutrition 2009, 25, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T. Dietary anthocyanin-rich plants: Biochemical basis and recent progress in health benefits studies. Mol. Nutr. Food Res. 2012, 56, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Shah, S.A.; Kim, M.O. 17beta-Estradiol via SIRT1/Acetyl-p53/NF-kB Signaling Pathway Rescued Postnatal Rat Brain Against Acute Ethanol Intoxication. Mol. Neurobiol. 2018, 55, 3067–3078. [Google Scholar] [CrossRef]
- Rahman, M.M.; Ichiyanagi, T.; Komiyama, T.; Sato, S.; Konishi, T. Effects of anthocyanins on psychological stress-induced oxidative stress and neurotransmitter status. J. Agric. Food Chem. 2008, 56, 7545–7550. [Google Scholar] [CrossRef]
- Ye, J.; Meng, X.; Yan, C.; Wang, C. Effect of purple sweet potato anthocyanins on beta-amyloid-mediated PC-12 cells death by inhibition of oxidative stress. Neurochem. Res. 2010, 35, 357–365. [Google Scholar] [CrossRef]
- Kang, T.H.; Hur, J.Y.; Kim, H.B.; Ryu, J.H.; Kim, S.Y. Neuroprotective effects of the cyanidin-3-O-beta-d-glucopyranoside isolated from mulberry fruit against cerebral ischemia. Neurosci. Lett. 2006, 391, 122–126. [Google Scholar] [CrossRef]
- Krikorian, R.; Nash, T.A.; Shidler, M.D.; Shukitt-Hale, B.; Joseph, J.A. Concord grape juice supplementation improves memory function in older adults with mild cognitive impairment. Br. J. Nutr. 2010, 103, 730–734. [Google Scholar] [CrossRef]
- Joseph, J.A.; Denisova, N.A.; Arendash, G.; Gordon, M.; Diamond, D.; Shukitt-Hale, B.; Morgan, D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr. Neurosci. 2003, 6, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Park, H.Y.; Kim, M.O. Anthocyanins protect against kainic acid-induced excitotoxicity and apoptosis via ROS-activated AMPK pathway in hippocampal neurons. CNS Neurosci. Ther. 2014, 20, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Kim, M.O. Protection of the developing brain with anthocyanins against ethanol-induced oxidative stress and neurodegeneration. Mol. Neurobiol. 2015, 51, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins Reversed D-Galactose-Induced Oxidative Stress and Neuroinflammation Mediated Cognitive Impairment in Adult Rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Jo, M.G.; Badshah, H.; Kim, M.O. Anthocyanins protect against LPS-induced oxidative stress-mediated neuroinflammation and neurodegeneration in the adult mouse cortex. Neurochem. Int. 2016, 100, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Chung, J.I.; Kim, M.O. Anthocyanins Improve Hippocampus-Dependent Memory Function and Prevent Neurodegeneration via JNK/Akt/GSK3beta Signaling in LPS-Treated Adult Mice. Mol. Neurobiol. 2019, 56, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Leung, Y.; Wang, M.; Chang, R.C. Nutraceuticals and their preventive or potential therapeutic value in Parkinson’s disease. Nutr. Rev. 2012, 70, 373–386. [Google Scholar] [CrossRef]
- Albarracin, S.L.; Stab, B.; Casas, Z.; Sutachan, J.J.; Samudio, I.; Gonzalez, J.; Gonzalo, L.; Capani, F.; Morales, L.; Barreto, G.E. Effects of natural antioxidants in neurodegenerative disease. Nutr. Neurosci. 2012, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yan, J.; Yang, T.; Yang, X.; Bezard, E.; Zhao, B. Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson’s disease through inhibition of ROS-NO pathway. Biol. Psychiatry 2007, 62, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Zbarsky, V.; Datla, K.P.; Parkar, S.; Rai, D.K.; Aruoma, O.I.; Dexter, D.T. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Radic. Res. 2005, 39, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Yu, M.S.; Ho, Y.S.; Wang, M.; Chang, R.C. Dietary oxyresveratrol prevents parkinsonian mimetic 6-hydroxydopamine neurotoxicity. Free Radic. Biol. Med. 2008, 45, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Ahmad, A.; Ishrat, T.; Khan, M.B.; Hoda, M.N.; Khuwaja, G.; Raza, S.S.; Khan, A.; Javed, H.; Vaibhav, K.; et al. Resveratrol attenuates 6-hydroxydopamine-induced oxidative damage and dopamine depletion in rat model of Parkinson’s disease. Brain Res. 2010, 1328, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Sze, S.C.; Ng, T.B.; Lee, C.K.; Leung, G.P.; Shaw, P.C.; Tong, Y.; Zhang, Y.B. Anti-Parkinsonian drug discovery from herbal medicines: What have we got from neurotoxic models? J. Ethnopharmacol. 2012, 139, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cassidy, A.; Schwarzschild, M.A.; Rimm, E.B.; Ascherio, A. Habitual intake of dietary flavonoids and risk of Parkinson disease. Neurology 2012, 78, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Strathearn, K.E.; Yousef, G.G.; Grace, M.H.; Roy, S.L.; Tambe, M.A.; Ferruzzi, M.G.; Wu, Q.L.; Simon, J.E.; Lila, M.A.; Rochet, J.C. Neuroprotective effects of anthocyanin- and proanthocyanidin-rich extracts in cellular models of Parkinsons disease. Brain Res. 2014, 1555, 60–77. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-Loaded PEG-Gold Nanoparticles Enhanced the Neuroprotection of Anthocyanins in an Abeta1-42 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef]
- Opinion, S. Scientific Opinion on the re-evaluation of anthocyanins (E 163) as a food additive. EFSA J. 2013, 11, 3145. [Google Scholar] [CrossRef]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C.S. The Case for Anthocyanin Consumption to Promote Human Health: A Review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef]
- Niki, E.; Yamamoto, Y.; Takahashi, M.; Yamamoto, K.; Yamamoto, Y.; Komuro, E.; Miki, M.; Yasuda, H.; Mino, M. Free radical-mediated damage of blood and its inhibition by antioxidants. J. Nutr. Sci. Vitaminol. (Tokyo) 1988, 34, 507–512. [Google Scholar] [CrossRef]
- Rossetto, M.; Vanzani, P.; Mattivi, F.; Lunelli, M.; Scarpa, M.; Rigo, A. Synergistic antioxidant effect of catechin and malvidin 3-glucoside on free radical-initiated peroxidation of linoleic acid in micelles. Arch. Biochem. Biophys. 2002, 408, 239–245. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Kanowski, S.; Hoerr, R. Ginkgo biloba extract EGb 761 in dementia: Intent-to-treat analyses of a 24-week, multi-center, double-blind, placebo-controlled, randomized trial. Pharmacopsychiatry 2003, 36, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. An open trial of high-dosage antioxidants in early Parkinson’s disease. Am. J. Clin. Nutr. 1991, 53, 380s–382s. [Google Scholar] [CrossRef] [PubMed]








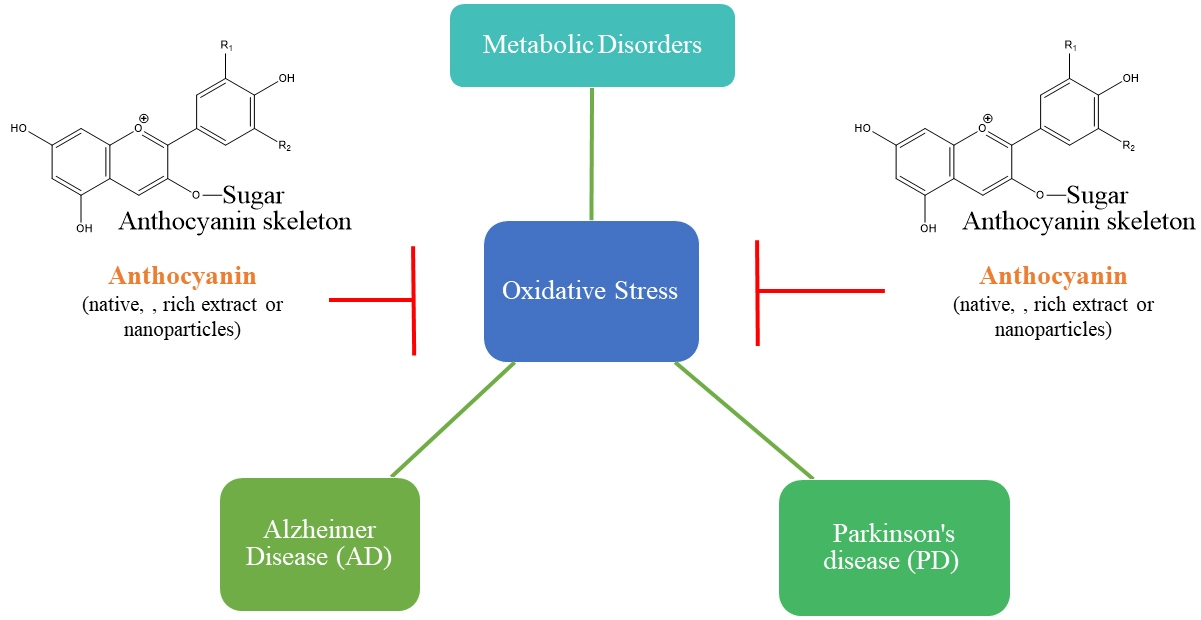{kind=link}
{kind=link}
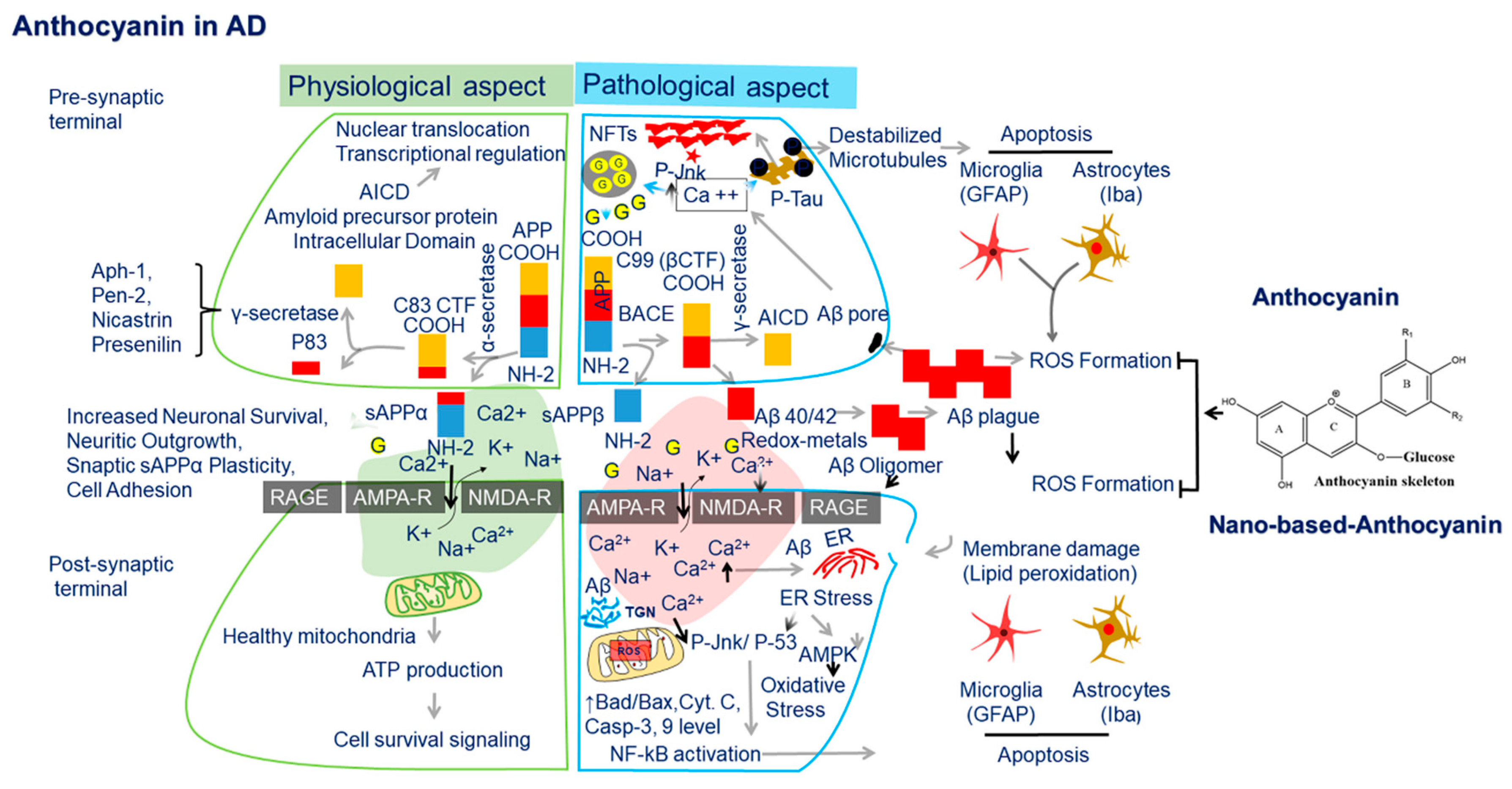{kind=link}
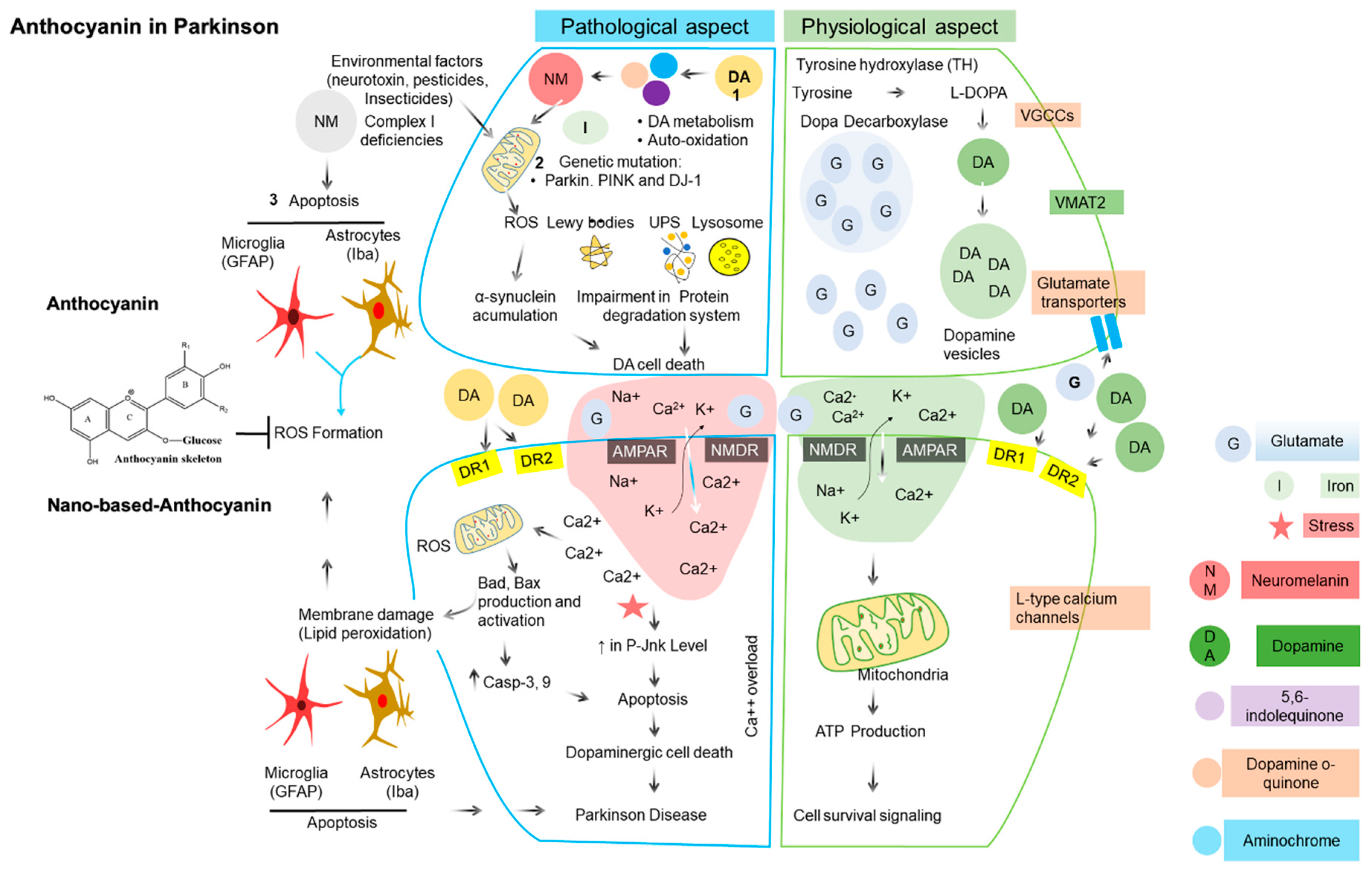{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Group | Classification of AD Drugs | Mechanism of Action | Side Effect | Ref. No. |
|---|---|---|---|---|---|
| 1 | Anticholinesterase Inhibitors (AChEI): (Currently approved Drugs for AD). | Donepezil(Aricept), Galantamine (Razadyne), Rivastigmine (Exelon), Tacrine(Cognex). | Increased Ach level by Inhibiting AchE. | Nausea, Diarrhea, Vomiting, Loss of appetite, Abdominal pain, Increased Frequency of bowel movements, Bradycardia, Hepatotoxicity (Tacrine/withdrawn from market). | [116] |
| 2 | NMDA Receptor Antagonist | Memantine(Namenda) | Increased Ca++influx (overload) thus blocking glutamatergic overstimulation. | Headache, Constipation, Confusion, and Dizziness. | [127] |
| 3 | Antihypertensive Drugs -Diuretics -Angiotensin -1 Receptor Blockers (ARB): -Angiotensin-Converting Enzyme Inhibitors (ACE-I): -Calcium Channel Blockers (CCB): Note. Dihydropyridine are more effective than nondihydropyridine agents in reducing incidence of AD). -Beta Blockers | Diuretics: A.Thiazide Diuretics: i) Thiazides: -Hydrochlorohiazie (HCTZ or HCT) (Apo-Hydro) -Benzthiazide (Dytide). ii) Thiazides like: -Chlorthalidone(Thalitone), -Metolazone (Zaroxolyn), -Indapamide (Lozol). B. Loop Diuretics: -Furosemide (Lasix), -Ethacrynic acid (Edecrin). C. Potassium-sparing Diuretics: i) Aldosterone Antagonist: -Spironolactone(Aldactone) -Eplerenone (Inspra). ii) Na+ Channel Blocker: -Amiloride (Midamor) -Triamterene (Dyrenium). D.Carbonic Anhydrase Inhibitors: -Acetazolamide (Diamox), -Dorzolamide (Trusopt) Osmotic diuresis: (Mannitol, Glycerol, Isosorbide and urea). ARB: -Candesartan (Atacand), -Valsartan (Diovan), -Losartan (Cozaar). ACE-I:(Centrally acting): -Captopril(Capoten): -Ramipril (Altace), -Lisinopril(Zestril, Prinivil), -Fosinopril (Monopril). ACE-I:(Non-Centrally acting): -Benazepril(Lotensin), -Enalapril(Vasotec), -Quinapril (Accupril). CCB: Dihydropyridine: -Amlodipine(Norvasc) -Nifedipine (Adalat). -Benzothiazepines: -Diltiazem (Cardizem LA). Phenylalkylamines: -Verapamil (Isoptin). BB: -Propranolol(Inderal), -Carvedilol(Coreg), -Nebivolol(Bystolic). | Diuretics: Decreased CSF amyloid-beta (Aβ1-42) level due to Hypokalemia associated with diuretics. ARB:- Decreased Aβ production and increased Aβ degradation. ACE-I: (Controversial) -Increased K+-mediated acetylcholine release, -Degradation of Aβ, Antioxidant potential and vascular protective effects. CCB: -Decreased intracellular calcium level by inhibiting calcium channels. -Decreased Aβ production. BB: -Decreased γ secretase activity i.e., Aß production. | Hyponatremia, dizziness, thirst, muscle cramps Hypokalemia (except potassium sparing diuretics). Dizziness, headache, drowsiness, nausea, cough (low incidence than ACEi), hyperkalemia, muscle and bone pain. ACE-I: Dry cough (due to increase in bradykinin level), hyperkalemia and fatigue. CCB: Constipation, flushing, drowsiness, edema and drowsiness. Beta Blockers: Coldness of extremities (hands and feet), dry mouth, skin and eyes, diarrhea and constipation. Others: Congestive Heart Failure, Vivid dreams, Parasthesias, Depression, Bronchial Bronchospasm, Night mares and Sexual dysfunction. | [128,129,130,131,132,133] |
| 4 | NSAIDs. (Anti-inflammatory drugs) | COX-2 Inhibitor: -Celecoxib(Celebrex) -Rofecoxib(Vioxx) -Valdecoxib (Bextra) COX-1 Inhibitor: -Ibuprofen (Brufen) -Aspirin(acetylsalicylicacid) -Naproxen(Anaprox) -Flurbiprofen (Anazin) | -Decreased neuroinflammation. | Gastrointestinal and Renal toxicity. | [134] |
| 5 | Secretase inhibitors | BACE inhibitors -Verubecestat (MK-8931), -Lanabecestat (AZD3293 or LY3314814), -Elenbecestat (E2609). Gamma secretase inhibitor: -Semagacestat (LY450139) | -Decreased Aβ production. | -Skin disorders, Small-bowel obstruction, Paranoia, and Increased agitation | [135,136] |
| 6 | Insulin | Insulin Rapid Acting: -Insulin Lispro (Humalog) -Insulin Aspart (Novolog) -Insulin Glulisine (Apidra) Short-Acting: -Regular Human (Humulin R) Intermediate-Acting: -NPH Human (Humulin N) Long Acting: -Insulin Determir (Levemir) -Insulin Glargine (Lantus) | -Glucose homeostasis -increased Aβ clearance by enhancing IDE and α-secretase activity. | -Weight gain, Hypoglycemia, Local reactions (allergic reactions), Mitogenic properties. | [137,138] |
| 7 | Cytokines | -Etanercept (Enbrel R) (TNF-α modulator) | -Potent antagonist of TNF alpha. | -Headache, Stomach pain, Weakness and Cough. | [139] |
| 8 | HuperzineA (Lycopodium alkaloid) | -HuperzineA | -Potent inhibitor of AChE -Anti-oxidant -Anti-apoptotic -Anti-inflammatory. | -Nausea, Vomiting, Sweating, Blurred vision. | [140] |
| 9 | Polyphenols | -Curcumin, Resveratrol | -Anti-inflammatory -Anti-oxidant -Aβ clearance –Chelating agent. | -Diarrhea, rash, Headache and yellow stool | [127,141] |
| 10 | Herbal supplements | -Ginkgobiloba -Panax gingseng -Withania somnifera | Ginkgobiloba: Inhibition of mitochondria dependent apoptosis OR Aβ aggregation inhibition. Panax gingseng: Reduction in Aβ peptide level. Withania Somnifera: Inhibition of AChE, reduction in of Aβ level, Antioxidant and anti-inflammatory activity. | -Nausea, Vomiting, Restlessness. | [142,143,144] |
| 11 | Hormones | -Melatonin/Estrogen (Estrace) | Melatonin: Anti-oxidant, Antiifibrillogenic and Attenuates Aβ toxicity Estrogen: Increased growth, survival and cholinergic activity, antioxidant and enhanced non-amyloidogenic metabolism of APP. | -Melatonin: -Depression, Headache, Sleepiness and irritability. -Estrogen: Weight gain and Post-menopausal breast cancer. | [145,146,147] |
| 12 | MAO-Is: (MAO-Ai and MAO-Bi) | MAO-Ai and MAO-Bi (non-selective): -Phenelzine (Nardil), -Tranylcypromine (Parnate), -Isocarboxazid (Marplan). MAO-Ai (Selective): -Clorgyline (irreversible), -Moclobemide (reversible). MAO-Bi (Selective): -Selegiline(Deprenyl) | -Accelerates nonamyloidogenic pathway. -Inhibited Aβ and tau pathophysiology. -Increased different neurotransmitters level in brain. | Significant Weight gain, Severe Orthostatic hypotension, Hypertensive Sexual Dysfunction. | [148,149] |
| 13 | Lipid-lowering drugs | Statins: -Lovastatin (Mevacor), -Simvastatin (Zocor), -Rosuvastatin (Crestor), Fibrates: -Clofibrate (Atromid-S), -Gemfibrozil (Lopid) Others: -Ezetimbe, torcetrapib, -implitapide. niacin | Statins: Inhibit HMG-CoA reductase ezyme, and thus Suppress cholesterol biosynthesis. Fibrates: Stimulate β-oxidation of fatty acids -Lowers plasma level of fatty acid and triacylglycerol. | Hepatotoxicity Carcinogenic Myopathy Nephrotoxicity, For-example, Cerivastatin (Removed from worldwide market due to serious myopathy/Rhabdomyolysis). | [150,151] |
| 14 | AD Immunotherapy | -Use of anti-Aβ protein antibodies (vaccine). | [127] |
| S. No. | Classification | Drugs | Mechanism of Action | Side Effect | Ref. No. |
|---|---|---|---|---|---|
| 1. | Dopamine agonists | Ergot derived dopamine agonists: -Bromocriptine (Parlodel) -Pergolide (Permax) -Cabergoline (Dostinex) Non-Ergot derived dopamine agonists: -Pramipexole (Mirapex) -Ropinirole (Requip) | The antiparkinsonian effects of Dopamine agonists (DA) is due to their direct activation of dopaminergic receptor (D1 and D2). | Ergot-derived-dopamine agonists: Hallucination, Delusions, orthostatic, hypotension, exacerbation of dyskinesias, skin inflammation, erythromelalgia, digital vasospasm, paraesthesias and pleural effusion or pulmonary infiltrates. Non-Ergot-derived dopamine agonists: Nausea, Hypotension, Confusion and somnolence and Exacerbation of dyskinesias. | [152,153]. |
| 2. | COMT inhibitors | Entacapone (Comtan®), Tolcapone (Tasmar®). | -Inhibition of COMT enzyme in periphery, thus reducing levodopa degradation into 3-O methyldopa and increased levodopa availability in brain where it is converted into dopamine. | Tolcapone (Tasmar®) (black box warning), Fulminant liver failure, vivid dreams, diarrhea, urine discoloration(orange), drowsiness, visualhallucinations and dyskinesia. | [154,155] |
| 3. | DOPA decarboxylase inhibitor (DDCi) | Levodopa (L-DOPA), Carbidopa (Lodosyn), Or Combination of both. | -Inhibition of DOPA decarboxylase in periphery, that caused levodopa degradation into dopamine (unable to cross BBB), and thus enable exogenously administered DOPA/levodopa to reach in brain in sufficient quantities, where it is converted to dopamine. | Motor fluctuations, Dyskinesias (“peak-dose dyskinesias”, “biphasic dyskinesias” and “wearing-off” dyskinesias | [153,156] |
| 4. | MAO-B Inhibitor | Selegiline(Eldepryl, Emsam, Zelapar)-Rasagiline (Azilect) -Safinamide (Xadago), -Tranylcypromine (Parnat, non-selective) | These drugs work by inactivating MAO enzyme that caused inactivation of dopamine neurotransmitter. | High blood pressure, nausea, constipation Headache, difficulty falling asleep, blurred vision, cheese reaction (tranylcypromine). | [157] |
| 5. | Anticholinergic drugs | Trihexyphenidyl (Artane) Benztropine (Cogentin) Procyclidine (Kemadrin) Biperiden (Akineton) | Reduction of persistent tremor or ameliorate dystonia or dyskinesias associated with DOPA decarboxylase inhibitor, or reduction in extrapyramidal side effects induced by antipsychotic agents. | Anticholinergic side effects: Dry mouth, Constipation, Urinary retention, Bowel obstruction | [158] |
| 6. | Antiviral drug | Amantadine (Symmetrel) | -Antagonists of NMDA receptor as well as dopamine re-uptake inhibitor, therefore increase extracellular dopamine levels. | Depression, anxiety, hallucination, dry mouth and constipation. | [159] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients 2019, 11, 1195. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11061195
Ullah R, Khan M, Shah SA, Saeed K, Kim MO. Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients. 2019; 11(6):1195. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11061195
Chicago/Turabian StyleUllah, Rahat, Mehtab Khan, Shahid Ali Shah, Kamran Saeed, and Myeong Ok Kim. 2019. "Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration" Nutrients 11, no. 6: 1195. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11061195