The Microbial Metabolite Trimethylamine N-Oxide Links Vascular Dysfunctions and the Autoimmune Disease Rheumatoid Arthritis


Abstract
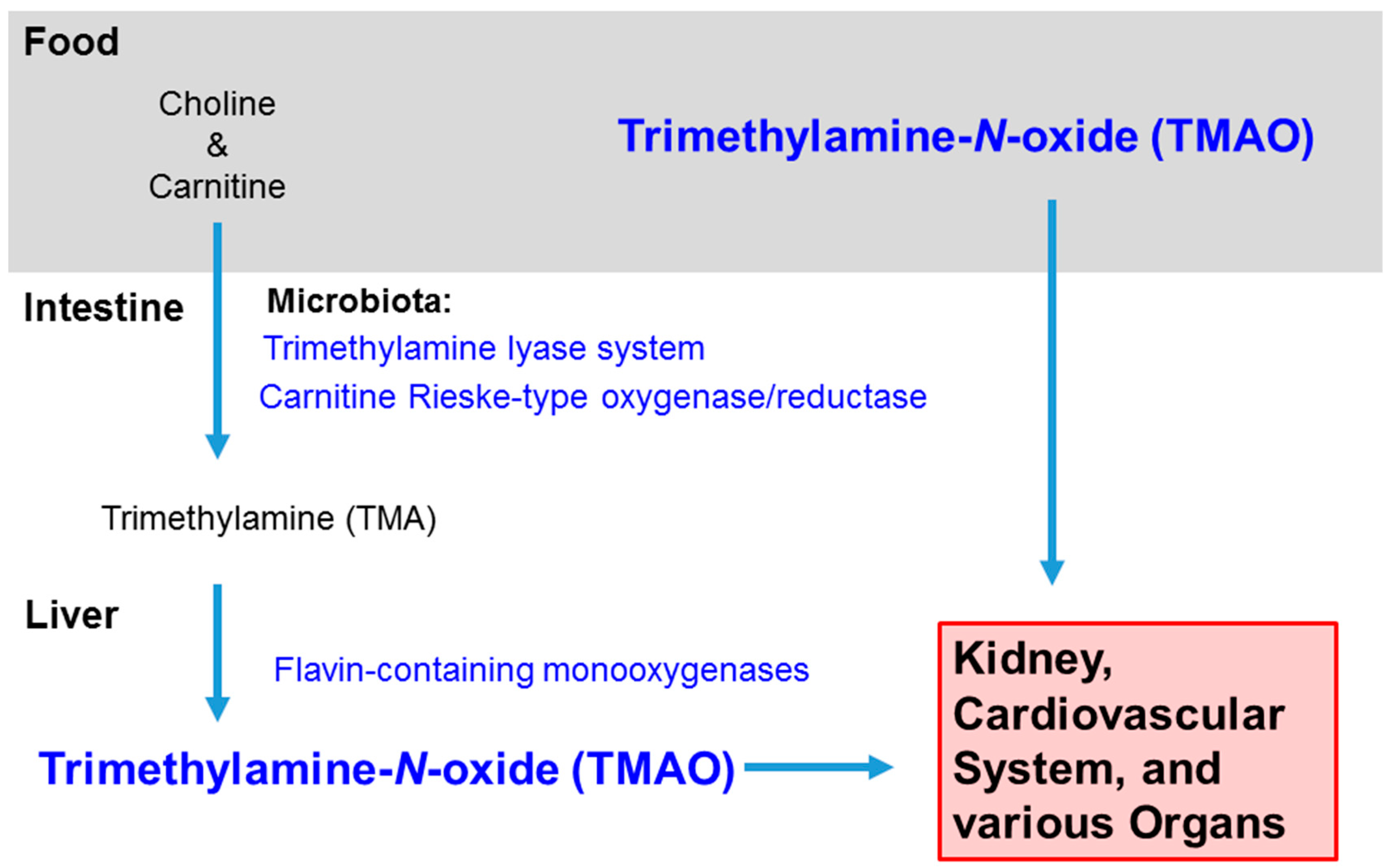
:1. Introduction
2. Microbiome Alters the Dietary Composition
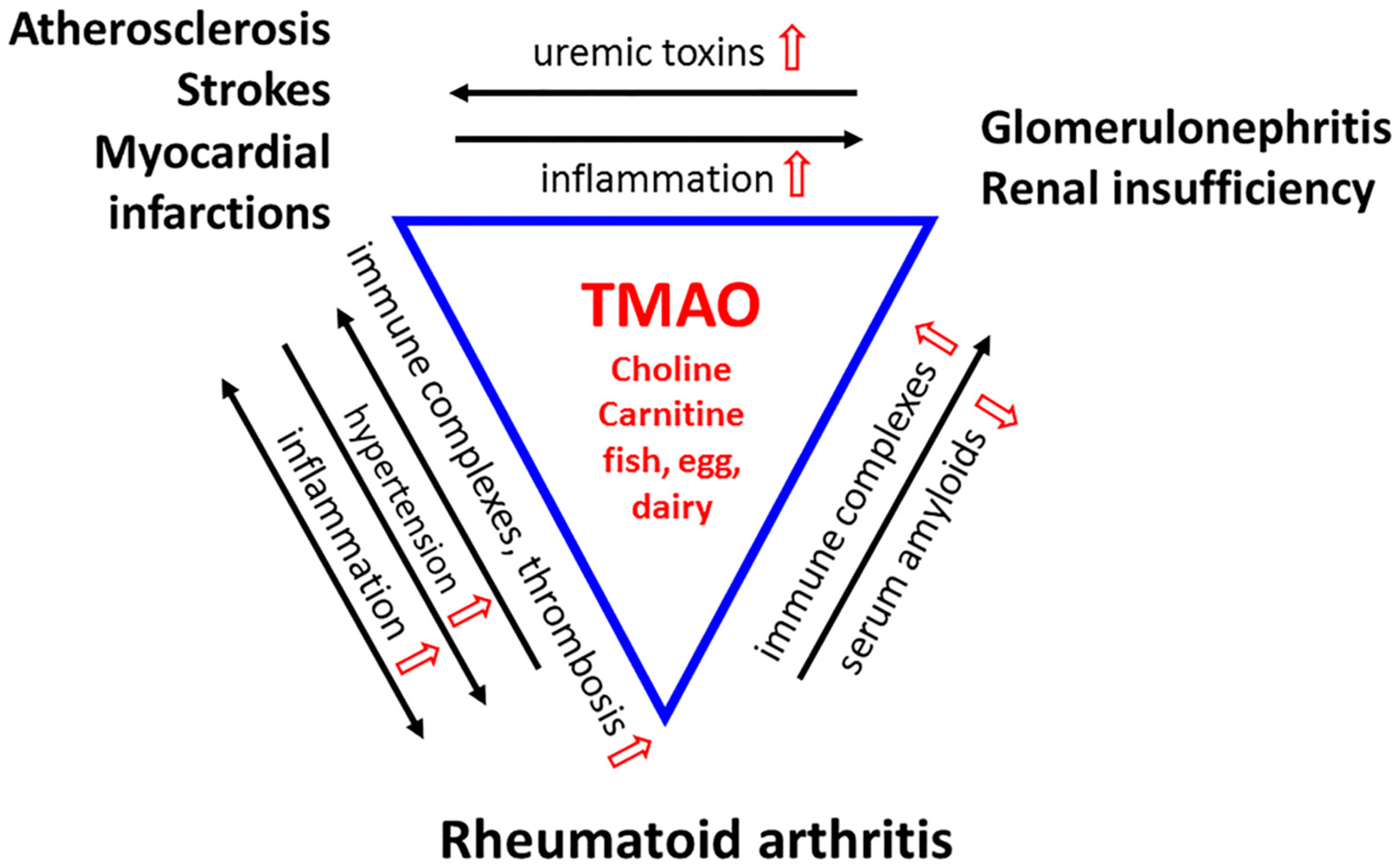
3. TMAO Promotes the Etiological Mechanisms of CVD in RA
4. Microbial Dysbiosis and TMAO Promote Rheumatoid Arthritis
5. TMAO Can Harm or Protect the Renal Function of RA Patients
6. Microbiota Remodeling Reverses TMAO-Mediated Pathologies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, P.; Flint, H.J. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Gao, X.; Liu, X.; Xu, J.; Xue, C.; Xue, Y.; Wang, Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014, 118, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients with Stroke and Is Related to Proinflammatory Monocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Bogiatzi, C.; Gloor, G.; Allen-Vercoe, E.; Reid, G.; Wong, R.G.; Urquhart, B.L.; Dinculescu, V.; Ruetz, K.N.; Velenosi, T.J.; Pignanelli, M.; et al. Metabolic products of the intestinal microbiome and extremes of atherosclerosis. Atherosclerosis 2018, 273, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Wang, Z. Cross-Talk between Gut Microbiota and Heart via the Routes of Metabolite and Immunity. Gastroenterol. Res. Pract. 2018, 2018, 6458094. [Google Scholar] [CrossRef] [PubMed]
- Zetterholm, S.G.; Verville, G.A.; Boutwell, L.; Boland, C.; Prather, J.C.; Bethea, J.; Cauley, J.; Warren, K.E.; Smith, S.A.; Magers, D.H.; et al. Noncovalent Interactions between Trimethylamine N-Oxide (TMAO), Urea, and Water. J. Phys. Chem. B 2018, 122, 8805–8811. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodriguez, E.; Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Fernandez-Fernandez, B.; Kanbay, M.; Tejedor, A.; Lazaro, A.; Ruiz-Ortega, M.; et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins 2018, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Prokopienko, A.J.; West, R.E., 3rd; Nolin, T.D.; Stubbs, J.R. Decreased Kidney Function is Associated with Enhanced Hepatic Flavin Monooxygenase Activity and Increased Circulating Trimethylamine N-Oxide Concentrations in Mice. Drug Metab. Dispos. 2018, 46, 1304–1309. [Google Scholar] [CrossRef]
- Missailidis, C.; Hallqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef]
- Moran-Ramos, S.; Lopez-Contreras, B.E.; Canizales-Quinteros, S. Gut Microbiota in Obesity and Metabolic Abnormalities: A Matter of Composition or Functionality? Arch. Med. Res. 2017, 48, 735–753. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions Between Gut Microbiota and Host Metabolism Predisposing to Obesity and Diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Sun, D.; Smith, S.R.; Bray, G.A.; Sacks, F.M.; Qi, L. Changes in Gut Microbiota–Related Metabolites and Long-term Successful Weight Loss in Response to Weight-Loss Diets: The POUNDS Lost Trial. Diabetes Care 2018, 41, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Tan, X.-Y.; Li, Q.-J.; Liao, G.-C.; Fang, A.-P.; Zhang, D.-M.; Chen, P.-Y.; Wang, X.-Y.; Luo, Y.; Long, J.-A.; et al. Trimethylamine N-oxide, a gut microbiota-dependent metabolite of choline, is positively associated with the risk of primary liver cancer: A case-control study. Nutr. Metab. 2018, 15, 81. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Ayesh, R.; Barrett, T.; Smith, R. Trimethylamine and foetor hepaticus. Scand. J. Gastroenterol. 1999, 34, 524–528. [Google Scholar] [PubMed]
- Bäckhed, F. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the Landscape of Gut Microbial Community Composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef]
- Gibson, G.R. Fibre and effects on probiotics (the prebiotic concept). Clin. Nutr. Suppl. 2004, 1, 25–31. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef]
- Zhao, X.; Han, Q.; Liu, Y.; Sun, C.; Gang, X.; Wang, G. The Relationship between Branched-Chain Amino Acid Related Metabolomic Signature and Insulin Resistance: A Systematic Review. J. Diabetes Res. 2016, 2016, 2794591. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-L.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.-D.; Zhang, Q.-Y.; Mi, M.-T. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio 2016, 7, e02210-15. [Google Scholar] [CrossRef] [PubMed]
- Canyelles, M.; Tondo, M.; Cedó, L.; Farràs, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Lam-Galvez, B.R.; Kirsop, J.; Wang, Z.; Levison, B.S.; Gu, X.; Copeland, M.F.; Bartlett, D.; Cody, D.B.; Dai, H.J.; et al. L-Carnitine in Omnivorous Diets Induces an Atherogenic Gut Microbial Pathway in Humans. J. Clin. Investig. 2019, 129, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio 2015, 6, e02481-14. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Manor, O.; Zubair, N.; Conomos, M.P.; Xu, X.; Rohwer, J.E.; Krafft, C.E.; Lovejoy, J.C.; Magis, A.T. A Multi-omic Association Study of Trimethylamine N-Oxide. Cell Rep. 2018, 24, 935–946. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary Metabolism, the Gut Microbiome, and Heart Failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Ussher, J.R.; Lopaschuk, G.D.; Arduini, A. Gut microbiota metabolism of l-carnitine and cardiovascular risk. Atherosclerosis 2013, 231, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Bockus, A.B.; Seibel, B.A. Synthetic capacity does not predict elasmobranchs’ ability to maintain trimethylamine oxide without a dietary contribution. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2018, 217, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Dyer, W.J. Amines in Fish Muscle. VI. Trimethylamine Oxide Content of Fish and Marine Invertebrates. J. Fish. Res. Board Can. 1952, 8, 314–324. [Google Scholar] [CrossRef]
- Bachand, G.D.; Jain, R.; Ko, R.; Bouxsein, N.F.; VanDelinder, V. Inhibition of Microtubule Depolymerization by Osmolytes. Biomacromolecules 2018, 19, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H.; Speers-Roesch, B.; Atchinson, S.; Reist, J.D.; Majewski, A.R.; Treberg, J.R. Osmolyte Adjustments as a Pressure Adaptation in Deep-Sea Chondrichthyan Fishes: An Intraspecific Test in Arctic Skates (Amblyraja hyperborea) along a Depth Gradient. Physiol. Biochem. Zool. 2018, 91, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Boserman, P.; van der Vegt, N.F.A.; Shea, J.E. Trimethylamine N-Oxide Counteracts Urea Denaturation by Inhibiting Protein-Urea Preferential Interaction. J. Am. Chem. Soc. 2018, 140, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Jethva, P.N.; Udgaonkar, J.B. The Osmolyte TMAO Modulates Protein Folding Cooperativity by Altering Global Protein Stability. Biochemistry 2018, 57, 5851–5863. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-Oxide (TMAO) Response to Animal Source Foods Varies among Healthy Young Men and is Influenced by their Gut Microbiota Composition: A Randomized Controlled Trial. Mol. Nutr. Food Res. 2017, 61, 1600324. [Google Scholar] [CrossRef]
- Smith, J.; Wishnok, J.; Deen, W. Metabolism and Excretion of Methylamines in Rats. Toxicol. Appl. Pharmacol. 1994, 125, 296–308. [Google Scholar] [CrossRef]
- Veeravalli, S.; Karu, K.; Scott, F.; Fennema, D.; Phillips, I.R.; Shephard, E.A. Effect of Flavin-Containing Monooxygenase Genotype, Mouse Strain, and Gender on Trimethylamine N-Oxide Production, Plasma Cholesterol Concentration, and an Index of Atherosclerosis. Drug Metab. Dispos. 2018, 46, 20–25. [Google Scholar] [CrossRef]
- Ascher, S.; Reinhardt, C. The gut microbiota: An emerging risk factor for cardiovascular and cerebrovascular disease. Eur. J. Immunol. 2018, 48, 564–575. [Google Scholar] [CrossRef]
- Fennema, D.; Phillips, I.R.; Shephard, E.A. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab. Dispos. 2016, 44, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R.; Zadlo-Dobrowolska, A. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Corbin, K.D.; Da Costa, K.-A.; Zhang, S.; Zhao, X.; Galanko, J.A.; Blevins, T.; Bennett, B.J.; O’Connor, A.; Zeisel, S.H. Effect of egg ingestion on trimethylamine-N-oxide production in humans: A randomized, controlled, dose-response study. Am. J. Clin. Nutr. 2014, 100, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.C.; Zhang, A.Q.; Smith, R.L. Dimethylamine and Diet. Food Chem. Toxicol. 2008, 46, 1734–1738. [Google Scholar] [CrossRef]
- Zhang, A.; Mitchell, S.; Smith, R. Dietary Precursors of Trimethylamine in Man: A Pilot Study. Food Chem. Toxicol. 1999, 37, 515–520. [Google Scholar] [CrossRef]
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-Oxide: A Carnitine-Derived Metabolite That Prolongs the Hypertensive Effect of Angiotensin II in Rats. Can. J. Cardiol. 2014, 30, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Zeisel, S.H.; DaCosta, K.A.; Fox, J.G. Endogenous formation of dimethylamine. Biochem. J. 1985, 232, 403–408. [Google Scholar] [CrossRef] [Green Version]
- Walker, V. The Fish Odour Syndrome. BMJ 1993, 307, 639–640. [Google Scholar] [CrossRef]
- Stubbs, J.R.; Stedman, M.R.; Liu, S.; Long, J.; Franchetti, Y.; West, R.E.; Prokopienko, A.J.; Mahnken, J.D.; Chertow, G.M.; Nolin, T.D. Trimethylamine N-Oxide and Cardiovascular Outcomes in Patients with ESKD Receiving Maintenance Hemodialysis. Clin. J. Am. Soc. Nephrol. 2019, 14, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Mafune, A.; Iwamoto, T.; Tsutsumi, Y.; Nakashima, A.; Yamamoto, I.; Yokoyama, K.; Yokoo, T.; Urashima, M. Associations among Serum Trimethylamine-N-Oxide (TMAO) Levels, Kidney Function and Infarcted Coronary Artery Number in Patients Undergoing Cardiovascular Surgery: A Cross-Sectional Study. Clin. Exp. Nephrol. 2016, 20, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Fletcher, C. Trimethylamine N-Oxide: Breathe New Life. Br. J. Pharmacol. 2018, 175, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Pham, Q.D.; Wolde-Kidan, A.; Gupta, A.; Schlaich, A.; Schneck, E.; Netz, R.R.; Sparr, E. Effects of Urea and TMAO on Lipid Self-Assembly under Osmotic Stress Conditions. J. Phys. Chem. B 2018, 122, 6471–6482. [Google Scholar] [CrossRef] [PubMed]
- Kerley, C.P. Dietary Patterns and Components to Prevent and Treat Heart Failure: A Comprehensive Review of Human Studies. Nutr. Res. Rev. 2019, 32, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Wang, Z.; Li, X.S.; Fan, Y.; Wu, Y.; Tang, W.H.W.; Hazen, S.L. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J. Am. Heart Assoc. 2016, 5, e002816. [Google Scholar] [CrossRef]
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-Oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef]
- Chelu, M.G.; Li, N. Biomarkers (plasma trimethylamine-N-oxide) to predict atrial fibrillation: Are we there yet? Int. J. Cardiol. 2018, 267, 116–117. [Google Scholar] [CrossRef]
- Cho, C.E.; Caudill, M.A. Trimethylamine-N-Oxide: Friend, Foe, or Simply Caught in the Cross-Fire? Trends Endocrinol. Metab. 2017, 28, 121–130. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Balarini, C.M.; Oliveira, M.Z.; Mc Pereira, T.; Silva, N.F.; Vasquez, E.C.; Meyrelles, S.S.; Gava, A.L. Hypercholesterolemia promotes early renal dysfunction in apolipoprotein E-deficient mice. Lipids Health Dis. 2011, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Johnson, C.; Zhou, J.; Wang, L.; Li, Y.-F.; Lu, Y.; Nanayakkara, G.; Fu, H.; Shao, Y.; Sanchez, C.; et al. Uremic toxins are conditional danger- or homeostasis-associated molecular patterns. Front. Biosci. 2018, 23, 348–387. [Google Scholar] [Green Version]
- Jain, A.; Pasare, C. Innate control of adaptive immunity: Beyond the three-signal paradigm. J. Immunol. 2017, 198, 3791–3800. [Google Scholar] [CrossRef] [PubMed]
- Van Tuijl, J.; Joosten, L.A.B.; Netea, M.G.; Bekkering, S.; Riksen, N.P. Immunometabolism orchestrates training of innate immunity in atherosclerosis. Cardiovasc. Res. 2019, 115, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Ogawa, E.; Okuyama, R.; Kanno, H. In Vasculitis of Small Muscular Arteries, Activation of Vessel-Infiltrating CD8 T Cells seems to be Antigen-Independent. Virchows Arch. 2018, 472, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Sha, X.; Meng, S.; Li, X.; Xi, H.; Maddaloni, M.; Pascual, D.W.; Shan, H.; Jiang, X.; Wang, H.; Yang, X.-F. Interleukin-35 Inhibits Endothelial Cell Activation by Suppressing MAPK-AP-1 Pathway. J. Biol. Chem. 2015, 290, 19307–19318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018, 17, 286. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, F.; Xu, D.; Zhang, Z.; Xu, F.; Tao, X.; Qiu, L.; Wei, H. Enterococcus faecium WEFA23 from infants lessens high-fat-diet-induced hyperlipidemia via cholesterol 7-alpha-hydroxylase gene by altering the composition of gut microbiota in rats. J. Dairy Sci. 2018, 101, 7757–7767. [Google Scholar] [CrossRef]
- Liu, S.; You, L.; Zhao, Y.; Chang, X. Wild Lonicera caerulea berry polyphenol extract reduces cholesterol accumulation and enhances antioxidant capacity in vitro and in vivo. Food Res. Int. 2018, 107, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Najar, A.G.; Yaghoobi, M.M.; Jahani, Y.; Vahabzadeh, Z. Trimethylamine-N-Oxide Treatment Induces Changes in the ATP-Binding Cassette Transporter A1 and Scavenger Receptor A1 in Murine Macrophage J774A.1 Cells. Inflammation 2016, 39, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.A. Effect of Trimethylamine N-Oxide on Interfacial Electrostatics at Phospholipid Monolayer—Water Interfaces and Its Relevance to Cardiovascular Disease. J. Phys. Chem. Lett. 2016, 7, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; Davis, R.C.; Civelek, M.; Orozco, L.; Wu, J.; Qi, H.; Pan, C.; Packard, R.R.S.; Eskin, E.; Yan, M.; et al. Genetic Architecture of Atherosclerosis in Mice: A Systems Genetics Analysis of Common Inbred Strains. PLoS Genet. 2015, 11, e1005711. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xie, Z.; Sun, M.; Wang, X.; Li, J.; Cui, J.; Zhang, F.; Yin, L.; Huang, D.; Hou, J.; et al. Plasma trimethylamine N-oxide is associated with vulnerable plaque characteristics in CAD patients as assessed by optical coherence tomography. Int. J. Cardiol. 2018, 265, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Kiouptsi, K.; Reinhardt, C. Contribution of the commensal microbiota to atherosclerosis and arterial thrombosis. Br. J. Pharmacol. 2018, 175, 4439–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, X.; Ran, L.; Lang, H.; Yi, L.; Mi, M. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef]
- Boini, K.M.; Hussain, T.; Li, P.-L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Ke, Y.; Li, D.; Zhao, M.; Liu, C.; Liu, J.; Zeng, A.; Shi, X.; Cheng, S.; Pan, B.; Zheng, L.; et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic. Biol. Med. 2018, 116, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Buffa, J.A.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.M.; Gupta, N.; Gregory, J.C.; Org, E.; Fu, X.; et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thromb. Haemost. 2018, 16, 1857–1872. [Google Scholar] [CrossRef] [Green Version]
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the Prevalence of Arthritis and Other Rheumatic Conditions in the United States. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef]
- National Collaborating Centre for Chronic Conditions (UK). Rheumatoid Arthritis: National Clinical Guideline for Management and Treatment in Adults; Royal College of Physicians: London, UK, 2009. [Google Scholar]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Vaahtovuo, J.; Munukka, E.; Korkeamäki, M.; Luukkainen, R.; Toivanen, P. Fecal microbiota in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1500–1505. [Google Scholar] [PubMed]
- Chung, Y.-L.; Rider, L.G.; Bell, J.D.; Summers, R.M.; Zemel, L.S.; Rennebohm, R.M.; Passo, M.H.; Hicks, J.; Miller, F.W.; Scott, D.L.; et al. Muscle metabolites, detected in urine by proton spectroscopy, correlate with disease damage in juvenile idiopathic inflammatory myopathies. Arthritis Rheum. 2005, 53, 565–570. [Google Scholar] [CrossRef]
- Blum, A.; Adawi, M. Rheumatoid arthritis (RA) and cardiovascular disease. Autoimmun. Rev. 2019, 18, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Cardiac and vascular complications in rheumatoid arthritis. Reumatologia 2019, 57, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualtierotti, R.; Ughi, N.; Marfia, G.; Ingegnoli, F. Practical Management of Cardiovascular Comorbidities in Rheumatoid Arthritis. Rheumatol. Ther. 2017, 4, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oláh, C.; Kardos, Z.; Sepsi, M.; Sas, A.; Kostyál, L.; Bhattoa, H.P.; Hodosi, K.; Kerekes, G.; Tamási, L.; Valikovics, A.; et al. Assessment of intracranial vessels in association with carotid atherosclerosis and brain vascular lesions in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Inui, K.; Sugioka, Y.; Sugioka, K.; Matsumura, Y.; Takahashi, S.; Tada, M.; Mamoto, K.; Wakitani, S.; Koike, T.; et al. High titer of anti-citrullinated peptide antibody is a risk factor for severe carotid atherosclerotic plaque in patients with rheumatoid arthritis: The TOMORROW study. Int. J. Rheum. Dis. 2017, 20, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Jagpal, A.; Navarro-Millán, I. Cardiovascular co-morbidity in patients with rheumatoid arthritis: A narrative review of risk factors, cardiovascular risk assessment and treatment. BMC Rheumatol. 2018, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Nomura, M.; Nakamura, K. Arthritis in mice induced by a single immunisation with collagen. Ann. Rheum. Dis. 1996, 55, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.K.; Rosloniec, E.F.; Cremer, M.A.; Kang, A.H. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997, 61, 1861–1878. [Google Scholar] [CrossRef]
- Jones, G.W.; Hill, D.G.; Sime, K.; Williams, A.S. In Vivo Models for Inflammatory Arthritis. Methods Mol. Biol. 2018, 1725, 101–118. [Google Scholar]
- Vincent, T.L.; Williams, R.O.; Maciewicz, R.; Silman, A.; Garside, P.; Arthritis Research UK animal Models Working Group. Mapping Pathogenesis of Arthritis through Small Animal Models. Rheumatology 2012, 51, 1931–1941. [Google Scholar] [CrossRef]
- Roberts, A.; Thompson, J.S. Genetic factors in the development of atheroma and on serum total cholesterol levels in inbred mice and their hybrids. Prog. Biochem. Pharmacol. 1977, 13, 298–305. [Google Scholar]
- Roberts, A.; Thompson, J.S. Inbred mice and their hypbrids as an animal model for atherosclerosis research. Adv. Exp. Med. Biol. 1976, 67, 313–327. [Google Scholar] [PubMed]
- Paigen, B.; Morrow, A.; Brandon, C.; Mitchell, D.; Holmes, P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 1985, 57, 65–73. [Google Scholar] [CrossRef]
- Svetlicky, N.; Kivity, S.; Odeh, Q.; Shovman, O.; Gertel, S.; Amital, H.; Gendelman, O.; Volkov, A.; Barshack, I.; Bar-Meir, E.; et al. Anti-citrullinated-protein-antibody-specific intravenous immunoglobulin attenuates collagen-induced arthritis in mice. Clin. Exp. Immunol. 2015, 182, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Monach, P.A.; Mathis, D.; Benoist, C. The K/BxN Arthritis Model. Curr. Protoc. Immunol. 2008, 81, 15.22.1–15.22.12. [Google Scholar]
- Schoels, M.; Bombardier, C.; Aletaha, D. Diagnostic and Prognostic Value of Antibodies and Soluble Biomarkers in Undifferentiated Peripheral Inflammatory Arthritis: A Systematic Review. J. Rheumatol. Suppl. 2011, 87, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.M.; Chen, W.M.; Tung, S.Y.; Wei, K.L.; Shen, C.H.; Chang, T.S.; Chang, P.J. Prevalence and Predictive Value of High-Positive Rheumatoid Factor and Anti-Citrullinated Protein Antibody Levels in Nonarthritic Patients with Chronic Hepatitis C Infection. Int. J. Rheum. Dis. 2019, 22, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Van Venrooij, W.J. Citrullinated proteins: Sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res. Ther. 2004, 6, 107–111. [Google Scholar] [CrossRef]
- Sokolove, J.; Brennan, M.J.; Sharpe, O.; Lahey, L.J.; Kao, A.H.; Krishnan, E.; Edmundowicz, D.; Lepus, C.M.; Wasko, M.C.; Robinson, W.H. Brief Report: Citrullination within the Atherosclerotic Plaque: A Potential Target for the Anti-Citrullinated Protein Antibody Response in Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 1719–1724. [Google Scholar] [CrossRef]
- Geraldino-Pardilla, L.; Giles, J.T.; Sokolove, J.; Zartoshti, A.; Robinson, W.H.; Budoff, M.; Detrano, R.; Bokhari, S.; Bathon, J.M. Association of Anti-Citrullinated Peptide Antibodies with Coronary Artery Calcification in Rheumatoid Arthritis. Arthritis Rheum. 2017, 69, 1276–1281. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; Van Der Heijden, A.; Lora, M.; Senshu, T.; Van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 2004, 6, R142–R150. [Google Scholar] [CrossRef]
- Bang, H.; Egerer, K.; Gauliard, A.; Lüthke, K.; Rudolph, P.E.; Fredenhagen, G.; Berg, W.; Feist, E.; Burmester, G.-R.; Burmester, G. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum. 2007, 56, 2503–2511. [Google Scholar] [CrossRef]
- György, B.; Tóth, E.; Tarcsa, E.; Falus, A.; Buzás, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef]
- Van Venrooij, W.J.; Pruijn, G.J. How citrullination invaded rheumatoid arthritis research. Arthritis Res. Ther. 2014, 16, 103. [Google Scholar] [CrossRef]
- Taleb, S. Inflammation in Atherosclerosis. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef]
- Khodabandehlou, K.; Masehi-Lano, J.J.; Poon, C.; Wang, J.; Chung, E.J. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp. Biol. Med. 2017, 242, 799–812. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Zwerina, J.; Hayer, S.; Redlich, K.; Bobacz, K.; Kollias, G.; Smolen, J.S.; Schett, G. Activation of p38 MAPK is a key step in tumor necrosis factor–mediated inflammatory bone destruction. Arthritis Rheum. 2006, 54, 463–472. [Google Scholar] [CrossRef]
- Makarov, S.S. NF-kappaB in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206. [Google Scholar] [CrossRef]
- Dong, X.; Zheng, Z.; Lin, P.; Fu, X.; Li, F.; Jiang, J.; Zhu, P. ACPAs Promote IL-1beta Production in Rheumatoid Arthritis by Activating the NLRP3 Inflammasome. Cell. Mol. Immunol. 2019. [Google Scholar] [CrossRef]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann-Geller, B.; Koppert, S.; Kesel, N.; Hasseli, R.; Ullrich, S.; Lefevre, S.; Frommer, K.; Gehrke, T.; Schonburg, M.; Rehart, S.; et al. Interactions between Rheumatoid Arthritis Synovial Fibroblast Migration and Endothelial Cells. Immunol. Cell Biol. 2019, 97, 178–189. [Google Scholar] [CrossRef]
- He, H.; Lian, X.; Tang, Z. Research progress of trimethylamine-N-oxide in the pathogenesis of atherosclerosis. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2017, 42, 986–990. [Google Scholar]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [Green Version]
- Dragoljevic, D.; Kraakman, M.J.; Nagareddy, P.R.; Ngo, D.; Shihata, W.; Kammoun, H.L.; Whillas, A.; Lee, M.K.S.; Al-Sharea, A.; Pernes, G.; et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur. Heart J. 2018, 39, 2158–2167. [Google Scholar] [CrossRef] [Green Version]
- Archer, A.M.; Saber, R.; Rose, S.; Shaffer, A.; Misharin, A.V.; Tsai, F.; Iii, G.K.H.; Dominguez, S.; Eren, M.; Vaughan, D.E.; et al. ApoE deficiency exacerbates the development and sustainment of a semi-chronic K/BxN serum transfer-induced arthritis model. J. Transl. Med. 2016, 14, 170. [Google Scholar] [CrossRef] [Green Version]
- Habets, K.L.; Trouw, L.A.; Levarht, E.N.; Korporaal, S.J.; Habets, P.A.; De Groot, P.; Huizinga, T.W.; Toes, R.E. Anti-citrullinated protein antibodies contribute to platelet activation in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 209. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.-S.; Lin, C.-L.; Peng, C.-L.; Chen, Y.-F.; Lu, C.-C.; Sung, F.-C.; Kao, C.-H. Rheumatoid arthritis and risk of acute myocardial infarction—A nationwide retrospective cohort study. Int. J. Cardiol. 2013, 168, 4750–4754. [Google Scholar] [CrossRef]
- Chung, W.S.; Peng, C.L.; Lin, C.L.; Chang, Y.J.; Chen, Y.F.; Chiang, J.Y.; Sung, F.C.; Kao, C.H. Rheumatoid Arthritis Increases the Risk of Deep Vein Thrombosis and Pulmonary Thromboembolism: A Nationwide Cohort Study. Ann. Rheum. Dis. 2014, 73, 1774–1780. [Google Scholar] [CrossRef]
- Masoud, S.; Lim, P.B.; Kitas, G.D.; Panoulas, V. Sudden cardiac death in patients with rheumatoid arthritis. World J. Cardiol. 2017, 9, 562–573. [Google Scholar] [CrossRef]
- Helin, H.J.; Korpela, M.M.; Mustonen, J.T.; Pasternack, A.I. Renal biopsy findings and clinicopathologic correlations in rheumatoid arthritis. Arthritis Rheum. 1995, 38, 242–247. [Google Scholar] [CrossRef]
- Paudyal, S.; Yang, F.M.; Rice, C.; Chen, C.-C.; Skelton, M.; Bethel, M.; Brown, S.; Nahman, N.S.; Carbone, L. End-stage renal disease in patients with rheumatoid arthritis. Semin. Arthritis Rheum. 2017, 46, 418–422. [Google Scholar] [CrossRef]
- Kapoor, T.; Bathon, J. Renal Manifestations of Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2018, 44, 571–584. [Google Scholar] [CrossRef]
- Anders, H.-J.; Vielhauer, V. Renal co-morbidity in patients with rheumatic diseases. Arthritis Res. Ther. 2011, 13, 222. [Google Scholar] [CrossRef]
- Shepshelovich, J.; Goldstein-Magal, L.; Globerson, A.; Yen, P.M.; Rotman-Pikielny, P.; Hirschberg, K. Protein synthesis inhibitors and the chemical chaperone TMAO reverse endoplasmic reticulum perturbation induced by overexpression of the iodide transporter pendrin. J. Cell Sci. 2005, 118, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- Bchetnia, M.; Lacroix, J.; Farez, T.; Larouche, M.; Powell, J.; McCuaig, C.; Dupere, A.; Morin, C.; Legendre-Guillemin, V.; Laprise, C. Reduction in Keratin Aggregates in Epidermolysis Bullosa Simplex Keratinocytes after Pretreatment with Trimethylamine N-Oxide. Exp. Dermatol. 2016, 25, 229–230. [Google Scholar] [CrossRef]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 124. [Google Scholar] [CrossRef]
- Wu, W.-K.; Panyod, S.; Ho, C.-T.; Kuo, C.-H.; Wu, M.-S.; Sheen, L.-Y. Dietary allicin reduces transformation of L-carnitine to TMAO through impact on gut microbiota. J. Funct. Foods 2015, 15, 408–417. [Google Scholar] [CrossRef]
- Bresciani, L.; Dall’Asta, M.; Favari, C.; Calani, L.; Del Rio, D.; Brighenti, F. An in vitro exploratory study of dietary strategies based on polyphenol-rich beverages, fruit juices and oils to control trimethylamine production in the colon. Food Funct. 2018, 9, 6470–6483. [Google Scholar] [CrossRef]
- Qiu, L.; Yang, D.; Tao, X.; Yu, J.; Xiong, H.; Wei, H. Enterobacter Aerogenes ZDY01 Attenuates Choline-Induced Trimethylamine N-Oxide Levels by Remodeling Gut Microbiota in Mice. J. Microbiol. Biotechnol. 2017, 27, 1491–1499. [Google Scholar]



{kind=link}
{kind=link}
| Mechanism of Actions | TMAO | CVD | RA |
|---|---|---|---|
| Microbial dysbiosis | + | + | + |
| Genetics: Major histocompatibility molecules | + | + | |
| Activated peptidyl arginine deaminase-4 (PAD-4) | + | ||
| Activated B cells | ? | + | |
| Induced production of anti-citrullinated peptide antibodies | + | ||
| Activated complement systems | ? | + | |
| Activated T cells | + | + | |
| Induced production of Interluekin-17 | + | + | |
| Disrupted reverse cholesterol transport (RCT) | + | + | |
| Interrupted bile synthesis and metabolism | + | + | |
| Disrupted cholesterol uptake and efflux balance | + | + | |
| Upregulated scavenger receptor A1 | + | ||
| Activated macrophages in synovial lining or intimal layer | + | + | + |
| Activated mitogen-activated protein kinase (MAPK) | + | + | + |
| Activated Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) | + | + | + |
| Activated NLRP3 inflammasome | + | + | + |
| Activated caspase-1 | + | + | + |
| Induced proinflammatory cytokines IL-1β, tumor necrosis factor-α, and interleukin-6 | + | + | + |
| Released reactive oxygen species (ROS) | + | + | + |
| Production of chemokines | + | + | + |
| Liberation of eicosanoids | + | + | |
| Upregulated cell adhesion molecules | + | + | + |
| Hypertension | + | + | + |
| Endothelial dysfunction: Inactivated endothelial nitric oxide synthase | + | + | + |
| Increased vascular permeability | + | + | + |
| Activated platelets and increased thrombotic events | + | + | + |
| Increased incidences of myocardial infarction and strokes | + | + | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, M.M.; Yang, X.; Wang, H.; Saaoud, F.; Sun, Y.; Fong, D. The Microbial Metabolite Trimethylamine N-Oxide Links Vascular Dysfunctions and the Autoimmune Disease Rheumatoid Arthritis. Nutrients 2019, 11, 1821. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11081821
Chan MM, Yang X, Wang H, Saaoud F, Sun Y, Fong D. The Microbial Metabolite Trimethylamine N-Oxide Links Vascular Dysfunctions and the Autoimmune Disease Rheumatoid Arthritis. Nutrients. 2019; 11(8):1821. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11081821
Chicago/Turabian StyleChan, Marion M., Xiaofeng Yang, Hong Wang, Fatma Saaoud, Yu Sun, and Dunne Fong. 2019. "The Microbial Metabolite Trimethylamine N-Oxide Links Vascular Dysfunctions and the Autoimmune Disease Rheumatoid Arthritis" Nutrients 11, no. 8: 1821. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11081821