A Special Amino-Acid Formula Tailored to Boosting Cell Respiration Prevents Mitochondrial Dysfunction and Oxidative Stress Caused by Doxorubicin in Mouse Cardiomyocytes


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
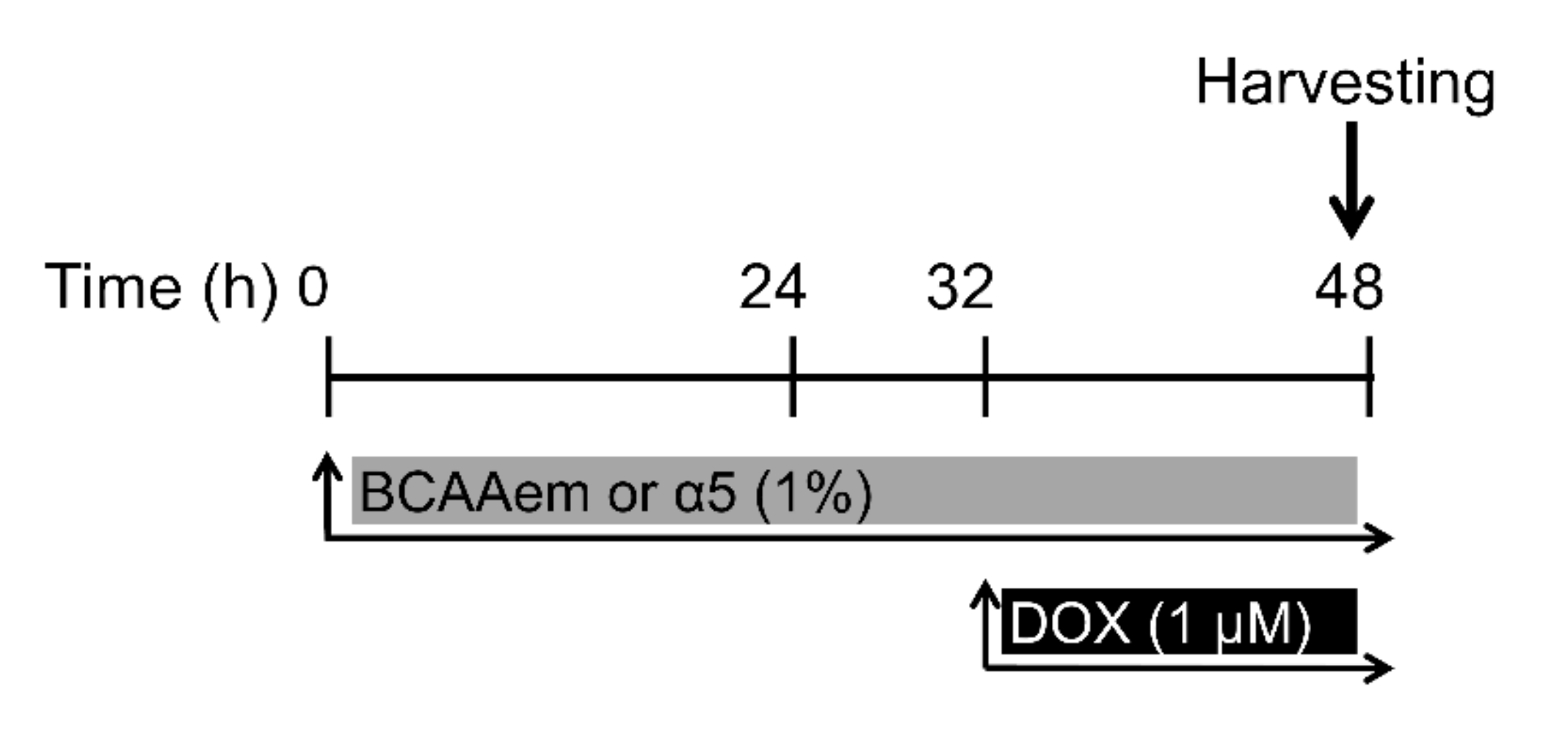
2.1. Cell Cultures and Treatments
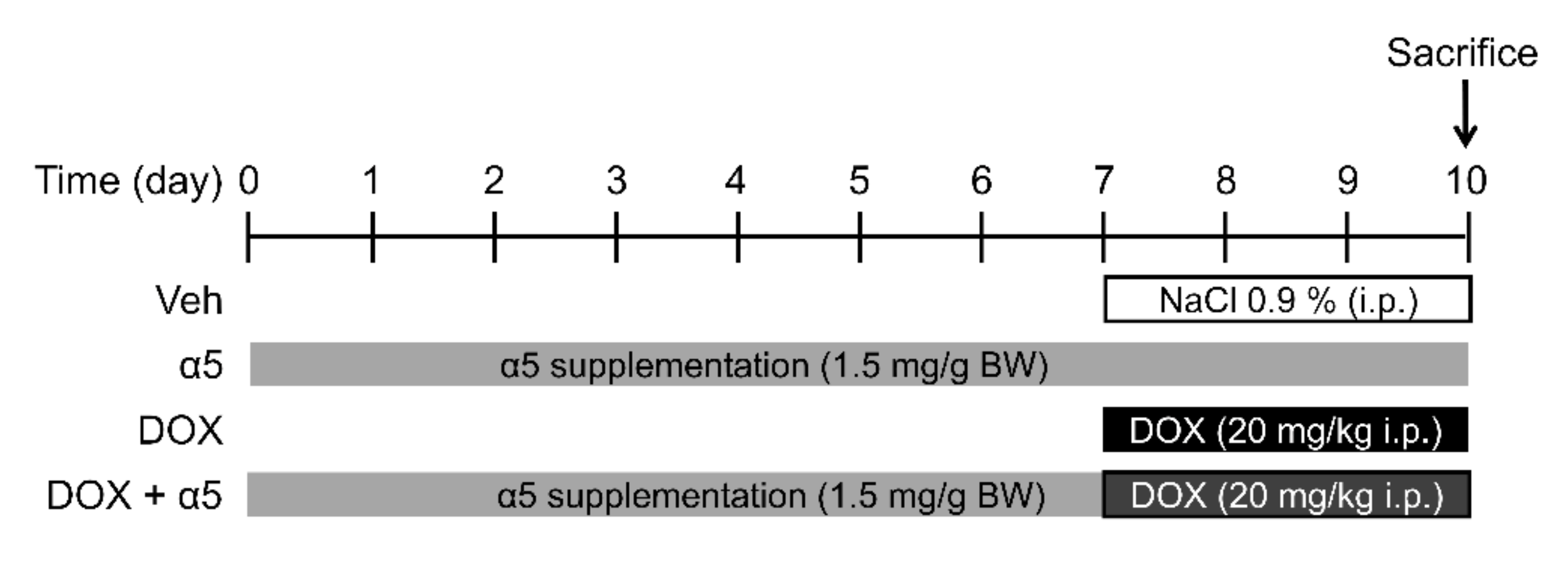
2.2. Animals and Treatments
2.3. Quantitative RT-PCR Analysis
2.4. Western Blot Analysis
2.5. Mitochondrial DNA
2.6. Citrate Synthase Activity
2.7. Oxygen Consumption
2.8. Mitochondrial Oxidative Stress
2.9. Viability Assay
2.10. Acid Phosphatase Assay
2.11. Statistical Analysis and Data Presentation
3. Results
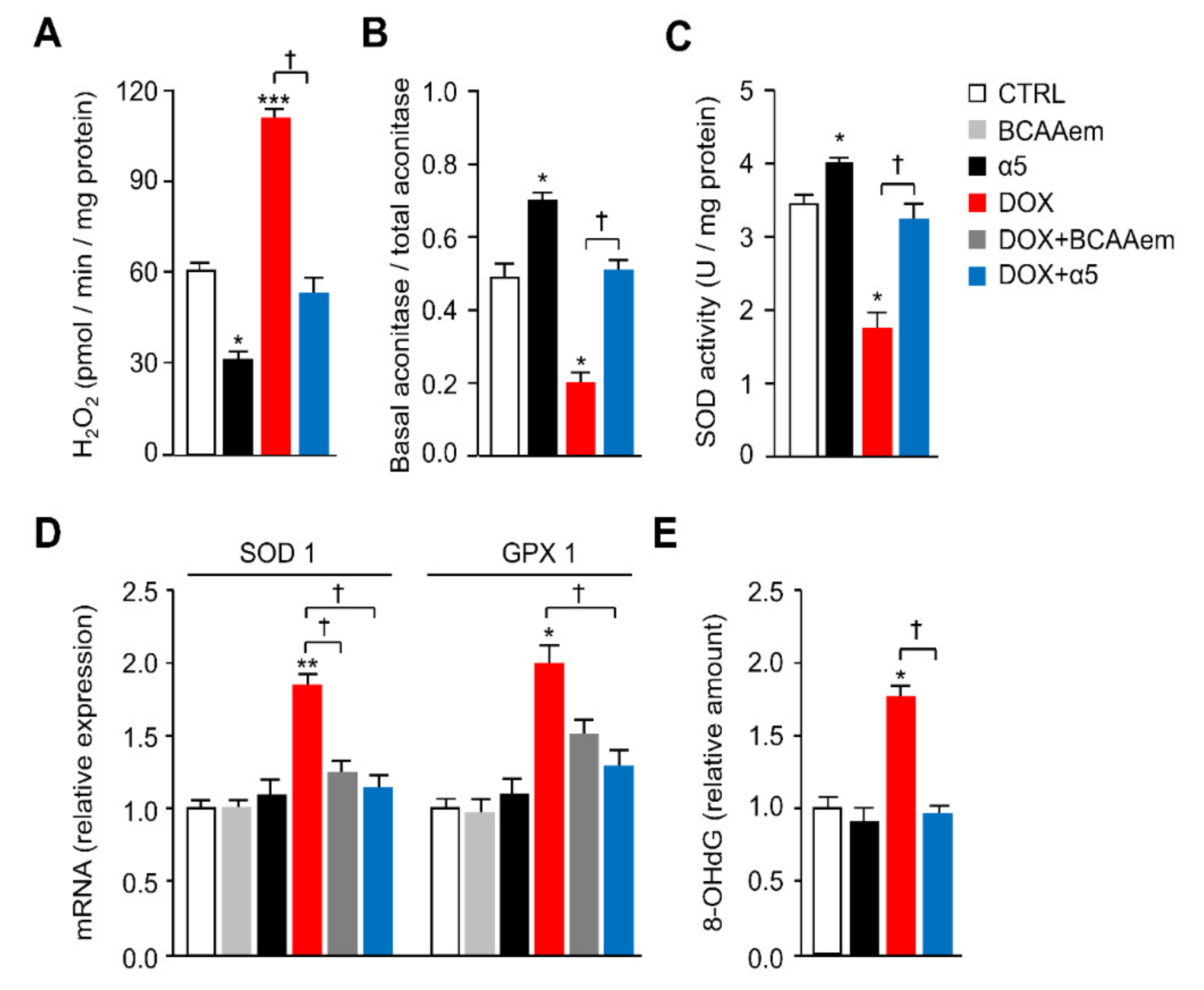
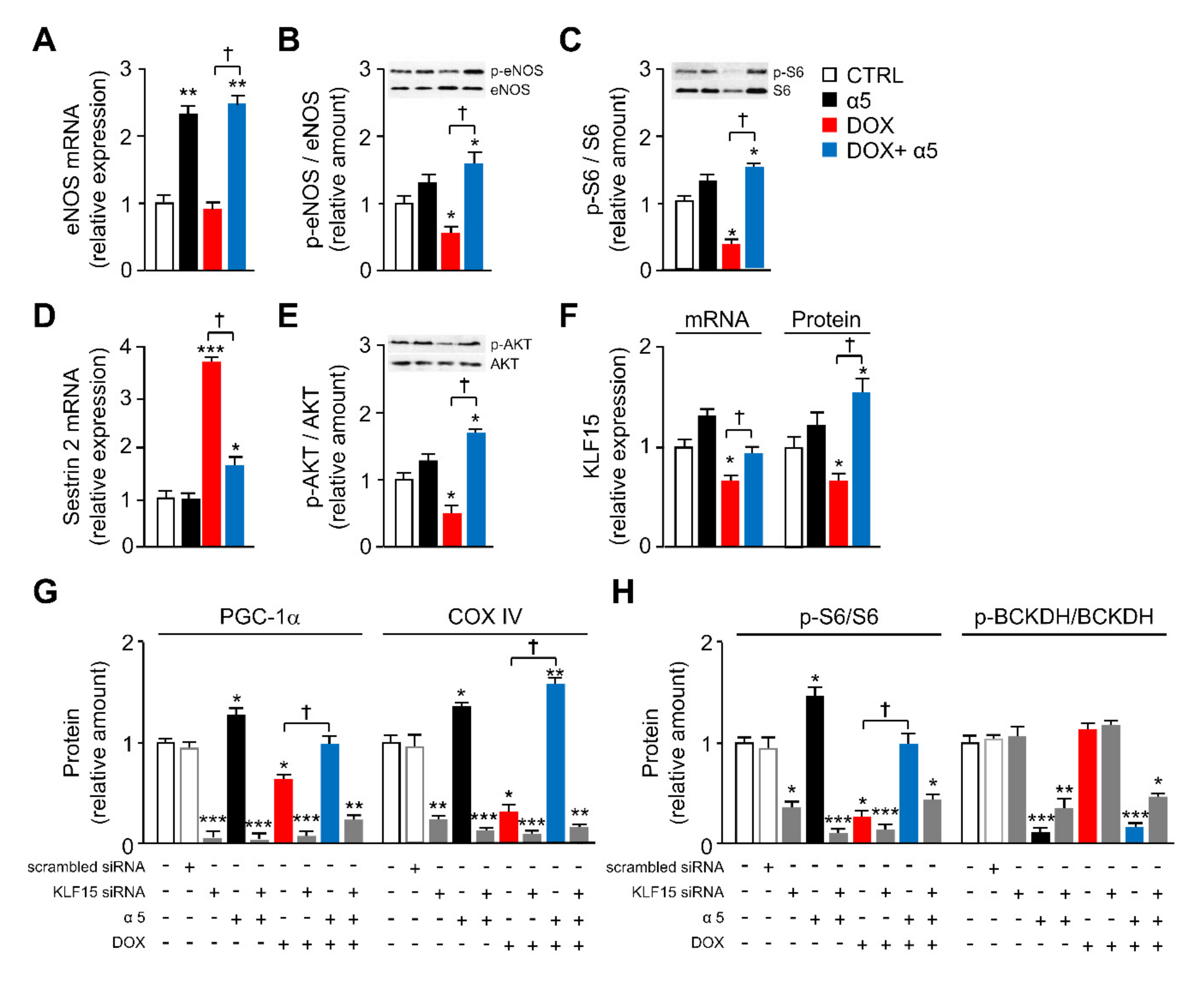
3.1. Specific Amino-Acid Mixtures Prevent Mitochondrial Dysfunction in HL-1 Cardiomyocytes Acutely Exposed to DOX
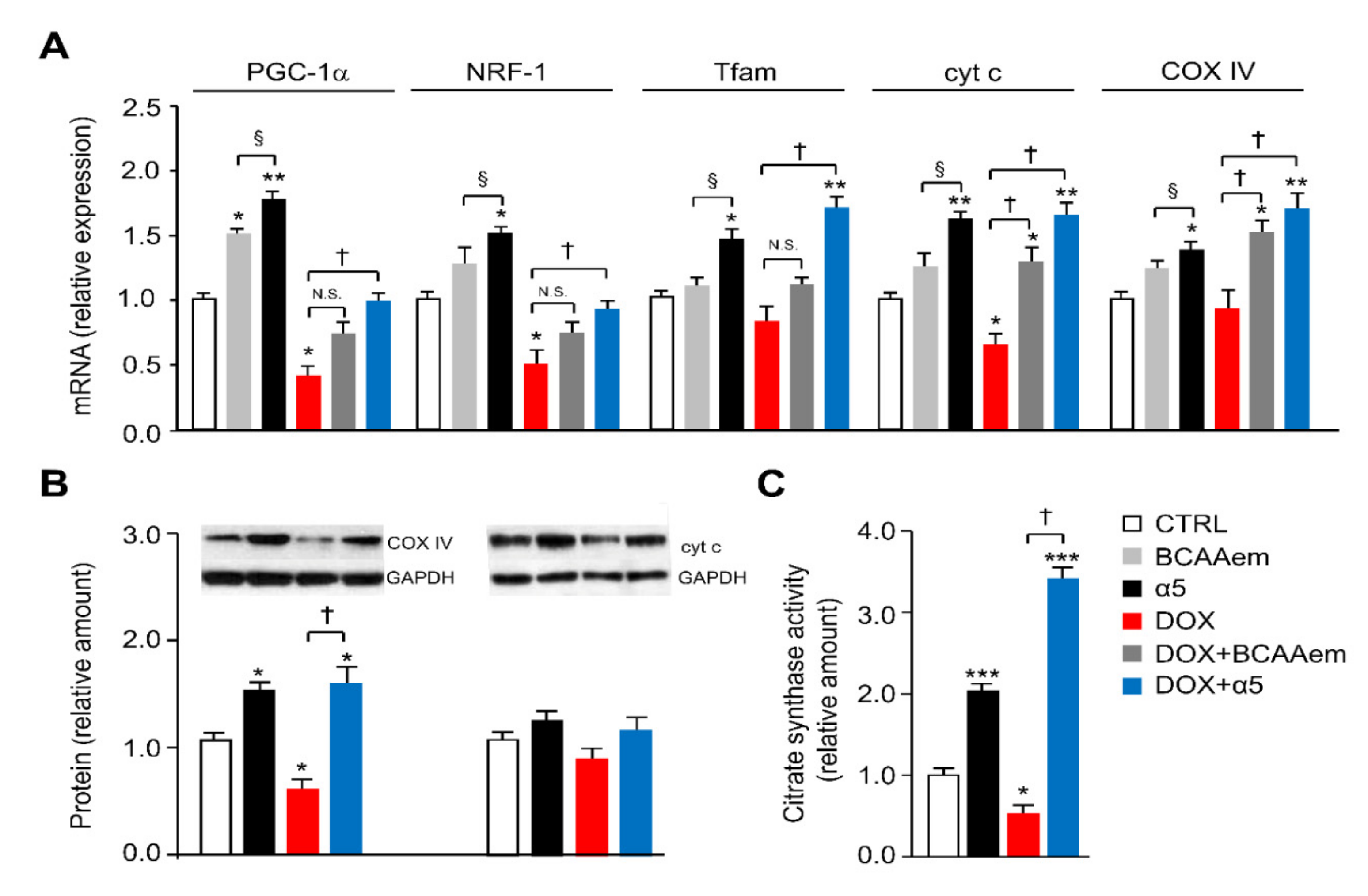
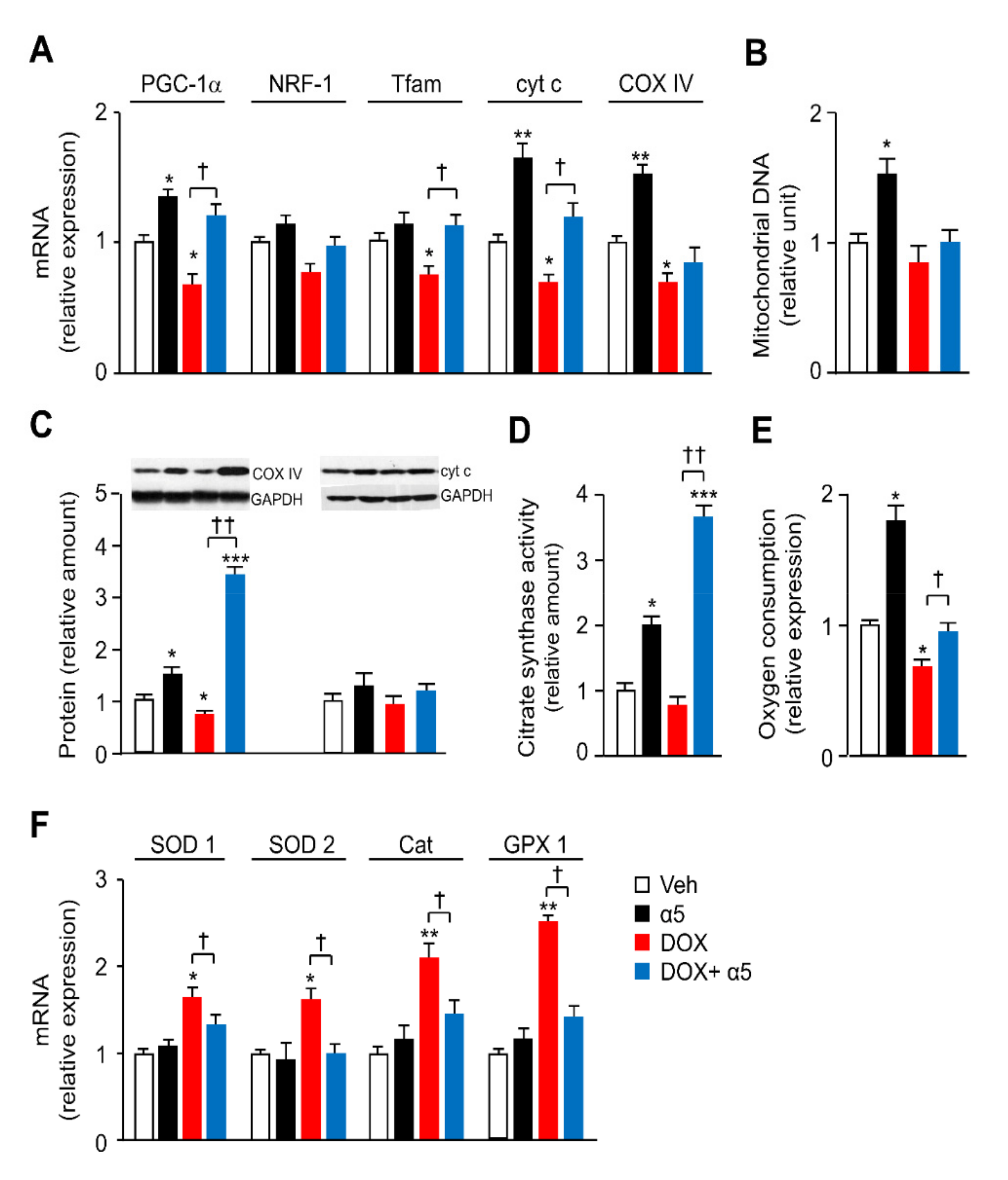
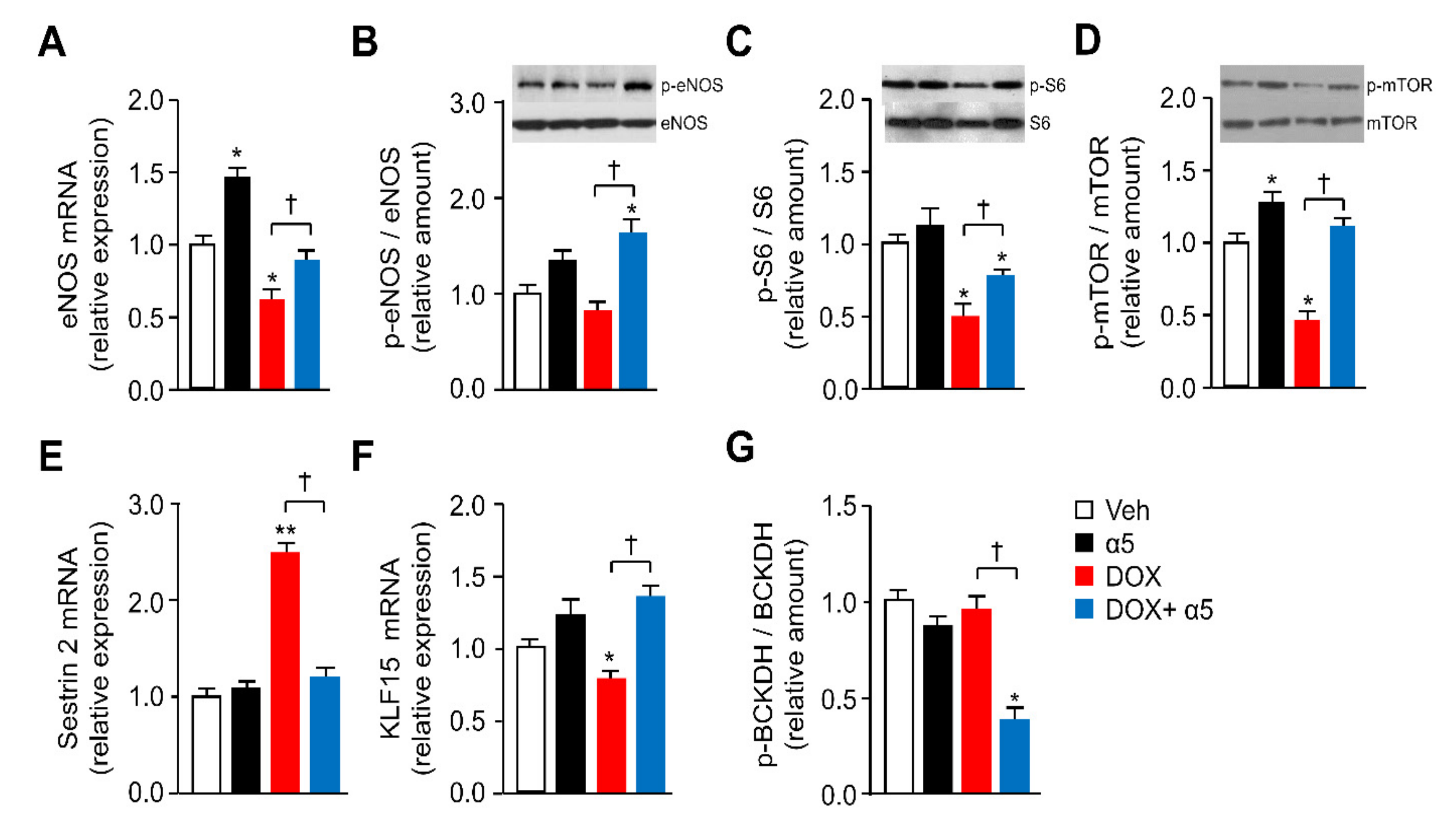
3.2. α5 Mixture Prevents Mitochondrial Dysfunction in Heart of DOX-Treated Mice
3.3. Different Signaling Pathways are Implicated in the Protective Effects of α5 Supplementation
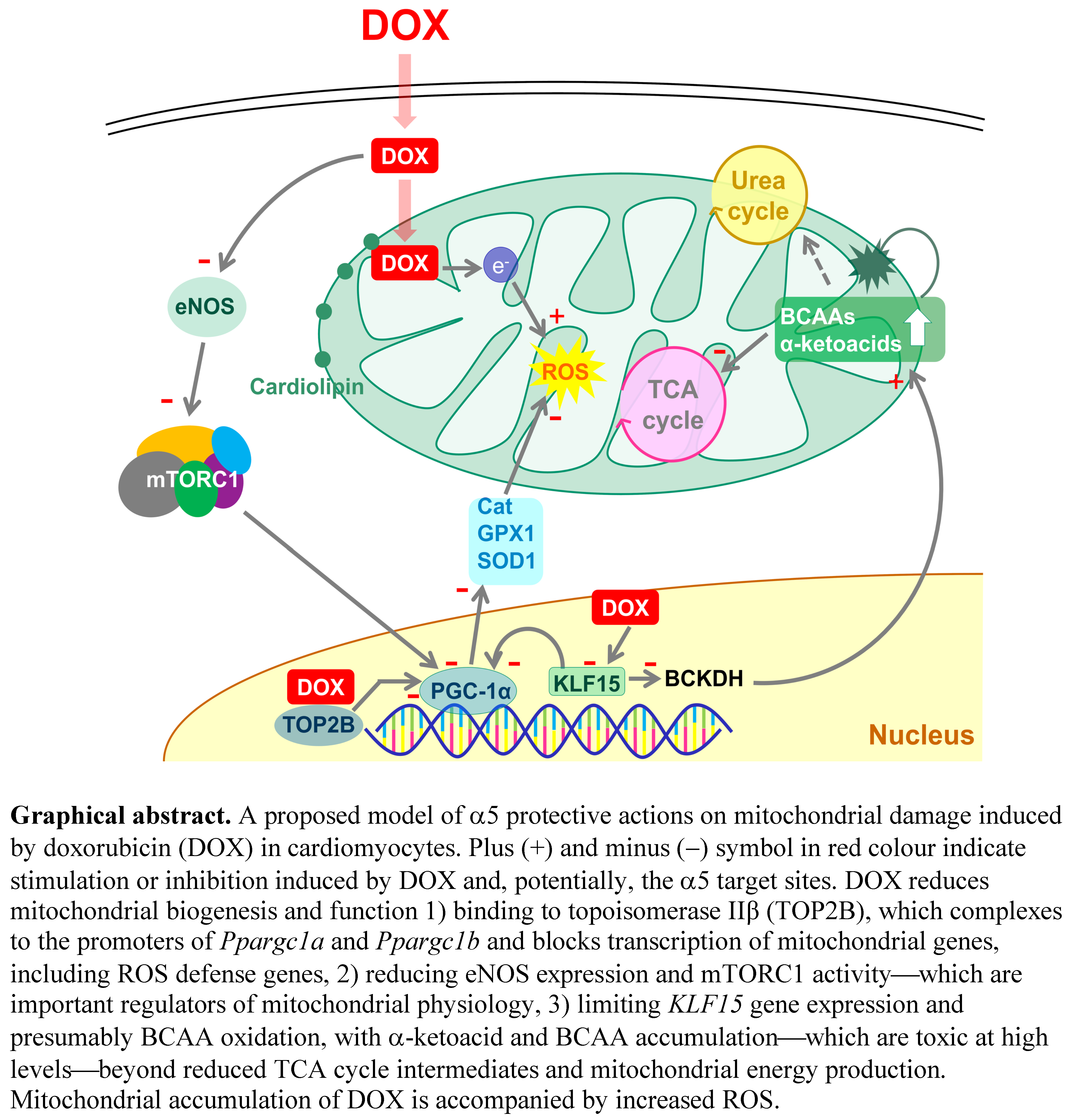
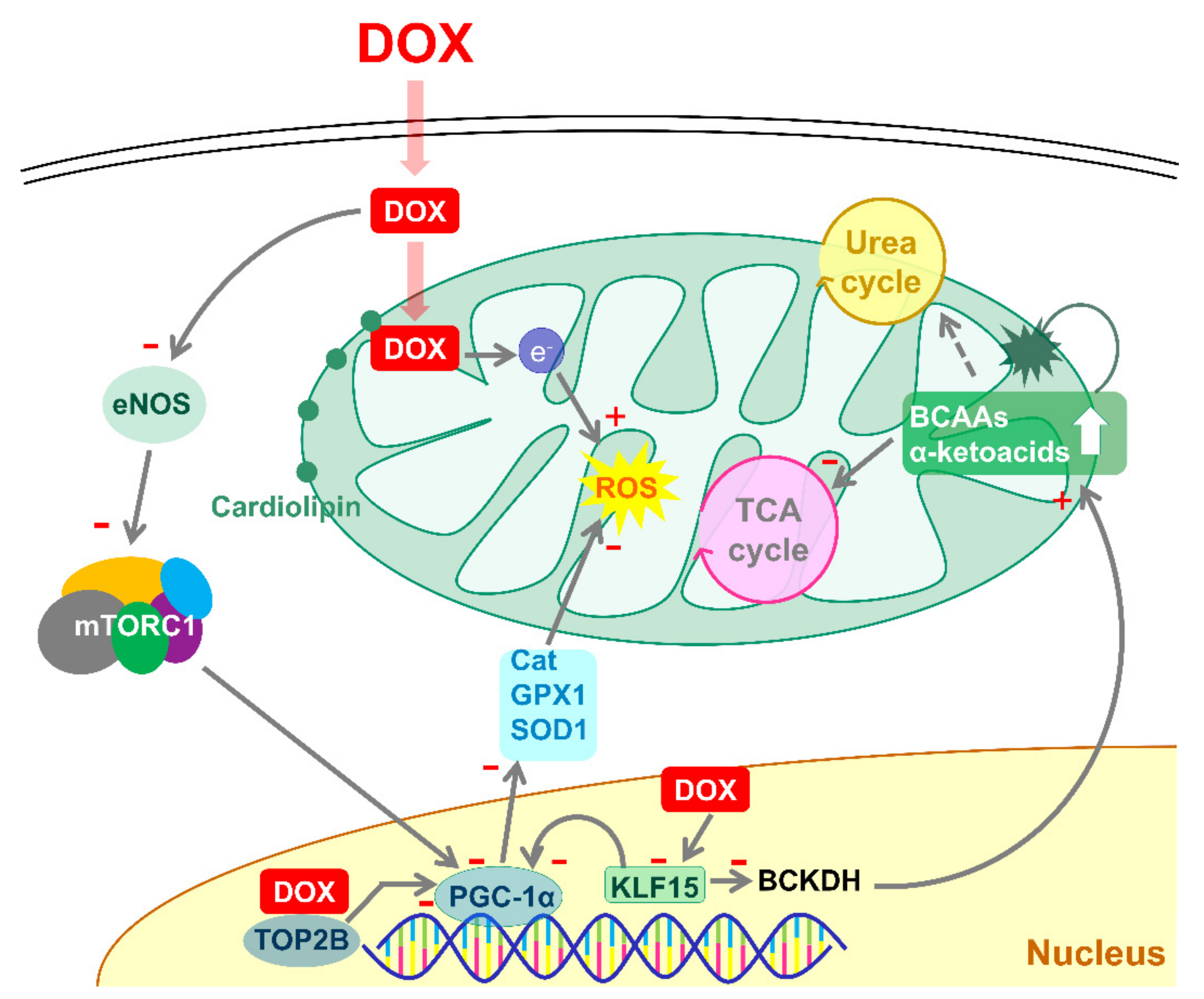
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smuder, A.J. Exercise stimulates beneficial adaptations to diminish doxorubicin-induced cellular toxicity. Am. J. Physiol. Integr. Comp. Physiol. 2019, 315, R662–R672. [Google Scholar] [CrossRef] [PubMed]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Soonpaa, M.H.; Chen, H.; Shen, W.; Payne, R.M.; Liechty, E.A.; Caldwell, R.L.; Shou, W.; Field, L.J. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 2009, 119, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Salvatorelli, E.; Menna, P.; Chello, M.; Covino, E.; Minotti, G. Modeling Human Myocardium Exposure to Doxorubicin Defines the Risk of Heart Failure from Low-Dose Doxorubicin. J. Pharmacol. Exp. Ther. 2017, 362, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, J.H. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983, 43, 460–472. [Google Scholar]
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef]
- Kavazis, A.N.; Morton, A.B.; Hall, S.E.; Smuder, A.J. Effects of doxorubicin on cardiac muscle subsarcolemmal and intermyofibrillar mitochondria. Mitochondrion 2017, 34, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Pani, G.; Bedogni, B.; Anzevino, R.; Colavitti, R.; Palazzotti, B.; Borrello, S.; Galeotti, T. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 2000, 60, 4654–4660. [Google Scholar]
- Xiong, Y.; Liu, X.; Lee, C.-P.; Chua, B.H.L.; Ho, Y.-S. Attenuation of doxorubicin-induced contractile and mitochondrial dysfunction in mouse heart by cellular glutathione peroxidase. Free Radic. Biol. Med. 2006, 41, 46–55. [Google Scholar] [CrossRef]
- Nisoli, E. Calorie Restriction Promotes Mitochondrial Biogenesis by Inducing the Expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.S.; Donthamsetty, S.; White, B.; Latendresse, J.R.; Mehendale, H.M. Mechanism of protection of moderately diet restricted rats against doxorubicin-induced acute cardiotoxicity. Toxicol. Appl. Pharmacol. 2007, 225, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; D’Antona, G.; Nisoli, E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: An evolutionary perspective. Aging 2011, 3, 464–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-Chain Amino Acid Supplementation Promotes Survival and Supports Cardiac and Skeletal Muscle Mitochondrial Biogenesis in Middle-Aged Mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Chen, T.; Zhao, D.; Zheng, J.; Liu, Z. Ginkgolide B Exerts Cardioprotective Properties against Doxorubicin-Induced Cardiotoxicity by Regulating Reactive Oxygen Species, Akt and Calcium Signaling Pathways In Vitro and In Vivo. PLoS ONE 2016, 11, e0168219. [Google Scholar] [CrossRef] [Green Version]
- Palanivel, R.; Eguchi, M.; Shuralyova, I.; Coe, I.; Sweeney, G. Distinct effects of short- and long-term leptin treatment on glucose and fatty acid uptake and metabolism in HL-1 cardiomyocytes. Metabolism 2006, 55, 1067–1075. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef]
- Tedesco, L.; Valerio, A.; Cervino, C.; Cardile, A.; Pagano, C.; Vettor, R.; Pasquali, R.; Carruba, M.O.; Marsicano, G.; Lutz, B.; et al. Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesis through endothelial nitric oxide synthase expression in white adipocytes. Diabetes 2008, 57, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Frontini, A.; Bertolotti, P.; Tonello, C.; Valerio, A.; Nisoli, E.; Cinti, S.; Giordano, A. Leptin-dependent STAT3 phosphorylation in postnatal mouse hypothalamus. Brain Res. 2008, 1215, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Gueguen, N.; Wetterwald, C.; Simard, G.; Malthièry, Y.; Ducluzeau, P.-H. Effects of the cannabinoid CB1 antagonist rimonabant on hepatic mitochondrial function in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1162–E1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, L.M.; Xie, H.; Koza, R.A.; Mynatt, R.; Hulver, M.W.; Bray, G.A.; Smith, S.R. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 2005, 54, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, L.; Corsetti, G.; Ruocco, C.; Ragni, M.; Rossi, F.; Carruba, M.O.; Valerio, A.; Nisoli, E. A specific amino acid formula prevents alcoholic liver disease in rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G566–G582. [Google Scholar] [CrossRef]
- Nisoli, E. Mitochondrial Biogenesis in Mammals: The Role of Endogenous Nitric Oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Valerio, A.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Pisconti, A.; Palomba, L.; Cantoni, O.; Clementi, E.; Moncada, S.; et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J. Clin. Investig. 2006, 116, 2791–2798. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Crescenzo, R.; D’Andrea, E.; Ferraro, M.; Bianco, F.; Liverini, G.; Iossa, S. Skeletal muscle subsarcolemmal mitochondrial dysfunction in high-fat fed rats exhibiting impaired glucose homeostasis. Int. J. Obes. 2007, 31, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Pervin, S.; Singh, R.; Hernandez, E.; Wu, G.; Chaudhuri, G. Nitric oxide in physiologic concentrations targets the translational machinery to increase the proliferation of human breast cancer cells: Involvement of mammalian target of rapamycin/eIF4E pathway. Cancer Res. 2007, 67, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.T.; Sinai, P.; Kain, S.R. An acid phosphatase assay for quantifying the growth of adherent and nonadherent cells. Anal. Biochem. 1996, 241, 103–108. [Google Scholar] [CrossRef]
- Chen, C.T.; Wang, Z.H.; Hsu, C.C.; Lin, H.H.; Chen, J.H. Taiwanese and Japanese yam (Dioscorea spp.) extracts attenuate doxorubicin-induced cardiotoxicity in mice. J. Food Drug Anal. 2017, 25, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Antona, G.; Tedesco, L.; Ruocco, C.; Corsetti, G.; Ragni, M.; Fossati, A.; Saba, E.; Fenaroli, F.; Montinaro, M.; Carruba, M.O.; et al. A Peculiar Formula of Essential Amino Acids Prevents Rosuvastatin Myopathy in Mice. Antioxid. Redox Signal. 2016, 25, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilli, A.; Visigalli, R.; Sala, R.; Gazzola, G.C.; Parolari, A.; Tremoli, E.; Bonomini, S.; Simon, A.; Closs, E.I.; Dall’Asta, V.; et al. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc. Res. 2008, 78, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Mao, X.; Huang, Z.; Wang, F.; Wu, G.; Qiao, S. Arginine enhances embryo implantation in rats through PI3K/PKB/mTOR/NO signaling pathway during early pregnancy. Reproduction 2013, 145, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, B.; Pumiglia, K. mTORc1 activity is necessary and sufficient for phosphorylation of eNOSS1177. Physiol. Rep. 2018, 6, e13733. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Dunlop, E.A.; Ekim, B.; Maj, N.E.; Tee, A.R.; Fingar, D.C. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem. 2010, 285, 7866–7879. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Huang, Y.; Semple, I.; Kim, M.; Zhang, Z.; Lee, J.H. Cardioprotective roles of sestrin 1 and sestrin 2 against doxorubicin cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H39–H48. [Google Scholar] [CrossRef]
- Fan, L.; Hsieh, P.N.; Sweet, D.R.; Jain, M.K. Krüppel-like factor 15: Regulator of BCAA metabolism and circadian protein rhythmicity. Pharmacol. Res. 2018, 130, 123–126. [Google Scholar] [CrossRef]
- Liu, B.; Xu, L.; Yu, X.; Li, W.; Sun, X.; Xiao, S.; Guo, M.; Wang, H. Protective effect of KLF15 on vascular endothelial dysfunction induced by TNF-α. Mol. Med. Rep. 2018, 18, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Uddin, G.M.; Zhang, L.; Shah, S.; Fukushima, A.; Wagg, C.S.; Gopal, K.; Al Batran, R.; Pherwani, S.; Ho, K.L.; Boisvenue, J.; et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc. Diabetol. 2019, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhen, H.; Kitaura, Y.; Kadota, Y.; Ishikawa, T.; Kondo, Y.; Xu, M.; Morishita, Y.; Ota, M.; Ito, T.; Shimomura, Y. mTORC1 is involved in the regulation of branched-chain amino acid catabolism in mouse heart. FEBS Open Bio 2016, 6, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, Y.; Honda, T.; Shiraki, M.; Murakami, T.; Sato, J.; Kobayashi, H.; Mawatari, K.; Obayashi, M.; Harris, R.A. Branched-chain amino acid catabolism in exercise and liver disease. J. Nutr. 2006, 136, 250S–253S. [Google Scholar] [CrossRef] [Green Version]
- Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J. Biol. Chem. 2011, 286, 6128–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Dong, W.; Shao, J.; Wang, Y.; Zhou, M.; Sun, H. Branched-Chain Amino Acid Negatively Regulates KLF15 Expression via PI3K-AKT Pathway. Front. Physiol. 2017, 8, 853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Luis, M.; Lin, X.; Wang, M.; Cai, L.; Cen, C.; Biskup, E. Anthracycline-induced cardiotoxicity in the chemotherapy treatment of breast cancer: Preventive strategies and treatment (Review). Mol. Clin. Oncol. 2019, 11, 15–23. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New insights into the role of mtor signaling in the cardiovascular system. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Noack, C.; Zafiriou, M.-P.; Schaeffer, H.-J.; Renger, A.; Pavlova, E.; Dietz, R.; Zimmermann, W.H.; Bergmann, M.W.; Zelarayán, L.C. Krueppel-like factor 15 regulates Wnt/β-catenin transcription and controls cardiac progenitor cell fate in the postnatal heart. EMBO Mol. Med. 2012, 4, 992–1007. [Google Scholar] [CrossRef]
- Sugi, K.; Hsieh, P.N.; Ilkayeva, O.; Shelkay, S.; Moroney, B.; Baadh, P.; Haynes, B.; Pophal, M.; Fan, L.; Newgard, C.B.; et al. Kruppel-like factor 15 is required for the cardiac adaptive response to fasting. PLoS ONE 2018, 13, e0192376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaraj, D.; Scheer, F.A.J.L.; Ripperger, J.A.; Haldar, S.M.; Lu, Y.; Prosdocimo, D.A.; Eapen, S.J.; Eapen, B.L.; Cui, Y.; Mahabeleshwar, G.H.; et al. Klf15 orchestrates circadian nitrogen homeostasis. Cell Metab. 2012, 15, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisch, S.; Gray, S.; Heymans, S.; Haldar, S.M.; Wang, B.; Pfister, O.; Cui, L.; Kumar, A.; Lin, Z.; Sen-Banerjee, S.; et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 7074–7079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Haldar, S.M.; Lu, Y.; Ibrahim, O.A.; Fisch, S.; Gray, S.; Leask, A.; Jain, M.K. The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J. Mol. Cell. Cardiol. 2008, 45, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Lu, G.; Gao, C.; Wang, Y.; Sun, H. Tissue-specific and nutrient regulation of the branched-chain α-keto acid dehydrogenase phosphatase, protein phosphatase 2Cm (PP2Cm). J. Biol. Chem. 2012, 287, 23397–23406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.Y.; Chalasani, U.; Pan, N.; Friedline, R.H.; Prosdocimo, D.A.; Nam, M.; Azuma, Y.; Maganti, R.; Yu, K.; Velagapudi, A.; et al. KLF15 is a molecular link between endoplasmic reticulum stress and insulin resistance. PLoS ONE 2013, 8, e77851. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Liu, H.; Zhang, J.; Huang, J.; Zhu, C.; Lu, Y.; Shi, Y.; Wang, Y. Ursolic acid prevents doxorubicin-induced cardiac toxicity in mice through eNOS activation and inhibition of eNOS uncoupling. J. Cell. Mol. Med. 2019, 23, 2174–2183. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Kassem, H.H. Protective effect of nebivolol on doxorubicin-induced cardiotoxicity in rats. Arch. Med. Sci. 2018, 14, 1450–1458. [Google Scholar] [CrossRef]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell. Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef] [Green Version]
- Akolkar, G.; Bagchi, A.K.; Ayyappan, P.; Jassal, D.S.; Singal, P.K. Doxorubicin-induced nitrosative stress is mitigated by vitamin C via the modulation of nitric oxide synthases. Am. J. Physiol. Cell Physiol. 2017, 312, C418–C427. [Google Scholar] [CrossRef] [Green Version]
- Zeglinski, M.; Premecz, S.; Lerner, J.; Wtorek, P.; daSilva, M.; Hasanally, D.; Chaudhary, R.; Sharma, A.; Thliveris, J.; Ravandi, A.; et al. Congenital Absence of Nitric Oxide Synthase 3 Potentiates Cardiac Dysfunction and Reduces Survival in Doxorubicin- and Trastuzumab-Mediated Cardiomyopathy. Can. J. Cardiol. 2014, 30, 359–367. [Google Scholar] [CrossRef]
- Krajinovic, M.; Elbared, J.; Drouin, S.; Bertout, L.; Rezgui, A.; Ansari, M.; Raboisson, M.-J.; Lipshultz, S.E.; Silverman, L.B.; Sallan, S.E.; et al. Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharmacogen. J. 2016, 16, 530–535. [Google Scholar] [CrossRef]
- Mitchell, M.I.; Engelbrecht, A.-M. Circadian Rhythms and Breast Cancer: The Role of Per2 in Doxorubicin-Induced Cell Death. J. Toxicol. 2015, 2015, 392360. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, T.; Aygenli, F.; Emisoglu, H.; Ozcelik, G.; Canturk, A.; Yilmaz, S.; Ozturk, N. Opposite Carcinogenic Effects of Circadian Clock Gene BMAL1. Sci. Rep. 2018, 8, 16023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, Y.; Wang, X.; Li, H.; Zhang, H.; Gong, J.; Shen, S.; Yin, W.; Hu, H. Efficacy and safety of oral branched-chain amino acid supplementation in patients undergoing interventions for hepatocellular carcinoma: A meta-analysis. Nutr. J. 2015, 14, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudry, H.A.; Pan, M.; Karinch, A.M.; Souba, W.W. Branched-chain amino acid-enriched nutritional support in surgical and cancer patients. J. Nutr. 2006, 136 (Suppl. 1), 314S–318S. [Google Scholar] [CrossRef]
- McKay, B.P.; Larder, A.L.; Lam, V. Pre-Operative vs. Peri-Operative Nutrition Supplementation in Hepatic Resection for Cancer: A Systematic Review. Nutr. Cancer 2019, 71, 179–198. [Google Scholar] [CrossRef]
- Wubetu, G.Y.; Utsunomiya, T.; Ishikawa, D.; Ikemoto, T.; Yamada, S.; Morine, Y.; Iwahashi, S.; Saito, Y.; Arakawa, Y.; Imura, S.; et al. Branched chain amino acid suppressed insulin-initiated proliferation of human cancer cells through induction of autophagy. Anticancer Res. 2014, 34, 4789–4796. [Google Scholar]
- Nishikawa, H.; Kita, R.; Kimura, T.; Ohara, Y.; Takeda, H.; Sakamoto, A.; Saito, S.; Nishijima, N.; Nasu, A.; Komekado, H.; et al. Clinical significance of early interventional therapy of branched-chain amino acid granules in patients with hepatocellular carcinoma: Propensity score matching analysis. Int. J. Oncol. 2014, 45, 1082–1090. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition (%) | BCAAem | α5 |
|---|---|---|
| Leucine | 30.01 | 31.0885 |
| Lysine | 19.58 | 16.9030 |
| Isoleucine | 15.00 | 10.3628 |
| Valine | 15.00 | 10.3628 |
| Threonine | 8.40 | 7.2540 |
| Cysteine | 3.60 | 3.1089 |
| Histidine | 3.60 | 3.1089 |
| Phenylalanine | 2.40 | 2.0726 |
| Methionine | 1.20 | 1.0363 |
| Tyrosine | 0.72 | 0.6218 |
| Tryptophan | 0.48 | 2.0726 |
| Citric acid | - | 8.001 |
| Succinic acid | - | 2.00 |
| Malic acid | - | 2.00 |
| Vitamin B1 | - | 0.004 |
| Vitamin B6 | - | 0.0038 |
| Gene | Sense Primer (5′-3’) | Antisense Primer (5′-3’) | PCR Product (bp) | Ta (°C) |
|---|---|---|---|---|
| Cat | CACTGACGAGATGGCACACTTTG | TGGAGAACCGAACGGCAATAGG | 173 | 60 |
| COX IV | TGGGACTATGACAAGAATGAGTGG | TTAGCATGGACCATTGGATACGG | 113 | 60 |
| cyt c | ATAGGGGCATGTCACCTCAAAC | GTGGTTAGCCATGACCTGAAAG | 172 | 60 |
| eNOS | AGCGGCTGGTACATGAGTTC | GATGAGGTTGTCCTGGTGTCC | 116 | 60 |
| GPX1 | TCTGGGACCTCGTGGACTG | CACTTCGCACTTCTCAAACAATG | 156 | 60 |
| KLF15 | ACACCAAGAGCAGCCACCTC | TGAGATCGCCGGTGCCTTGA | 130 | 60 |
| mtDNA | CCACTTCATCTTACCATTTA | ATCTGCATCTGAGTTTAATC | 106 | 54 |
| NRF1 | ACAGATAGTCCTGTCTGGGGAAA | TGGTACATGCTCACAGGGATCT | 99 | 60 |
| PGC-1α | ACTATGAATCAAGCCACTACAGAC | TTCATCCCTCTTGAGCCTTTCG | 148 | 60 |
| SESN2 | GCCCCTGAGAAGCTCCGCAA | GAGTTCGGCCAGGGACCAGC | 129 | 60 |
| SOD1 | GGCTTCTCGTCTTGCTCTC | AACTGGTTCACCGCTTGC | 153 | 60 |
| SOD2 | GCCTCCCAGACCTGCCTTAC | GTGGTACTTCTCCTCGGTGGCG | 131 | 60 |
| TBP | ACCCTTCACCAATGACTCCTATG | TGACTGCAGCAAATCGCTTGG | 186 | 60 |
| Tfam | AAGACCTCGTTCAGCATATAACATT | TTTTCCAAGCCTCATTTACAAGC | 104 | 60 |
| 18S | CTGCCCTATCAACTTTCGATGGTAG | CCGTTTCTCAGGCTCCCTCTC | 100 | 60 |
| CTRL | α5 | DOX | DOX + α5 | |
|---|---|---|---|---|
| Body weight (g) Heart weight (g) Food intake (g) Water intake (g) | 24.84 ± 1.3 0.12 ± 0.01 5.2 ± 0.41 6.1 ± 0.7 | 24.46 ± 2 0.11 ± 0.02 5.34 ± 0.7 8.24 ± 0.88 * | 20.75 ± 0.82 * 0.085 ± 0.02 * 3.73 ± 0.6* 6.13 ± 1.0 | 20.46 ± 1.04 * 0.09 ± 0.02 * 3.59 ± 0.47 * 7.07 ± 1.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tedesco, L.; Rossi, F.; Ragni, M.; Ruocco, C.; Brunetti, D.; Carruba, M.O.; Torrente, Y.; Valerio, A.; Nisoli, E. A Special Amino-Acid Formula Tailored to Boosting Cell Respiration Prevents Mitochondrial Dysfunction and Oxidative Stress Caused by Doxorubicin in Mouse Cardiomyocytes. Nutrients 2020, 12, 282. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12020282
Tedesco L, Rossi F, Ragni M, Ruocco C, Brunetti D, Carruba MO, Torrente Y, Valerio A, Nisoli E. A Special Amino-Acid Formula Tailored to Boosting Cell Respiration Prevents Mitochondrial Dysfunction and Oxidative Stress Caused by Doxorubicin in Mouse Cardiomyocytes. Nutrients. 2020; 12(2):282. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12020282
Chicago/Turabian StyleTedesco, Laura, Fabio Rossi, Maurizio Ragni, Chiara Ruocco, Dario Brunetti, Michele O. Carruba, Yvan Torrente, Alessandra Valerio, and Enzo Nisoli. 2020. "A Special Amino-Acid Formula Tailored to Boosting Cell Respiration Prevents Mitochondrial Dysfunction and Oxidative Stress Caused by Doxorubicin in Mouse Cardiomyocytes" Nutrients 12, no. 2: 282. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12020282