
Autonomic Nervous System Neuroanatomical Alterations Could Provoke and Maintain Gastrointestinal Dysbiosis in Autism Spectrum Disorder (ASD): A Novel Microbiome–Host Interaction Mechanistic Hypothesis


{kind=link}
{kind=link}
Abstract
:1. Introduction
- -
- Why is dysbiosis so prevalent in ASD?
- -
- How does dysbiosis arise in the first place?
- -
- How is it maintained?
2. Materials and Methods
3. Results and Discussion
3.1. A Novel “Intestinal Microbiome–Host Interaction” Hypothesis
3.2. Autonomic Nervous System Dysfunction in Association with ASD
3.3. The Autonomic Nervous System and Intestinal Immune Homeostasis
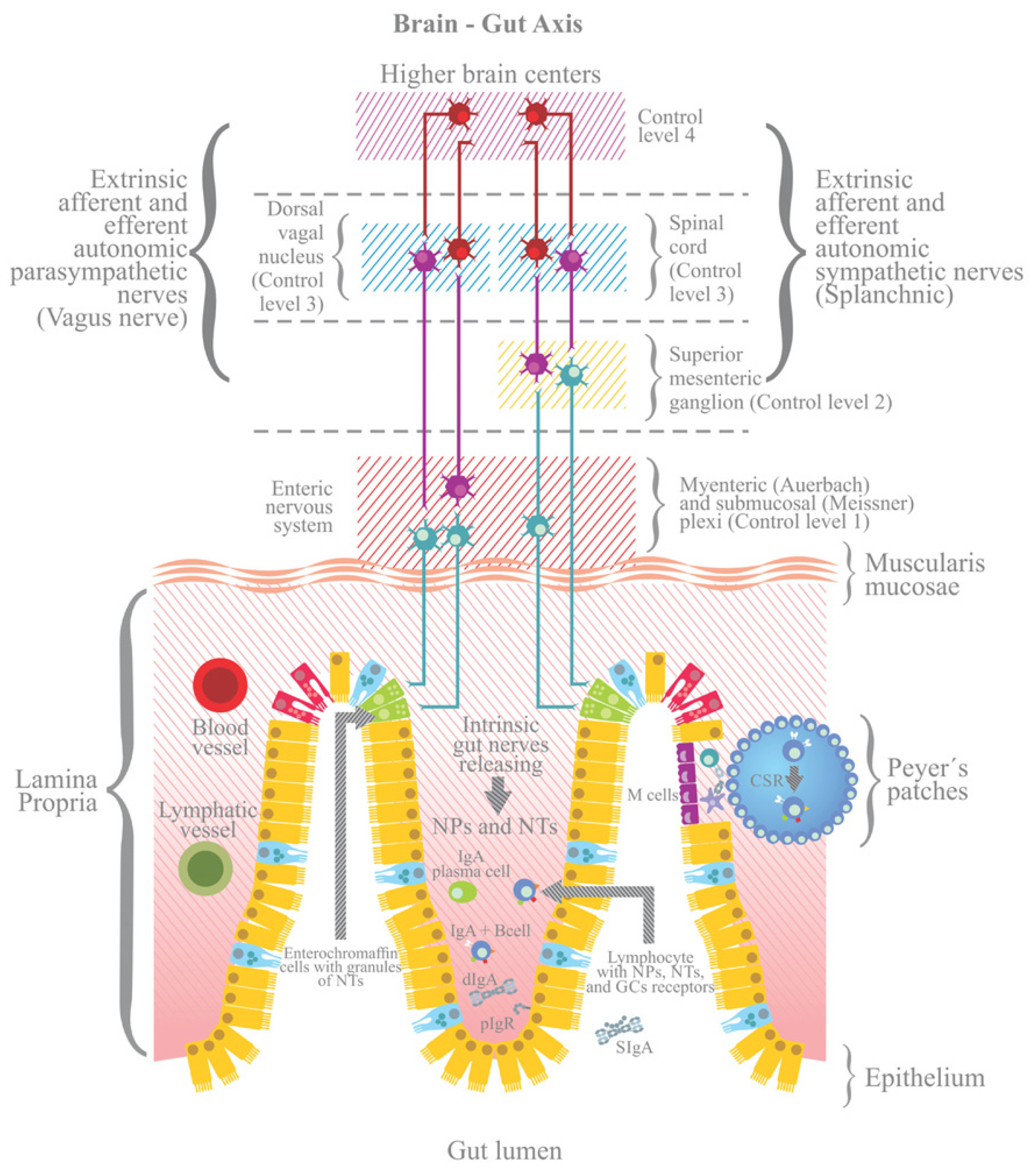
3.4. The ANS and the Neuronal Control of the Intestine
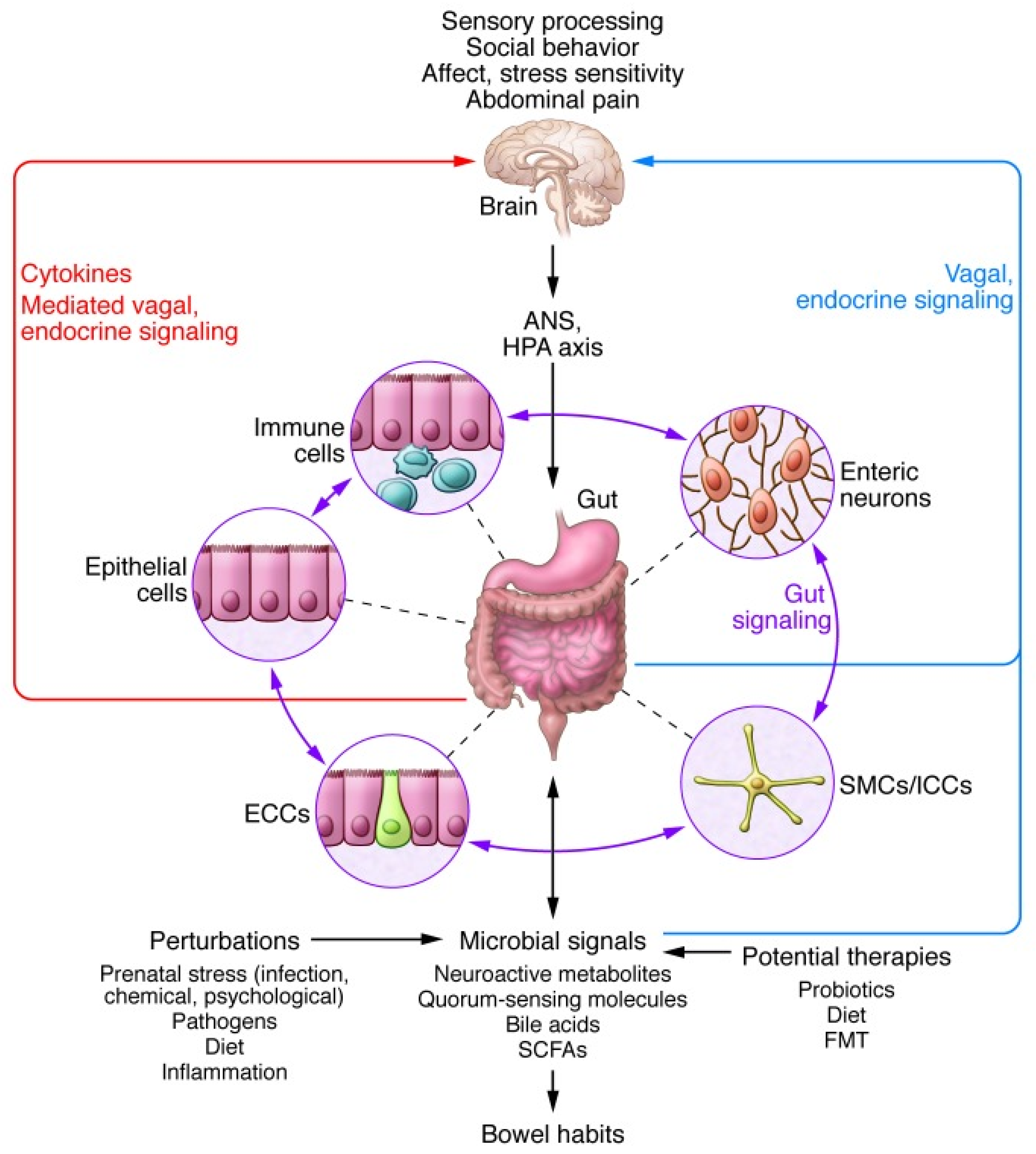
3.5. The Brain–Gut Axis: Microbiome–Host Interactions and Immuno–Modulation
3.6. The ANS and the Control of Intestinal Immune Homeostasis
3.7. Persistent Dysbiosis in the Absence of Chronic Low-Grade Inflammation
3.8. ANS Functioning Abnormalities and Gastrointestinal Dysfunctions
4. Conclusions
- -
- The neuro–anatomical alterations that underlie ASD provoke autonomic nervous system (ANS) functioning abnormalities characterized by an over-activation of the sympathetic branch of the ANS on a background of parasympathetic activity deficits;
- -
- This induces deregulation of the gut–brain axis;
- -
- This results in attenuation of the humoral and cellular components of the intestinal immune system, together with dysregulation of intestinal osmotic homeostasis and mucus production, with ensuing, persistent dysbiosis, which then sets-up a vicious cycle where immune and osmotic functioning abnormalities maintain a dysbiotic state which, in turn, maintains immune and osmotic dysregulation.
- -
- One of the possible origins of ASD-associated dysbiosis;
- -
- Its persistence;
- -
- Why improvements achieved by microbiota transfer therapy are generally transient;
- -
- Why, in spite of persistent dysbiosis, ASD is not associated with systemic chronic low-grade inflammation (as opposed to neuroinflammation), while all other psychiatric disorders also associated with persistent dysbiosis are;
- -
- The origins of the unusually high incidences of gastrointestinal dysfunctions;
- -
- The high frequency of comorbidities such as sleep disorders, anxiety, and depression;
- -
- Perhaps most importantly, the reasons for the very high heterogeneities in terms of forms and types of dysbioses as well as in frequencies of comorbidities associated with clinical ASD.
Author Contributions
Funding
Conflicts of Interest
References
- Coury, D.L.; Ashwood, P.; Fasano, A.; Fuchs, G.; Geraghty, M.; Kaul, A.; Mawe, G.; Patterson, P.; Jones, N.E. Gastrointestinal conditions in children with autism spectrum disorder: Developing a research agenda. Pediatrics 2012, 130, S160–S168. [Google Scholar] [CrossRef] [Green Version]
- Horvath, K.; Perman, J.A. Autistic disorder and gastrointestinal disease. Curr. Opin. Pediatr. 2002, 14, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; et al. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. 2011, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiola, F.; Ianiro, G.; Franceschi, F.; Fagiuoli, S.; Gasbarrini, G.; Gasbarrini, A. Gut microbiota in autism and mood disorders. World J. Gastroenterol. 2016, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseribafrouei, A.; Hestad, K.; Avershina, E.; Sekelja, M.; Linlokken, A.; Wilson, R.; Rudi, K. Correlation between the human fecal microbiota and depression. Neurogastroenterol. Motil. 2014, 26, 1155–1162. [Google Scholar] [CrossRef]
- Kelly, J.R.; Borre, Y.; O’Brian, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G.; et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jia, H.; Zhou, C.; Yang, Y.; Zhao, Y.; Yang, M.; Zou, Z. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm. Biomed. Anal. 2017, 138, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2002, 35, S6–S16. [Google Scholar] [CrossRef]
- Song, Y.; Liu, C.; Finegold, S.M. Real-time PCR quantitation of clostridia in feces of autistic children. Appl. Environ. Microbiol. 2004, 70, 6459–6465. [Google Scholar] [CrossRef] [Green Version]
- Parracho, H.M.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism–comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; Labaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.L.; Hornig, M.; Parekh, T.; Lipkin, W.I. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [Green Version]
- De Palma, G.; Blennerhassett, P.; Lu, J.; Deng, Y.; Park, A.J.; Green, W.; Denou, E.; Silva, M.A.; Santacruz, A.; Sanz, Y.; et al. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat. Commun. 2015, 6, 7735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tye, C.; Runicles, A.K.; Whitehouse, A.J.O.; Alvares, G.A. Characterizing the Interplay Between Autism Spectrum Disorder and Comorbid Medical Conditions: An Integrative Review. Front. Psychiatry 2018, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Kent, R.S. Front-matter. In Anxiety in Children and Adolescents with Autism Spectrum Disorder; Kerns, C.M., Renno, P., Storch, E.A., Kendall, P.C., Wood, J.J., Eds.; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2017; pp. 1–3. [Google Scholar]
- Hudson, C.C.; Hall, L.; Harkness, K.L. Prevalence of Depressive Disorders in Individuals with Autism Spectrum Disorder: A Meta-Analysis. J. Abnorm. Child Psychol. 2019, 47, 165–175. [Google Scholar] [CrossRef]
- Klukowski, M.; Wasilewska, J.; Lebensztejn, D. Sleep and gastrointestinal disturbances in autism spectrum disorder in children. Dev. Period. Med. 2015, 19, 157–161. [Google Scholar]
- Liu, X.; Hubbard, J.A.; Fabes, R.A.; Adam, J.B. Sleep disturbances and correlates of children with autism spectrum disorders. Child Psychiatry Hum. Dev. 2006, 37, 179–191. [Google Scholar] [CrossRef]
- Louis, P. Does the human gut microbiota contribute to the etiology of autism spectrum disorders? Dig. Dis. Sci. 2012, 57, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Samonis, G.; Gikas, A.; Anaissie, E.J.; Vrenzos, G.; Maraki, S.; Tselentis, Y.; Bodey, G.P. Prospective evaluation of effects of broad-spectrum antibiotics on gastrointestinal yeast colonization of humans. Antimicrob. Agents Chemother. 1993, 37, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2013, 62, 1591–1601. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Sathe, N.; Andrews, J.C.; McPheeters, M.L.; Warren, Z.E. Nutritional and Dietary Interventions for Autism Spectrum Disorder: A Systematic Review. Pediatrics 2017, 139. [Google Scholar] [CrossRef]
- Li, Y.J.; Li, Y.M.; Xiang, D.X. Supplement intervention associated with nutritional deficiencies in autism spectrum disorders: A systematic review. Eur. J. Nutr. 2018, 57, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Vargason, T.; Santiago, M.; Hahn, J.; Krajmalnik-Brown, R. Distinct Fecal and Plasma Metabolites in Children with Autism Spectrum Disorders and Their Modulation after Microbiota Transfer Therapy. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Borody, T.J.; Kang, D.W.; Khoruts, A.; Krajmalnik-Brown, R.; Sadowsky, M.J. Microbiota transplant therapy and autism: Lessons for the clinic. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1033–1037. [Google Scholar] [CrossRef]
- Parker, K.J.; Oztan, O.; Libove, R.A.; Sumiyoshi, R.D.; Jackson, L.P.; Karhson, D.S.; Summers, J.E.; Hinman, K.E.; Motonaga, K.S.; Phillips, J.M.; et al. Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc. Natl. Acad. Sci. USA 2017, 114, 8119–8124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Kagitani-Shimono, K.; Mohri, I.; Yamamoto, T.; Sanefuji, W.; Nakamura, A.; Oishi, M.; Kimura, T.; Onaka, T.; Ozono, K.; et al. Long-term administration of intranasal oxytocin is a safe and promising therapy for early adolescent boys with autism spectrum disorders. J. Child. Adolesc. Psychopharmacol. 2013, 23, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Moy, S.S.; Teng, B.L.; Nikolova, V.D.; Riddick, N.V.; Simpson, C.D.; Van Deusen, A.; Janzen, W.P.; Sassano, M.F.; Pedersen, C.A.; Jarstfer, M.B. Prosocial effects of an oxytocin metabolite, but not synthetic oxytocin receptor agonists, in a mouse model of autism. Neuropharmacology 2019, 144, 301–311. [Google Scholar] [CrossRef]
- Yamasue, H.; Okada, T.; Munesue, T.; Kuroda, M.; Fujioka, T.; Uno, Y.; Matsumoto, K.; Kuwabara, H.; Mori, D.; Okamoto, Y.; et al. Effect of intranasal oxytocin on the core social symptoms of autism spectrum disorder: A randomized clinical trial. Mol. Psychiatry 2018, 25. [Google Scholar] [CrossRef]
- Kruppa, J.A.; Gossen, A.; Oberwelland Weiss, E.; Kohls, G.; Grossheinrich, N.; Cholemkery, H.; Freitag, C.M.; Karges, W.; Wolfle, E.; Sinzig, J.; et al. Neural modulation of social reinforcement learning by intranasal oxytocin in male adults with high-functioning autism spectrum disorder: A randomized trial. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2019, 44, 749–756. [Google Scholar] [CrossRef]
- Huang, T.T.; Lai, J.B.; Du, Y.L.; Xu, Y.; Ruan, L.M.; Hu, S.H. Current Understanding of Gut Microbiota in Mood Disorders: An Update of Human Studies. Front. Genet. 2019, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Immune Dysfunction: Epidemiological Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Rosenblat, J.D.; Brietzke, E.; Mansur, R.B.; Maruschak, N.A.; Lee, Y.; McIntyre, R.S. Inflammation as a neurobiological substrate of cognitive impairment in bipolar disorder: Evidence, pathophysiology and treatment implications. J. Affect. Disord. 2015, 188, 149–159. [Google Scholar] [CrossRef]
- Rosenblat, J.D. Targeting the immune system in the treatment of bipolar disorder. Psychopharmacology 2019, 236, 2909–2921. [Google Scholar] [CrossRef]
- Dipasquale, V.; Cutrupi, M.C.; Colavita, L.; Manti, S.; Cuppari, C.; Salpietro, C. Neuroinflammation in Autism Spectrum Disorders: Role of High Mobility Group Box 1 Protein. Int J. Mol. Cell Med. 2017, 6, 148–155. [Google Scholar] [CrossRef]
- Eissa, N.; Sadeq, A.; Sasse, A.; Sadek, B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front. Pharm. 2020, 11, 886. [Google Scholar] [CrossRef]
- Gadal, F.; Bozic, C.; Pillot-Brochet, C.; Malinge, S.; Wagner, S.; Le Cam, A.; Buffat, L.; Crepin, M.; Iris, F. Integrated transcriptome analysis of the cellular mechanisms associated with Ha-ras-dependent malignant transformation of the human breast epithelial MCF7 cell line. Nucleic Acids Res. 2003, 31, 5789–5804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadal, F.; Starzec, A.; Bozic, C.; Pillot-Brochet, C.; Malinge, S.; Ozanne, V.; Vicenzi, J.; Buffat, L.; Perret, G.; Iris, F.; et al. Integrative analysis of gene expression patterns predicts specific modulations of defined cell functions by estrogen and tamoxifen in MCF7 breast cancer cells. J. Mol. Endocrinol. 2005, 34, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Iris, F. Psychiatric systems medicine: Closer at hand than anticipated but not with the expected portrait. Pharmacopsychiatry 2012, 45, S12–S21. [Google Scholar] [CrossRef] [PubMed]
- Iris, F.; Filiou, M.; Turck, C.W. Differential Proteomics Analyses Reveal Anxiety-Associated Molecular and Cellular Mechanisms in Cingulate Cortex Synapses. Am. J. Psychiatry Neurosci. 2014, 2, 25–42. [Google Scholar] [CrossRef]
- Iris, F.; Gea, M.; Lampe, P.H.; Santamaria, P. [Production and implementation of predictive biological models]. Med. Sci. (Paris) 2009, 25, 608–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iris, F.J.M.; Gea, M.; Lampe, P.-H.; Querleux, B. Heuristic Modelling Applied to Epidermal Homeostasis. In Computational Biophysics of the Skin; Pan Stanford Publishing: Stanford, CA, USA, 2014; pp. 461–524. [Google Scholar]
- Nussbaumer, M.; Asara, J.M.; Teplytska, L.; Murphy, M.P.; Logan, A.; Turck, C.W.; Filiou, M.D. Selective Mitochondrial Targeting Exerts Anxiolytic Effects In Vivo. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 1751–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouillot, F.; Blois, H.; Iris, F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecur. Bioterror. 2010, 8, 155–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turck, C.W.; Iris, F. Proteome-based pathway modelling of psychiatric disorders. Pharmacopsychiatry 2011, 44, S54–S61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iris, F.; Beopoulos, A.; Gea, M. How scientific literature analysis yields innovative therapeutic hypothesis through integrative iterations. Curr. Opin. Pharm. 2018, 42, 62–70. [Google Scholar] [CrossRef]
- Furness, J.B. Integrated Neural and Endocrine Control of Gastrointestinal Function. Adv. Exp. Med. Biol. 2016, 891, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Fleshner, M.; Crane, C.R. Exosomes, DAMPs and miRNA: Features of Stress Physiology and Immune Homeostasis. Trends Immunol. 2017, 38, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.T.; Lu, J.T.; Ran, Z.H.; Zheng, Q. Exosome in intestinal mucosal immunity. J. Gastroenterol. Hepatol. 2016, 31, 1694–1699. [Google Scholar] [CrossRef] [Green Version]
- Willemze, R.A.; Welting, O.; van Hamersveld, P.; Verseijden, C.; Nijhuis, L.E.; Hilbers, F.W.; Meijer, S.L.; Heesters, B.A.; Folgering, J.H.A.; Darwinkel, H.; et al. Loss of intestinal sympathetic innervation elicits an innate immune driven colitis. Mol. Med. 2019, 25, 1. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Xiao, X.; Liu, Y.; Zhang, R.; Liu, J.; Liu, Q.; Wang, P.; Cheng, G. Mosquito C-type lectins maintain gut microbiome homeostasis. Nat. Microbiol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.C.E.; Shanahan, M.T.; Singh, A.P.; Sethupathy, P. Gut Microbial Influences on the Mammalian Intestinal Stem Cell Niche. Stem Cells Int. 2017, 2017, 5604727. [Google Scholar] [CrossRef]
- Wang, Y.; Hensley, M.K.; Tasman, A.; Sears, L.; Casanova, M.F.; Sokhadze, E.M. Heart Rate Variability and Skin Conductance During Repetitive TMS Course in Children with Autism. Appl. Psychophysiol. Biofeedback 2016, 41, 47–60. [Google Scholar] [CrossRef]
- Ming, X.; Patel, R.; Kang, V.; Chokroverty, S.; Julu, P.O. Respiratory and autonomic dysfunction in children with autism spectrum disorders. Brain Dev. 2016, 38, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Liu, J.; Liu, K.; Koh, M.; Tian, R.; Hobbie, C.; Fong, M.; Chen, Q.; Zhao, M.; Budjan, C.; et al. Altered Autonomic Functions and Gut Microbiome in Individuals with Autism Spectrum Disorder (ASD): Implications for Assisting ASD Screening and Diagnosis. J. Autism Dev. Disord 2021, 51, 144–157. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.; Fouquaet, I.; Boets, B.; Naulaers, G.; Steyaert, J. Autism spectrum disorder and pupillometry: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2021, 120, 479–508. [Google Scholar] [CrossRef]
- Tessier, M.P.; Pennestri, M.H.; Godbout, R. Heart rate variability of typically developing and autistic children and adults before, during and after sleep. Int. J. Psychophysiol. Off. J. Int. Organ. Psychophysiol. 2018, 134, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Burr, R.L. Interpretation of normalized spectral heart rate variability indices in sleep research: A critical review. Sleep 2007, 30, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.A.; Zhou, W.; Dailey, M.J. Evidence for a direct effect of the autonomic nervous system on intestinal epithelial stem cell proliferation. Physiol. Rep. 2018, 6, e13745. [Google Scholar] [CrossRef] [PubMed]
- Busch, R.A.; Heneghan, A.F.; Pierre, J.F.; Wang, X.; Kudsk, K.A. The enteric nervous system neuropeptide, bombesin, reverses innate immune impairments during parenteral nutrition. Ann. Surg. 2014, 260, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef]
- Heneghan, A.F.; Pierre, J.F.; Tandee, K.; Shanmuganayagam, D.; Wang, X.; Reed, J.D.; Steele, J.L.; Kudsk, K.A. Parenteral nutrition decreases paneth cell function and intestinal bactericidal activity while increasing susceptibility to bacterial enteroinvasion. JPEN J. Parenter. Enter. Nutr. 2014, 38, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Ayabe, T.; Wulff, H.; Darmoul, D.; Cahalan, M.D.; Chandy, K.G.; Ouellette, A.J. Modulation of mouse Paneth cell alpha-defensin secretion by mIKCa1, a Ca2+-activated, intermediate conductance potassium channel. J. Biol. Chem. 2002, 277, 3793–3800. [Google Scholar] [CrossRef] [Green Version]
- Satoh, Y.; Ishikawa, K.; Oomori, Y.; Takeda, S.; Ono, K. Bethanechol and a G-protein activator, NaF/AlCl3, induce secretory response in Paneth cells of mouse intestine. Cell Tissue Res. 1992, 269, 213–220. [Google Scholar] [CrossRef]
- Satoh, Y.; Habara, Y.; Ono, K.; Kanno, T. Carbamylcholine- and catecholamine-induced intracellular calcium dynamics of epithelial cells in mouse ileal crypts. Gastroenterology 1995, 108, 1345–1356. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signaling. Ann. N. Y. Acad. Sci. USA 1995, 766, 31–43. [Google Scholar] [CrossRef]
- Mathias, A.; Pais, B.; Favre, L.; Benyacoub, J.; Corthesy, B. Role of secretory IgA in the mucosal sensing of commensal bacteria. Gut Microbes 2014, 5, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaetzel, C.S. Cooperativity among secretory IgA, the polymeric immunoglobulin receptor, and the gut microbiota promotes host-microbial mutualism. Immunol. Lett. 2014, 162, 10–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Hoffert, U.; Hornef, M.W.; Henriques-Normark, B.; Axelsson, L.G.; Midtvedt, T.; Putsep, K.; Andersson, M. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 2008, 57, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjoberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Ghosh, D.; Huttner, K.M.; Paterson, Y.; Bevins, C.L. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 2003, 422, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Vaishnava, S.; Hooper, L.V. Multi-layered regulation of intestinal antimicrobial defense. Cell. Mol. Life Sci. CMLS 2008, 65, 3019–3027. [Google Scholar] [CrossRef]
- Sun, X.; Jobin, C. Nucleotide-binding oligomerization domain-containing protein 2 controls host response to Campylobacter jejuni in Il10-/- mice. J. Infect. Dis. 2014, 210, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarroch, E.E. Autonomic nervous system and neuroimmune interactions: New insights and clinical implications. Neurology 2019, 92, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef]
- Yamakawa, K.; Rajendran, P.S.; Takamiya, T.; Yagishita, D.; So, E.L.; Mahajan, A.; Shivkumar, K.; Vaseghi, M. Vagal nerve stimulation activates vagal afferent fibers that reduce cardiac efferent parasympathetic effects. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1579–H1590. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, D.M.; Ringel, Y.; Heyman, M.B.; Foster, J.A.; Bercik, P.; Shulman, R.J.; Versalovic, J.; Verdu, E.F.; Dinan, T.G.; Hecht, G.; et al. The intestinal microbiome, probiotics and prebiotics in neurogastroenterology. Gut Microbes 2013, 4, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; van Hylckama Vlieg, J.E. Fate, activity, and impact of ingested bacteria within the human gut microbiota. Trends Microbiol. 2015, 23, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furness, J.B. Novel gut afferents: Intrinsic afferent neurons and intestinofugal neurons. Auton. Neurosci. 2006, 125, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Chesne, J.; Cardoso, V.; Veiga-Fernandes, H. Neuro-immune regulation of mucosal physiology. Mucosal. Immunol. 2019, 12, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, W.J. The Gut’s Little Brain in Control of Intestinal Immunity. ISRN Gastroenterol. 2013, 2013, 630159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellono, N.W.; Bayrer, J.R.; Leitch, D.B.; Castro, J.; Zhang, C.; O’Donnell, T.A.; Brierley, S.M.; Ingraham, H.A.; Julius, D. Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 2017, 170, 185–198.e116. [Google Scholar] [CrossRef] [Green Version]
- Campos-Rodriguez, R.; Godinez-Victoria, M.; Abarca-Rojano, E.; Pacheco-Yepez, J.; Reyna-Garfias, H.; Barbosa-Cabrera, R.E.; Drago-Serrano, M.E. Stress modulates intestinal secretory immunoglobulin A. Front. Integr. Neurosci. 2013, 7, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abot, A.; Cani, P.D.; Knauf, C. Impact of Intestinal Peptides on the Enteric Nervous System: Novel Approaches to Control Glucose Metabolism and Food Intake. Front. Endocrinol. 2018, 9, 328. [Google Scholar] [CrossRef]
- Lach, G.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Anxiety, Depression, and the Microbiome: A Role for Gut Peptides. Neurotherapeutics 2018, 15, 36–59. [Google Scholar] [CrossRef] [Green Version]
- McVey Neufeld, K.A.; Perez-Burgos, A.; Mao, Y.K.; Bienenstock, J.; Kunze, W.A. The gut microbiome restores intrinsic and extrinsic nerve function in germ-free mice accompanied by changes in calbindin. Neurogastroenterol. Motil. 2015, 27, 627–636. [Google Scholar] [CrossRef] [PubMed]
- McVey Neufeld, K.A.; Mao, Y.K.; Bienenstock, J.; Foster, J.A.; Kunze, W.A. The microbiome is essential for normal gut intrinsic primary afferent neuron excitability in the mouse. Neurogastroenterol. Motil. 2013, 25, 183–188. [Google Scholar] [CrossRef]
- Bufe, B.; Schumann, T.; Kappl, R.; Bogeski, I.; Kummerow, C.; Podgorska, M.; Smola, S.; Hoth, M.; Zufall, F. Recognition of bacterial signal peptides by mammalian formyl peptide receptors: A new mechanism for sensing pathogens. J. Biol. Chem. 2015, 290, 7369–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bufe, B.; Zufall, F. The sensing of bacteria: Emerging principles for the detection of signal sequences by formyl peptide receptors. Biomol. Concepts 2016, 7, 205–214. [Google Scholar] [CrossRef]
- Cianciulli, A.; Acquafredda, A.; Cavallo, P.; Saponaro, C.; Calvello, R.; Mitolo, V.; Panaro, M.A. f-Met-Leu-Phe stimulates nitric oxide production in chick embryo neurons: The role of NF-kB. Immunopharmacol. Immunotoxicol. 2009, 31, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Monstein, H.J.; Grahn, N.; Truedsson, M.; Ohlsson, B. Oxytocin and oxytocin-receptor mRNA expression in the human gastrointestinal tract: A polymerase chain reaction study. Regul. Pept. 2004, 119, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, B.; Forsling, M.L.; Rehfeld, J.F.; Sjolund, K. Cholecystokinin stimulation leads to increased oxytocin secretion in women. Eur. J. Surg. 2002, 168, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, B.; Truedsson, M.; Djerf, P.; Sundler, F. Oxytocin is expressed throughout the human gastrointestinal tract. Regul. Pept. 2006, 135, 7–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, M.; Alster, P.; Lundeberg, T.; Uvnas-Moberg, K. Oxytocin increases nociceptive thresholds in a long-term perspective in female and male rats. Neurosci. Lett. 1996, 212, 87–90. [Google Scholar] [CrossRef]
- Uvnas-Moberg, K.; Arn, I.; Theorell, T.; Jonsson, C.O. Gastrin, somatostatin and oxytocin levels in patients with functional disorders of the gastrointestinal tract and their response to feeding and interaction. J. Psychosom. Res. 1991, 35, 525–533. [Google Scholar] [CrossRef]
- Alfven, G. Plasma oxytocin in children with recurrent abdominal pain. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Welch, M.G.; Tamir, H.; Gross, K.J.; Chen, J.; Anwar, M.; Gershon, M.D. Expression and developmental regulation of oxytocin (OT) and oxytocin receptors (OTR) in the enteric nervous system (ENS) and intestinal epithelium. J. Comp. Neurol. 2009, 512, 256–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain. Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wostmann, B.S. The germfree animal in nutritional studies. Annu. Rev. Nutr. 1981, 1, 257–279. [Google Scholar] [CrossRef]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Shroff, K.E.; Meslin, K.; Cebra, J.J. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect. Immun. 1995, 63, 3904–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furness, J.B.; Kunze, W.A.; Bertrand, P.P.; Clerc, N.; Bornstein, J.C. Intrinsic primary afferent neurons of the intestine. Prog. Neurobiol. 1998, 54, 1–18. [Google Scholar] [CrossRef]
- Mao, Y.K.; Kasper, D.L.; Wang, B.; Forsythe, P.; Bienenstock, J.; Kunze, W.A. Bacteroides fragilis polysaccharide A is necessary and sufficient for acute activation of intestinal sensory neurons. Nat. Commun. 2013, 4, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.; Borojevic, R.; Verdu, E.F.; Huizinga, J.D.; Ratcliffe, E.M. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol. Motil. 2014, 26, 98–107. [Google Scholar] [CrossRef]
- Straub, R.H.; Wiest, R.; Strauch, U.G.; Harle, P.; Scholmerich, J. The role of the sympathetic nervous system in intestinal inflammation. Gut 2006, 55, 1640–1649. [Google Scholar] [CrossRef]
- Hart, A.; Kamm, M.A. Review article: Mechanisms of initiation and perpetuation of gut inflammation by stress. Aliment. Pharm. 2002, 16, 2017–2028. [Google Scholar] [CrossRef]
- Savidge, T.C.; Newman, P.; Pothoulakis, C.; Ruhl, A.; Neunlist, M.; Bourreille, A.; Hurst, R.; Sofroniew, M.V. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 2007, 132, 1344–1358. [Google Scholar] [CrossRef]
- Jarillo-Luna, A.; Rivera-Aguilar, V.; Martinez-Carrillo, B.E.; Barbosa-Cabrera, E.; Garfias, H.R.; Campos-Rodriguez, R. Effect of restraint stress on the population of intestinal intraepithelial lymphocytes in mice. Brain Behav. Immun. 2008, 22, 265–275. [Google Scholar] [CrossRef]
- Martinez-Carrillo, B.E.; Godinez-Victoria, M.; Jarillo-Luna, A.; Oros-Pantoja, R.; Abarca-Rojano, E.; Rivera-Aguilar, V.; Yepez, J.P.; Sanchez-Torres, L.E.; Campos-Rodriguez, R. Repeated restraint stress reduces the number of IgA-producing cells in Peyer’s patches. Neuroimmunomodulation 2011, 18, 131–141. [Google Scholar] [CrossRef]
- Reyna-Garfias, H.; Miliar, A.; Jarillo-Luna, A.; Rivera-Aguilar, V.; Pacheco-Yepez, J.; Baeza, I.; Campos-Rodriguez, R. Repeated restraint stress increases IgA concentration in rat small intestine. Brain Behav. Immun. 2010, 24, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Mantis, N.J.; Rol, N.; Corthesy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal. Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Park, A.J.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.A.; Fahnestock, M.; Moine, D.; et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.K.; Sharba, S.; Navabi, N.; Linden, S.K. Colonic levels of vasoactive intestinal peptide decrease during infection and exogenous VIP protects epithelial mitochondria against the negative effects of IFNgamma and TNFalpha induced during Citrobacter rodentium infection. PLoS ONE 2018, 13, e0204567. [Google Scholar] [CrossRef]
- Coquenlorge, S.; Duchalais, E.; Chevalier, J.; Cossais, F.; Rolli-Derkinderen, M.; Neunlist, M. Modulation of lipopolysaccharide-induced neuronal response by activation of the enteric nervous system. J. Neuroinflamm. 2014, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Saurer, T.B.; Ijames, S.G.; Lysle, D.T. Neuropeptide Y Y1 receptors mediate morphine-induced reductions of natural killer cell activity. J. Neuroimmunol. 2006, 177, 18–26. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol 2016, 594, 5781–5790. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdes-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Reardon, C.; Duncan, G.S.; Brustle, A.; Brenner, D.; Tusche, M.W.; Olofsson, P.S.; Rosas-Ballina, M.; Tracey, K.J.; Mak, T.W. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 1410–1415. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Miller, W.L. The Hypothalamic-Pituitary-Adrenal Axis: A Brief History. Horm. Res. Paediatr. 2018, 89, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Cailotto, C.; Gomez-Pinilla, P.J.; Costes, L.M.; van der Vliet, J.; Di Giovangiulio, M.; Nemethova, A.; Matteoli, G.; Boeckxstaens, G.E. Neuro-anatomical evidence indicating indirect modulation of macrophages by vagal efferents in the intestine but not in the spleen. PLoS ONE 2014, 9, e87785. [Google Scholar] [CrossRef] [Green Version]
- Birrenbach, T.; Bocker, U. Inflammatory bowel disease and smoking: A review of epidemiology, pathophysiology, and therapeutic implications. Inflamm. Bowel. Dis. 2004, 10, 848–859. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelstoft, M.S.; Egerod, K.L.; Holst, B.; Schwartz, T.W. A gut feeling for obesity: 7TM sensors on enteroendocrine cells. Cell Metab. 2008, 8, 447–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, P.L.; Anini, Y. Direct and indirect mechanisms regulating secretion of glucagon-like peptide-1 and glucagon-like peptide-2. Can. J. Physiol. Pharm. 2003, 81, 1005–1012. [Google Scholar] [CrossRef]
- Sandoval, D.; Dunki-Jacobs, A.; Sorrell, J.; Seeley, R.J.; D’Alessio, D.D. Impact of intestinal electrical stimulation on nutrient-induced GLP-1 secretion in vivo. Neurogastroenterol. Motil. 2013, 25, 700–705. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.; Longuet, C.; Maida, A.; Bahrami, J.; Xu, E.; Baker, C.L.; Brubaker, P.L.; Drucker, D.J.; Adeli, K. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology 2009, 137, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Rowland, K.J.; Brubaker, P.L. The "cryptic" mechanism of action of glucagon-like peptide-2. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1–G8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, A.L.; Katz, S.; Fang, J.C.; Bernstein, C.N.; Abou-Assi, S.G.; Teduglutide Study, G. Teduglutide, a novel mucosally active analog of glucagon-like peptide-2 (GLP-2) for the treatment of moderate to severe Crohn’s disease. Inflamm. Bowel. Dis. 2010, 16, 962–973. [Google Scholar] [CrossRef]
- Jeppesen, P.B.; Gilroy, R.; Pertkiewicz, M.; Allard, J.P.; Messing, B.; O’Keefe, S.J. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 2011, 60, 902–914. [Google Scholar] [CrossRef]
- Hyland, N.P.; Sjoberg, F.; Tough, I.R.; Herzog, H.; Cox, H.M. Functional consequences of neuropeptide Y Y 2 receptor knockout and Y2 antagonism in mouse and human colonic tissues. Br. J. Pharm. 2003, 139, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Cox, H.M. Neuropeptide Y receptors; antisecretory control of intestinal epithelial function. Auton. Neurosci. 2007, 133, 76–85. [Google Scholar] [CrossRef]
- Stengel, A.; Goebel, M.; Wang, L.; Tache, Y.; Sachs, G.; Lambrecht, N.W. Differential distribution of ghrelin-O-acyltransferase (GOAT) immunoreactive cells in the mouse and rat gastric oxyntic mucosa. Biochem. Biophys. Res. Commun. 2010, 392, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Robinson, E.S.; Rivera, L.R.; McMillan, P.J.; Testro, A.; Nikfarjam, M.; Bravo, D.M.; Furness, J.B. Glucagon-like peptide 1 and peptide YY are in separate storage organelles in enteroendocrine cells. Cell Tissue Res. 2014, 357, 63–69. [Google Scholar] [CrossRef]
- Young, R.L. Sensing via intestinal sweet taste pathways. Front. Neurosci. 2011, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigalet, D.L.; Wallace, L.E.; Holst, J.J.; Martin, G.R.; Kaji, T.; Tanaka, H.; Sharkey, K.A. Enteric neural pathways mediate the anti-inflammatory actions of glucagon-like peptide 2. Am. J. Physiol Gastrointest. Liver Physiol. 2007, 293, G211–G221. [Google Scholar] [CrossRef]
- Shirazi-Beechey, S.P.; Moran, A.W.; Batchelor, D.J.; Daly, K.; Al-Rammahi, M. Glucose sensing and signalling; regulation of intestinal glucose transport. Proc. Nutr Soc. 2011, 70, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgren, O. Enteric nerves and diarrhoea. Pharm. Toxicol. 2002, 90, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Gwynne, R.M.; Ellis, M.; Sjovall, H.; Bornstein, J.C. Cholera toxin induces sustained hyperexcitability in submucosal secretomotor neurons in guinea pig jejunum. Gastroenterology 2009, 136, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Tropini, C.; Moss, E.L.; Merrill, B.D.; Ng, K.M.; Higginbottom, S.K.; Casavant, E.P.; Gonzalez, C.G.; Fremin, B.; Bouley, D.M.; Elias, J.E.; et al. Transient Osmotic Perturbation Causes Long-Term Alteration to the Gut Microbiota. Cell 2018, 173, 1742–1754.e1717. [Google Scholar] [CrossRef] [Green Version]
- Darkoh, C.; Plants-Paris, K.; Bishoff, D.; DuPont, H.L. Clostridium difficile Modulates the Gut Microbiota by Inducing the Production of Indole, an Interkingdom Signaling and Antimicrobial Molecule. mSystems 2019, 4, e00346-18. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beopoulos, A.; Gea, M.; Fasano, A.; Iris, F. Autonomic Nervous System Neuroanatomical Alterations Could Provoke and Maintain Gastrointestinal Dysbiosis in Autism Spectrum Disorder (ASD): A Novel Microbiome–Host Interaction Mechanistic Hypothesis. Nutrients 2022, 14, 65. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14010065
Beopoulos A, Gea M, Fasano A, Iris F. Autonomic Nervous System Neuroanatomical Alterations Could Provoke and Maintain Gastrointestinal Dysbiosis in Autism Spectrum Disorder (ASD): A Novel Microbiome–Host Interaction Mechanistic Hypothesis. Nutrients. 2022; 14(1):65. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14010065
Chicago/Turabian StyleBeopoulos, Athanasios, Manuel Gea, Alessio Fasano, and François Iris. 2022. "Autonomic Nervous System Neuroanatomical Alterations Could Provoke and Maintain Gastrointestinal Dysbiosis in Autism Spectrum Disorder (ASD): A Novel Microbiome–Host Interaction Mechanistic Hypothesis" Nutrients 14, no. 1: 65. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14010065