S-Adenosylmethionine Deficiency and Brain Accumulation of S-Adenosylhomocysteine in Thioacetamide-Induced Acute Liver Failure


Abstract
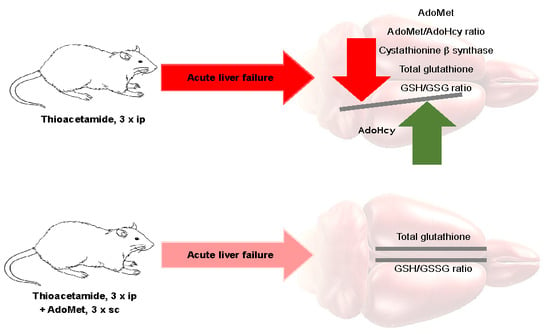
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
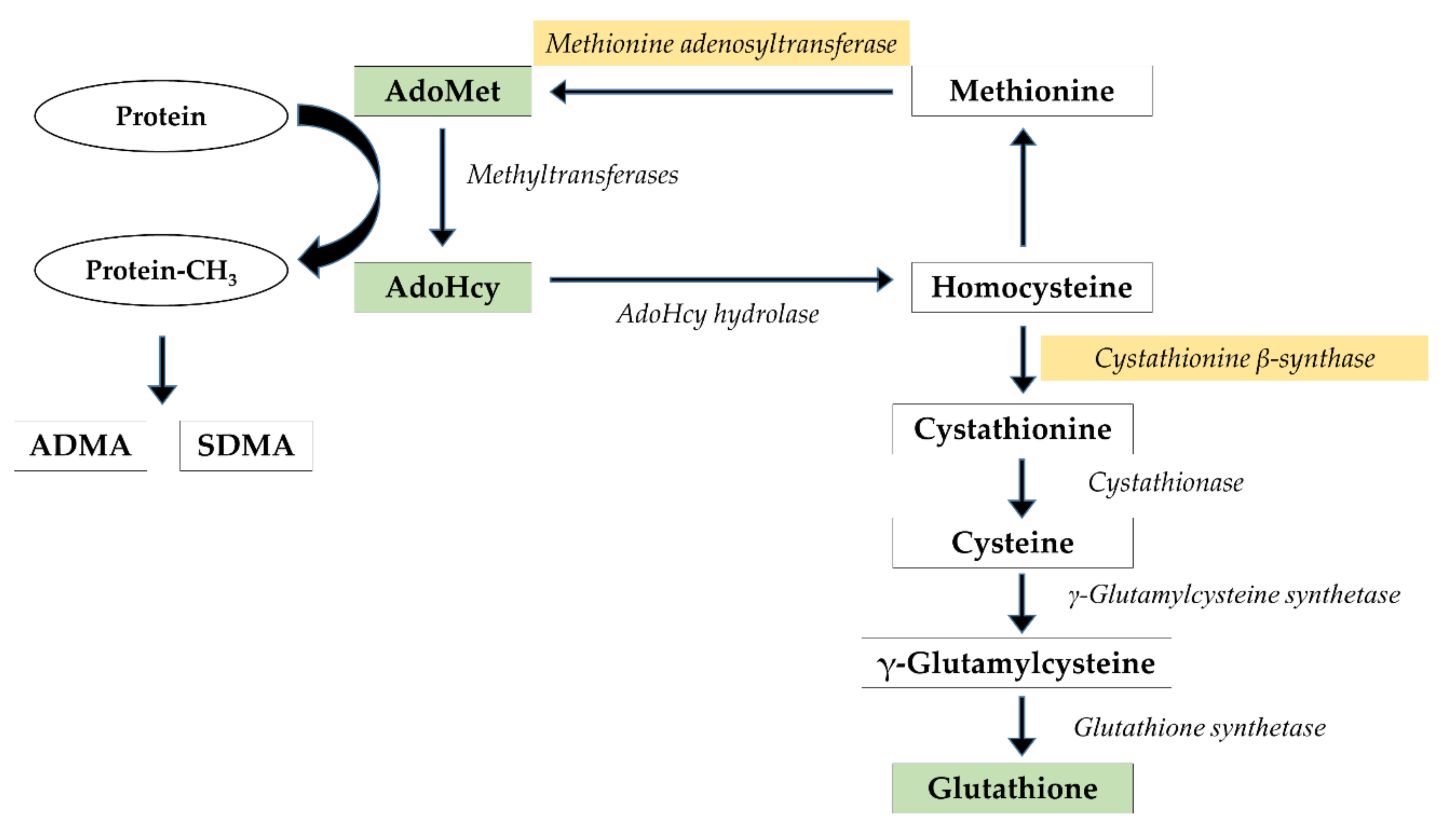
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals
2.3. The Acute Liver Failure Model and S-Adenosylmethionine Administration
2.4. Determination of the Tissue Level of AdoMet and AdoHcy by HPLC-FD
2.5. GSSG/GSH Determination
2.6. Immunoblotting of CBS and MAT1A Proteins
2.7. Statistical Analysis
3. Results
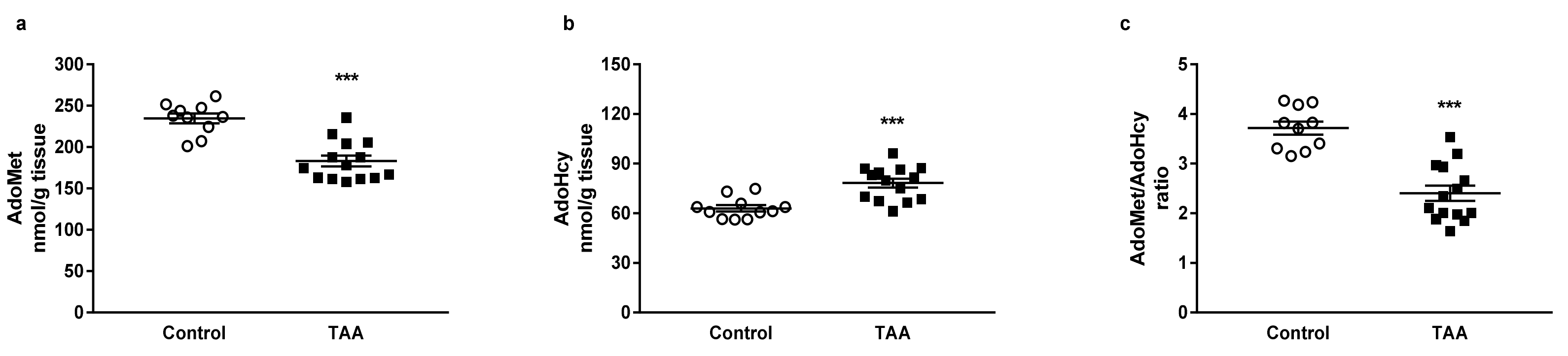
3.1. Cerebral and Hepatic AdoMet and AdoHcy Concentrations
3.1.1. Cerebral Cortex
3.1.2. Liver
3.2. CBS Protein Expression in the Rat Cerebral Cortex
3.3. CBS and MAT1A Protein Expression in the Rat Liver
3.4. The Effect of AdoMet (10 mg/kg sc) Treatment on the Total Glutathione Level and GSH/GSSG Ratio in the Cerebral Cortex
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vitvitsky, V.; Thomas, M.; Ghorpade, A.; Gendelman, H.E.; Banerjee, R. A Functional Transsulfuration Pathway in the Brain Links to Glutathione Homeostasis. J. Biol. Chem. 2006, 281, 35785–35793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudova, A.; Bauman, Z.; Braun, A.; Vitvitsky, V.; Lu, S.C.; Banerjee, R. S-adenosylmethionine stabilizes cystathionine β-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and Methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenen, W.A.M. S-Adenosylmethionine: jack of all trades and master of everything? Biochem. Soc. Trans. 2006, 34, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, C.S. S-Adenosyl–L-methionine: Its role in the treatment of liver disorders. Am. J. Clin. Nutr. 2002, 76, 1183–1187S. [Google Scholar] [CrossRef] [Green Version]
- Mato, J.M.; Lu, S.C. Role of S-adenosyl–L-methionine in liver health and injury. Hepatology 2007, 45, 1306–1312. [Google Scholar] [CrossRef]
- Guo, T.; Chang, L.; Xiao, Y.; Liu, Q. S-adenosyl–L-methionine for the treatment of chronic liver disease: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0122124. [Google Scholar] [CrossRef]
- Lieber, C.S. Role of S-adenosyl–L-methionine in the treatment of liver diseases. J. Hepatol. 1999, 30, 1155–1159. [Google Scholar] [CrossRef]
- Fontecave, M.; Atta, M.; Mulliez, E. S-adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 2004, 29, 243–249. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Mato, J.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Caudill, M.A.; Wang, J.C.; Melnyk, S.; Pogribny, I.P.; Jernigan, S.; Collins, M.D.; Santos-Guzman, J.; Swendseid, M.E.; Cogger, E.A.; James, S.J. Intracellular S-Adenosylhomocysteine Concentrations Predict Global DNA Hypomethylation in Tissues of Methyl-Deficient Cystathionine β-Synthase Heterozygous Mice. J. Nutr. 2001, 131, 2811–2818. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Dubchenko, E.A.; Kruglova, M.P.; Virus, E.D.; Bulgakova, P.O.; Alexandrin, V.V.; Fedoseev, A.N.; Boyko, A.N.; Grachev, S.V.; Kubatiev, A.A. Determination of S-adenosylmethionine and S-adenosylhomocysteine in blood plasma by UPLC with fluorescence detection. J. Chromatogr. B 2019, 1124, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.; Milewski, K.; Jaźwiec, R.; Zielińska, M. Intracerebral Administration of S-Adenosylhomocysteine or S-Adenosylmethionine Attenuates the Increases in the Cortical Extracellular Levels of Dimethylarginines Without Affecting cGMP Level in Rats with Acute Liver Failure. Neurotox. Res. 2017, 31, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.-Z.; Mato, J.M.; Kanel, G.; Lu, S.C. Differential effect of thioacetamide on hepatic methionine adenosyltransferase expression in the rat. Hepatology 1999, 29, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Mesa, M.L.; Carrizosa, R.; Martinez-Honduvilla, C.; Benito, M.; Fabregat, I. Changes in rat liver gene expression induced by thioacetamide: Protective role of S-adenosyl–L-methionine by a glutathione-dependent mechanism. Hepatology 1996, 23, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Ming, Z.; Fan, Y.J.; Yang, X.; Wayne Lautt, W. Synergistic protection by S-adenosylmethionine with vitamins C and E on liver injury induced by thioacetamide in rats. Free Radic. Biol. Med. 2006, 40, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Correa, J.A.; De La Cruz, J.P.; Martin-Aurioles, E.; Lopez-Egea, M.A.; Ortiz, P.; de la Cuesta, F.S. Effects of S-adenosyl-L-methionine on hepatic and renal oxidative stress in an experimental model of acute biliary obstruction in rats. Hepatology 1997, 26, 121–127. [Google Scholar] [CrossRef]
- Castro, R.; Struys, E.A.; Jansen, E.E.W.; Blom, H.J.; de Almeida, I.T.; Jakobs, C. Quantification of plasma S-adenosylmethionine and S-adenosylhomocysteine as their fluorescent 1,N6-etheno derivatives: An adaptation of previously described methodology. J. Pharm. Biomed. Anal. 2002, 29, 963–968. [Google Scholar] [CrossRef]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Corrales, F.; Giménez, A.; Alvarez, L.; Caballería, J.; Pajares, M.A.; Andreu, H.; Parés, A.; Mato, J.M.; Rodés, J. S-adenosylmethionine treatment prevents carbon tetrachloride—InducedS-adenosylmethionine synthetase inactivation and attenuates liver injury. Hepatology 1992, 16, 1022–1027. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.; Mingorance, J.; Martínez-Chantar, M.; Casado, M.; Martín-Sanz, P.; Boscá, L.; Mato, J. Regulation of rat liver S-adenosylmethionine synthetase during septic shock: Role of nitric oxide. Hepatology 1997, 25, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Cabrero, C.; Duce, A.M.; Ortiz, P.; Alemany, S.; Mato, J.M. Specific loss of the high-molecular-weight form ofS-adenosyl-L-methionine synthetase in human liver cirrhosis. Hepatology 1988, 8, 1530–1534. [Google Scholar] [CrossRef]
- Okada, G.; Sawai, Y.; Teraoka, H.; Tsukada, K. Differential effects of dimethylsulfoxide on S -adenosylmethionine synthetase from rat liver and hepatoma. FEBS Lett. 1979, 106, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Montanari, A.; Simoni, I.; Vallisa, D.; Trifirò, A.; Colla, R.; Abbiati, R.; Borghi, L.; Novarini, A. Free amino acids in plasma and skeletal muscle of patients with liver cirrhosis. Hepatology 1988, 8, 1034–1039. [Google Scholar] [CrossRef]
- Hardwick, D.F.; Applegarth, D.A.; Cockcroft, D.M.; Ross, P.M.; Calder, R.J. Pathogenesis of methionine-induced toxicity. Metabolism 1970, 19, 381–391. [Google Scholar] [CrossRef]
- Noureddin, M.; Sander-Struckmeier, S.; Mato, J.M. Early treatment efficacy of S-adenosylmethionine in patients with intrahepatic cholestasis: A systematic review. World J. Hepatol. 2020, 12, 46. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Hepatoprotective effects of S -adenosyl-L-methionine against alcohol- and cytochrome P450 2E1-induced liver injury. World J. Gastroenterol. 2010, 16, 1366. [Google Scholar] [CrossRef]
- Yang, H.; Ramani, K.; Xia, M.; Ko, K.S.; Li, T.W.H.; Oh, P.; Li, J.; Lu, S.C. Dysregulation of glutathione synthesis during cholestasis in mice: Molecular mechanisms and therapeutic implications. Hepatology 2009, 49, 1982–1991. [Google Scholar] [CrossRef] [Green Version]
- Vincenzi, B.; Russo, A.; Terenzio, A.; Galvano, A.; Santini, D.; Vorini, F.; Antonelli-Incalzi, R.; Vespasiani-Gentilucci, U.; Tonini, G. The use of same in chemotherapy-induced liver injury. Crit. Rev. Oncol. Hematol. 2018, 130, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.I.; García-Román, J.; Gómez-Ñañez, I.; García-Román, R. Chronic liver diseases and the potential use of S-adenosyl-l-methionine as a hepatoprotector. Eur. J. Gastroenterol. Hepatol. 2018, 30, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D. S-adenosylmethionine (SAMe) for neuropsychiatric disorders: A clinician-oriented review of research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [Green Version]
- Tallan, H.; Moore, S.; Stein, W. L-cystathionine in human brain. J. Biol. Chem. 1958, 230, 707–716. [Google Scholar]
- Banerjee, R.; Vitvitsky, V.; Garg, S.K. The undertow of sulfur metabolism on glutamatergic neurotransmission. Trends Biochem. Sci. 2008, 33, 413–419. [Google Scholar] [CrossRef]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the Antioxidant Glutathione in Neurons: Supply by Astrocytes of CysGly as Precursor for Neuronal Glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, A.M.; Hilgier, W.; Zielińska, M. S-Adenosylmethionine Deficiency and Brain Accumulation of S-Adenosylhomocysteine in Thioacetamide-Induced Acute Liver Failure. Nutrients 2020, 12, 2135. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12072135
Czarnecka AM, Hilgier W, Zielińska M. S-Adenosylmethionine Deficiency and Brain Accumulation of S-Adenosylhomocysteine in Thioacetamide-Induced Acute Liver Failure. Nutrients. 2020; 12(7):2135. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12072135
Chicago/Turabian StyleCzarnecka, Anna Maria, Wojciech Hilgier, and Magdalena Zielińska. 2020. "S-Adenosylmethionine Deficiency and Brain Accumulation of S-Adenosylhomocysteine in Thioacetamide-Induced Acute Liver Failure" Nutrients 12, no. 7: 2135. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12072135