
Dioscin Ameliorates Hyperuricemia-Induced Atherosclerosis by Modulating of Cholesterol Metabolism through FXR-Signaling Pathway

 ,
,
Abstract
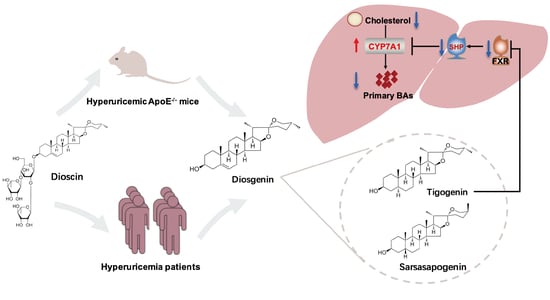
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal
2.3. Cell Culture and Treatment
2.4. Uric Acid and Lipids Analysis
2.5. Histological Oil Red O Staining
2.6. Atherosclerotic Lesions Analysis
2.7. Bile acid Profile Analysis
2.8. QRT-PCR
2.9. FXR Mediated Coactivator Recruitment Assay
2.10. Clinic Trial of Dioscin-Enriched Preparation in Hyperuricemia Individual
2.11. Statistical Analysis
3. Results
3.1. Hepatic CYP7A1 Expression Was Reduced in Hyperuricemia Mice Associated with an Elevation in Serum Cholesterol Levels
3.2. Uric Acid-Accelerated Atherosclerosis Progress in ApoE−/− Mice with Elevated Serum Cholesterol Level, Decreased Hepatic Bile Acid Level. The Effects at Least in Part from Activation of FXR and Down-Regulation of CYP7A1 in the Liver
3.2.1. Aortic Lesions Increased in PO-induced Hyperuricemic ApoE−/− Mice
3.2.2. Elevation of Serum Cholesterol Level was Aggravated in ApoE−/− Mice under Hyperuricemic Condition
3.2.3. Hepatic Bile Acid Levels Decreased in PO-induced Hyperuricemic ApoE−/− Mice, Associated with Decrease in Ileal-Conjugated Bile Acid Levels
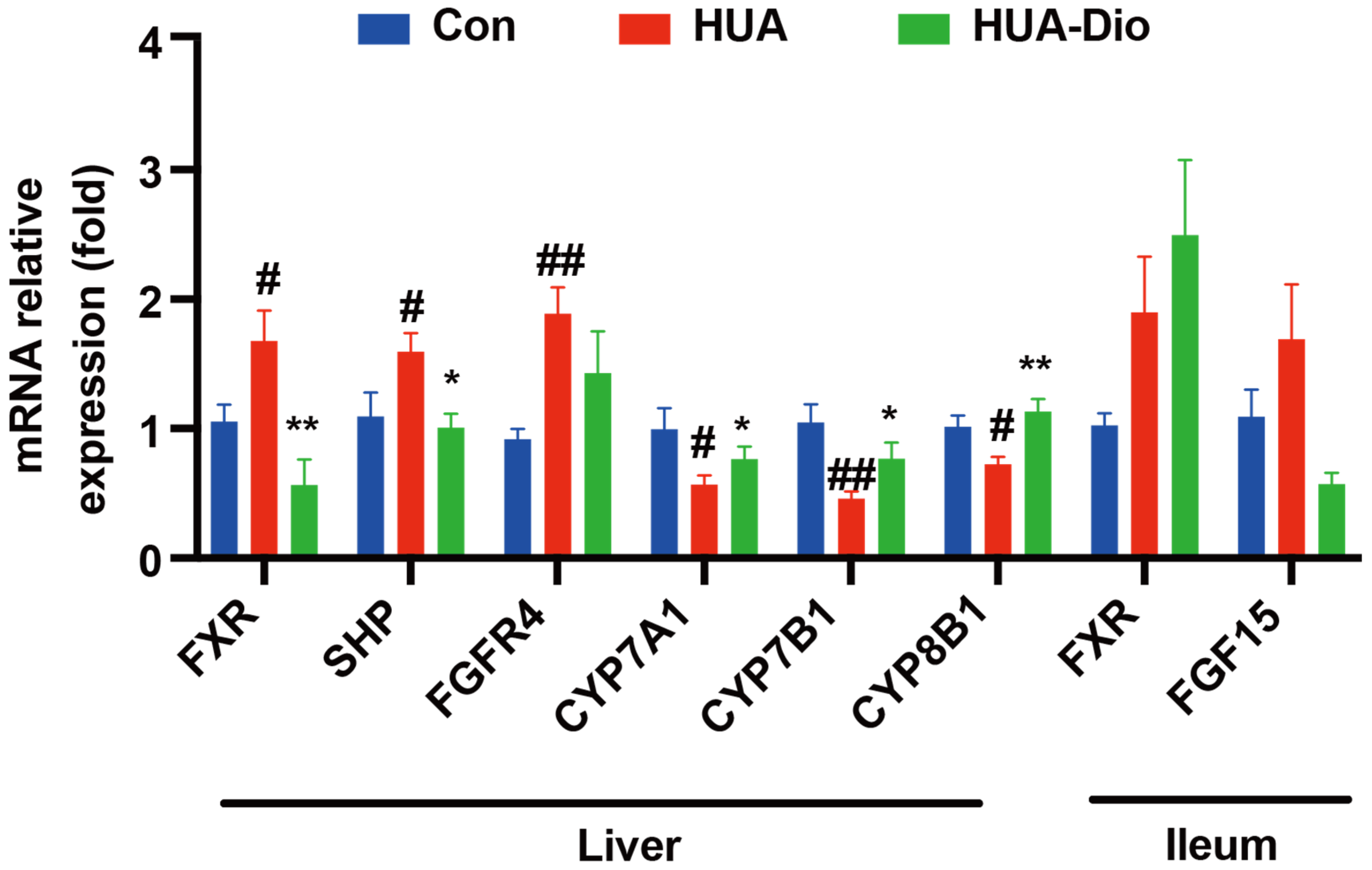
3.2.4. Hepatic FXR Was Activated and CYP7A1 Was Down-Regulated in PO-Induced Hyperuricemic ApoE−/− Mice
3.3. Dioscin Alleviated Hyperuricemia-Aggravated Atherosclerosis and Improved the Conversion of Cholesterols to Bile Acid in the Liver of ApoE−/− Mice
3.3.1. Dioscin Alleviated Hyperuricemia-Aggravated Atherosclerosis and Dysfunction of Lipid Metabolism
3.3.2. Dioscin Administration Modulated the Dysfunction of Bile Acid Profile in Hyperuricemic ApoE−/− Mice
3.3.3. Dioscin Treatment Inhibited Hepatic FXR Signaling
3.4. Dioscin Inhibited FXR through Its Metabolite Tigogenin
3.5. Treatment with Dioscin-Enriched Preparation Ameliorates Hypercholesterolemia in Hyperuricemia Patients
3.5.1. Description of Patients and Interventions
3.5.2. Demographic and Baseline Characteristics
3.5.3. Serum Uric Acid and Cholesterol Analysis
3.6. The BA Profile Changes after Oral Administration of Dioscin-Enriched Preparation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dalbeth, N.; Gosling, A.; Gaffo, A.; Abhishek, A. Gout. Lancet 2021, 397, 1843–1855. [Google Scholar] [CrossRef]
- Liu, R.; Han, C.; Wu, D.; Xia, X.; Gu, J.; Guan, H.; Shan, Z.; Teng, W. Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: A Systematic Review and Meta-Analysis. Biomed Res. Int. 2015, 2015, 762820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, D.; Kang, D.; Johnson, R. Uric acid and cardiovascular risk. N. Engl. J. Med. 2008, 359, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Sun, G.; Luo, A.; Lin, S.; Wang, J.; Zhang, Q.; Jiang, J.; Zhang, M.; Wang, F.; Tan, W. Serum uric acid is independently associated with aortic arch calcification in a cross-sectional study of middle-aged and elderly women. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 932–938. [Google Scholar] [CrossRef]
- Borghi, C.; Tykarski, A.; Widecka, K.; Filipiak, K.; Domienik-Karłowicz, J.; Kostka-Jeziorny, K.; Varga, A.; Jaguszewski, M.; Narkiewicz, K.; Mancia, G. Expert consensus for the diagnosis and treatment of patient with hyperuricemia and high cardiovascular risk. Cardiol. J. 2018, 25, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Doherty, M.; Pascual, E.; Barskova, V.; Becce, F.; Castañeda-Sanabria, J.; Coyfish, M.; Guillo, S.; Jansen, T.; Janssens, H.; et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann. Rheum. Dis. 2017, 76, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Taguchi, I.; Teragawa, H.; Ishizaka, N.; Kanzaki, Y.; Tomiyama, H.; Sata, M.; Sezai, A.; Eguchi, K.; Kato, T.; et al. Febuxostat does not delay progression of carotid atherosclerosis in patients with asymptomatic hyperuricemia: A randomized, controlled trial. PLoS Med. 2020, 17, e1003095. [Google Scholar] [CrossRef] [Green Version]
- Reinders, M.; Haagsma, C.; Jansen, T.; van Roon, E.; Delsing, J.; van de Laar, M.; Brouwers, J. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300-600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann. Rheum. Dis. 2009, 68, 892–897. [Google Scholar] [CrossRef]
- Kang, E.; Park, E.; Shin, A.; Song, J.; Kim, S. Cardiovascular risk associated with allopurinol vs. benzbromarone in patients with gout. Eur. Heart. J. 2021, 42, 4578–4588. [Google Scholar] [CrossRef]
- Liang, J.; Jiang, Y.; Huang, Y.; Song, W.; Li, X.; Huang, Y.; Ou, J.; Wei, Q.; Gu, J. The comparison of dyslipidemia and serum uric acid in patients with gout and asymptomatic hyperuricemia: A cross-sectional study. Lipids Health Dis. 2020, 19, 31. [Google Scholar] [CrossRef] [Green Version]
- Georgakis, M.; Malik, R.; Anderson, C.; Parhofer, K.; Hopewell, J.; Dichgans, M. Genetic determinants of blood lipids and cerebral small vessel disease: Role of high-density lipoprotein cholesterol. Brain 2020, 143, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.; Robinson, J.; Lichtenstein, A.; Bairey Merz, C.; Blum, C.; Eckel, R.; Goldberg, A.; Gordon, D.; Levy, D.; Lloyd-Jones, D.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Yang, H.; Song, B. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell. Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Jiang, L.; Yang, H.; Song, B. Intracellular Cholesterol Transport by Sterol Transfer Proteins at Membrane Contact Sites. Trends. Biochem. Sci. 2019, 44, 273–292. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell. Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jin, L.; Liu, J.; Wang, W.; Yu, H.; Li, J.; Chen, Q.; Wang, T. Effect and mechanism of dioscin from Dioscorea spongiosa on uric acid excretion in animal model of hyperuricemia. J. Ethnopharmacol. 2018, 214, 29–36. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, C.; Jin, Y.; Ma, X.; Xie, T.; Wang, J.; Liu, K.; Sun, H. Disocin prevents postmenopausal atherosclerosis in ovariectomized LDLR−/− mice through a PGC-1α/ERα pathway leading to promotion of autophagy and inhibition of oxidative stress, inflammation and apoptosis. Pharmacol. Res. 2019, 148, 104414. [Google Scholar] [CrossRef]
- Bao, R.; Liu, M.; Wang, D.; Wen, S.; Yu, H.; Zhong, Y.; Li, Z.; Zhang, Y.; Wang, T. Eurycoma longifoliaEffect of Stem Extract on Uric Acid Excretion in Hyperuricemia Mice. Front. Pharmacol. 2019, 10, 1464. [Google Scholar] [CrossRef]
- Rom, O.; Liu, Y.; Liu, Z.; Zhao, Y.; Wu, J.; Ghrayeb, A.; Villacorta, L.; Fan, Y.; Chang, L.; Wang, L.; et al. Glycine–based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci. Transl. Med. 2020, 12, eaaz2841. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, M.; Zhao, T.; Zhao, B.; Jia, L.; Zhu, Y.; Zhang, B.; Gao, X.; Li, G.; Li, X.; et al. Danhong injection inhibits the development of atherosclerosis in both Apoe−/− and Ldlr−/− mice. J. Cardiovasc. Pharmacol. 2014, 63, 441–452. [Google Scholar] [CrossRef]
- Chiang, J. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.; McDonald, J.; Luo, G.; Jones, S.; Goodwin, B.; Richardson, J.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Pang, Y.; He, X.; Zhang, Y.; Zhang, J.; Zhao, Z.; Yi, T.; Chen, H. UPLC-QTOF-MS identification of metabolites in rat biosamples after oral administration of Dioscorea saponins: A comparative study. J. Ethnopharma. 2015, 165, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Li, N.; Yao, Y.; Wang, K.; Yang, J.; Cui, Q.; Geng, B.; Chen, J.; Wang, Y.; Cheng, W.; et al. Longitudinal association between serum uric acid levels and multiterritorial atherosclerosis. J. Cell. Mol. Med. 2019, 23, 4970–4979. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Sun, Q.; Xu, H.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. Hyperuricemia induces lipid disturbances mediated by LPCAT3 upregulation in the liver. FASEB J. 2020, 34, 13474–13493. [Google Scholar] [CrossRef]
- Kuwabara, M.; Borghi, C.; Cicero, A.; Hisatome, I.; Niwa, K.; Ohno, M.; Johnson, R.; Lanaspa, M. Elevated serum uric acid increases risks for developing high LDL cholesterol and hypertriglyceridemia: A five-year cohort study in Japan. Int. J. Cardiol. 2018, 261, 183–188. [Google Scholar] [CrossRef]
- Peng, T.; Wang, C.; Kao, T.; Chan, J.; Yang, Y.; Chang, Y.; Chen, W. Relationship between hyperuricemia and lipid profiles in US adults. Biomed Res. Int. 2015, 2015, 127596. [Google Scholar] [CrossRef]
- Rodríguez-Castelán, J.; Méndez-Tepepa, M.; Rodríguez-Antolín, J.; Castelán, F.; Cuevas-Romero, E. Hypothyroidism affects lipid and glycogen content and peroxisome proliferator-activated receptor δ expression in the ovary of the rabbit. Reprod. Fertil. Dev. 2018, 30, 1380–1387. [Google Scholar] [CrossRef]
- Hager, M.; Narla, A.; Tannock, L. Dyslipidemia in patients with chronic kidney disease. Rev. Endocr. Metab. Disord. 2017, 18, 29–40. [Google Scholar] [CrossRef]
- Rana, R.; Shearer, A.; Fletcher, E.; Nguyen, N.; Guha, S.; Cox, D.; Abdelmalek, M.; Wang, Y.; Baleja, J.; Covic, L.; et al. PAR2 controls cholesterol homeostasis and lipid metabolism in nonalcoholic fatty liver disease. Mol. Metab. 2019, 29, 99–113. [Google Scholar] [CrossRef]
- DuBroff, R. A Reappraisal of the Lipid Hypothesis. Am. J. Med. 2018, 131, 993–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doumas, M.; Imprialos, K.; Dimakopoulou, A.; Stavropoulos, K.; Binas, A.; Athyros, V. The Role of Statins in the Management of Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2018, 24, 4587–4592. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.; Jardine, A.; Schmieder, R.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.; Chevaile, A.; Cobbe, S.; Grönhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhyay, S.; Anand, U.; Gadekar, V.; Jha, N.; Gupta, P.; Behl, T.; Kumar, M.; Radha; Shekhawat, M.; Dey, A. Dioscin: A review on pharmacological properties and therapeutic values. BioFactors 2022, 48, 22–55. [Google Scholar] [CrossRef]
- Li, Y.; Gao, M.; Yin, L.; Xu, L.; Qi, Y.; Sun, P.; Peng, J. Dioscin ameliorates methotrexate-induced liver and kidney damages via adjusting miRNA-145-5p-mediated oxidative stress. Free Radic. Biol. Med. 2021, 169, 99–109. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (S.D.) | Baseline | One Month Later | ||||
|---|---|---|---|---|---|---|
| Control (n = 11) | DA Capsule (n = 25) | p | Control (n = 11) | DA Capsule (n = 25) | p | |
| Age (years) | 40.31 ± 9.54 | 40.15 ± 8.88 | 0.961 | 40.44 ± 2.13 | 40.15 ± 8.882 | 0.961 |
| ALT (U/L) | 31.16 ± 19.22 | 32.87 ± 17.29 | 0.770 | 29.30 ± 20.76 | 32.71 ± 17.85 | 0.579 |
| AST (U/L) | 31.87 ± 28.52 | 20.87 ± 5.35 | 0.160 | 21.77 ± 6.26 | 22.72 ± 7.94 | 0.693 |
| Blood urea nitrogen (mmol/L) | 5.72 ± 1.08 | 5.84 ± 1.09 | 0.752 | 5.53 ± 0.88 | 5.23 ± 0.92 * | 0.319 |
| BMI (kg/m2) | 28.83 ± 3.42 | 28.90 ± 5.19 | 0.970 | 28.78 ± 3.66 | 28.29 ± 5.75 | 0.817 |
| Diastolic BP (mm Hg) | 88.10 ± 9.96 | 83.19 ± 16.25 | 0.400 | 85.30 ± 8.49 | 81.13 ± 16.05 | 0.457 |
| Fasting glucose (mmol/L) | 5.24 ± 0.69 | 5.27 ± 0.93 | 0.925 | 5.19 ± 0.81 | 5.35 ± 0.69 | 0.560 |
| HDL-C (mmol/L) | 1.08 ± 0.16 | 1.08 ± 0.21 | 0.996 | 1.03 ± 0.20 | 1.06 ± 0.15 | 0.650 |
| Heart rate (/min) | 67.17 ± 8.08 | 69.20 ± 11.79 | 0.603 | 68.08 ± 8.45 | 71.00 ± 10.46 | 0.420 |
| Height (cm) | 177.10 ± 5.71 | 176.59 ± 7.56 | 0.800 | 177.10 ± 5.71 | 176.59 ± 7.56 | 0.800 |
| Hemoglobin (g/L) | 150.40 ± 9.83 | 158.16 ± 9.24 | 0.016 | 151.53 ± 6.74 | 157.40 ± 7.77 | 0.020 |
| LDL-C (mmol/L) | 3.05 ± 0.66 | 3.21 ± 0.82 | 0.539 | 3.27 ± 0.69 * | 3.10 ± 0.76 | 0.470 |
| Serum creatinine (μmol/L) | 83.91 ± 14.03 | 87.87 ± 11.38 | 0.326 | 85.67 ± 14.51 | 85.66 ± 10.41 | 0.996 |
| Systolic BP (mm Hg) | 132.90 ± 16.45 | 127.56 ± 17.55 | 0.448 | 123.00 ± 18.74 * | 125.25 ± 22.05 | 0.971 |
| Total cholesterol (mmol/L) | 4.89 ± 0.87 | 5.08 ± 1.01 | 0.541 | 4.99 ± 0.93 | 4.86 ± 1.02 | 0.700 |
| Triglycerides (mmol/L) | 2.27 ± 1.65 | 2.66 ± 1.79 | 0.502 | 2.14 ± 1.03 | 2.60 ± 2.02 | 0.417 |
| Uric acid (μmol/L) | 497.45 ± 43.73 | 514.22 ± 57.33 | 0.394 | 463.15 ± 78.69 | 488.24 ± 82.61 * | 0.401 |
| Urine creatinine (mg/dL/24 h) | 15.44 ± 2.12 | 17.34 ± 7.19 | 0.331 | 15.10 ± 3.54 | 20.05 ± 12.29 | 0.109 |
| Urine uric acid (mg/24 h) | 631.68 ± 163.55 | 773.28 ± 344.18 | 0.153 | 639.81 ± 212.57 | 851.93 ± 421.22 | 0.067 |
| Urine volume (L) | 1.98 ± 0.70 | 2.12 ± 0.79 | 0.603 | 2.00 ± 0.57 | 2.20 ± 1.02 | 0.482 |
| VLDL (mmol/L) | 0.76 ± 0.39 | 0.80 ± 0.35 | 0.745 | 0.68 ± 0.24 | 0.71 ± 0.46 | 0.857 |
| WBC count (×109/mL) | 6.90 ± 1.31 | 6.22 ± 1.47 | 0.148 | 7.37 ± 1.36 | 6.36 ± 1.28 | 0.024 |
| Weight (kg) | 90.10 ± 8.27 | 91.50 ± 21.75 | 0.829 | 89.60 ± 8.54 | 90.07 ± 22.74 | 0.944 |
| γGGT (U/L) | 38.17 ± 27.44 | 47.10 ± 31.19 | 0.369 | 37.85 ± 23.99 | 45.90 ± 36.31 | 0.452 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, R.; Wang, W.; Chen, B.; Pan, J.; Chen, Q.; Liu, M.; Wang, D.; Wu, Y.; Yu, H.; Han, L.; et al. Dioscin Ameliorates Hyperuricemia-Induced Atherosclerosis by Modulating of Cholesterol Metabolism through FXR-Signaling Pathway. Nutrients 2022, 14, 1983. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14091983
Bao R, Wang W, Chen B, Pan J, Chen Q, Liu M, Wang D, Wu Y, Yu H, Han L, et al. Dioscin Ameliorates Hyperuricemia-Induced Atherosclerosis by Modulating of Cholesterol Metabolism through FXR-Signaling Pathway. Nutrients. 2022; 14(9):1983. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14091983
Chicago/Turabian StyleBao, Ruixia, Wei Wang, Beibei Chen, Jujie Pan, Qian Chen, Mengyang Liu, Dan Wang, Yuzheng Wu, Haiyang Yu, Lifeng Han, and et al. 2022. "Dioscin Ameliorates Hyperuricemia-Induced Atherosclerosis by Modulating of Cholesterol Metabolism through FXR-Signaling Pathway" Nutrients 14, no. 9: 1983. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14091983