Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment


{kind=link}
Abstract
:1. Introduction
2. Senescence
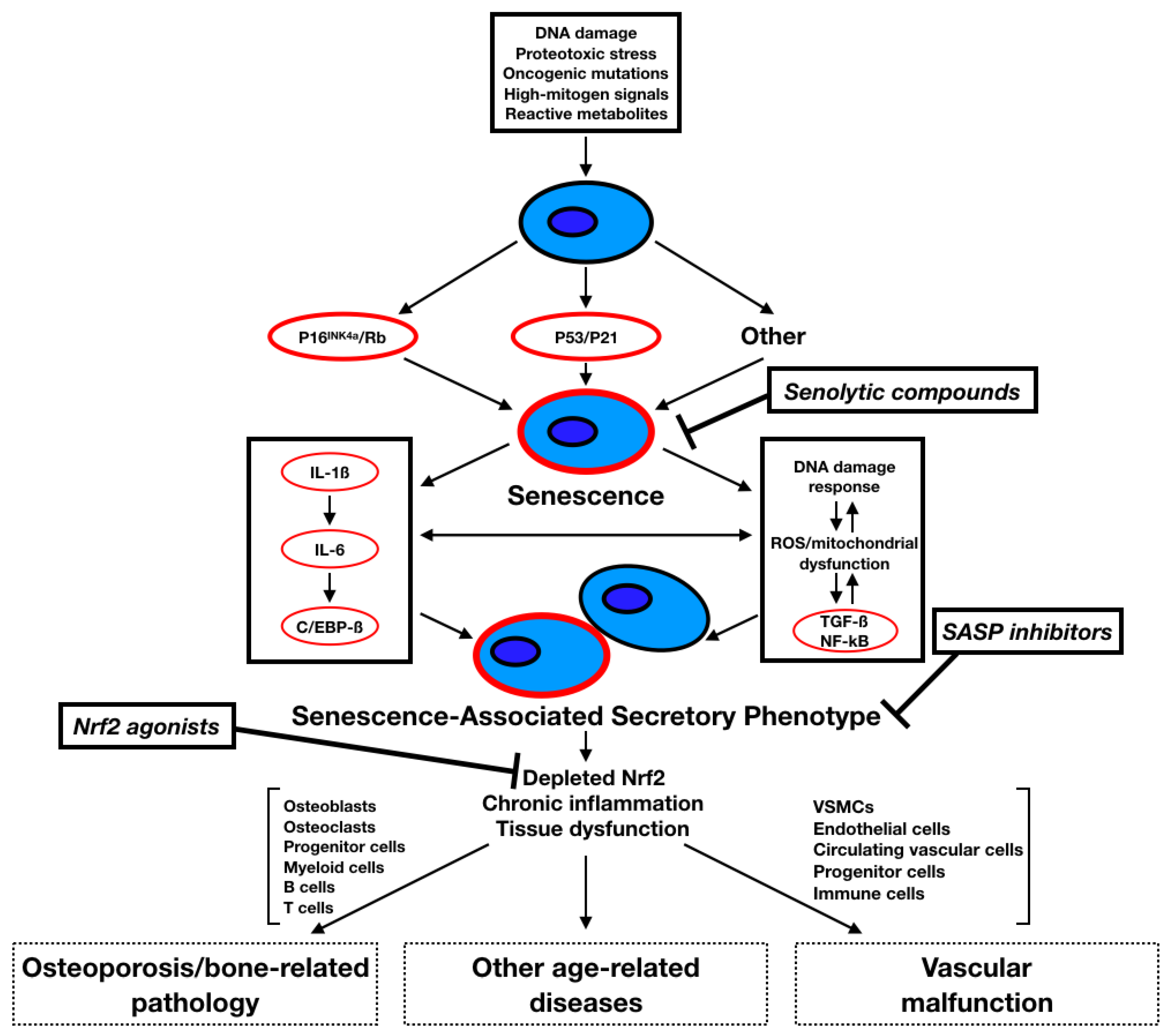
2.1. Introduction to Senescence
2.2. SASP
2.3. SCAPs
2.4. Biomarkers of Cellular Senescence
2.5. Senescence in CKD–MBD
3. Calcification
3.1. Introduction to Calcification
3.2. Tunica Media Calcification
3.3. Tunica Intima Calcification
4. Does Senescence Drive Vascular Calcification?
5. Targeting Vascular Calcification Therapeutically
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.T.; Sprott, R.L. Biomarkers of aging. Exp. Gerontol. 1988, 23, 223–239. [Google Scholar] [CrossRef]
- Shiels PG, R.-R.K. Biological Ageing, Inflammation and Nutrition: How Might They Impact on Systemic Sclerosis? Curr. Aging Sci. 2015, 8, 123–130. [Google Scholar] [CrossRef]
- Shiels, P.G.; McGuinness, D.; Eriksson, M.; Kooman, J.P.; Stenvinkel, P. The role of epigenetics in renal ageing. Nat. Rev. Nephrol. 2017, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.c.; Digiacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Ines, S.; Matej, D.; Cynthia, J.S.; Darren, J.B.; Jan, M.V.D. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2016, 13. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Luttropp, K.; McGuinness, D.; Witasp, A.; Rashid Qureshi, A.; Wernerson, A.; Nordfors, L.; Schalling, M.; Ripsweden, J.; Wennberg, L.; et al. CDKN2A/p16INK4 a expression is associated with vascular progeria in chronic kidney disease. Aging (Albany NY) 2017, 9, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD–MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Mukai, H.; Dai, L.; Chen, Z.; Lindholm, B.; Ripsweden, J.; Brismar, T.B.; Heimbürger, O.; Barany, P.; Qureshi, A.R.; Söderberg, M.; et al. Inverse J-shaped relation between coronary arterial calcium density and mortality in advanced chronic kidney disease. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension (Dallas, Tex.: 1979) 2001, 38, 938. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in CKD—Evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.; Shiels, P.G.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Larsson, T.E. Chronic Kidney Disease: A Clinical Model of Premature Aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD–MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities.(Review series). J. Clin. Investig. 2013, 123, 966. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Lucie, H.; Aurélien, M.; Jean-Marc, C.; Saïd, K.; Ziad, A.M. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef]
- Khosla, N.S.; Farr, L.J.; Kirkland, L.J. Inhibiting Cellular Senescence: A New Therapeutic Paradigm for Age-Related Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fougère, B.; Boulanger, E.; Nourhashémi, F.; Guyonnet, S.; Cesari, M. Chronic Inflammation: Accelerator of Biological Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348. [Google Scholar] [CrossRef]
- Fulop, G.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 2018, 40, 513–521. [Google Scholar] [CrossRef]
- Kuosmanen, S.M.; Sihvola, V.; Kansanen, E.; Kaikkonen, M.U.; Levonen, A.-L. MicroRNAs mediate the senescence-associated decline of NRF2 in endothelial cells. Redox Biol. 2018, 18, 77–83. [Google Scholar] [CrossRef]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; Von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; Von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, E.J.; Cachia, A.J.; Chung, H.R.; Wilund, K.R. Resveratrol Supplementation Reduces Aortic Atherosclerosis and Calcification and Attenuates Loss of Aerobic Capacity in a Mouse Model of Uremia. J. Med. Food 2014, 17, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.; Zhao, J.; McGowan, S.; Zhu, Y.; Brooks, R.; Grassi, D.; Gregg, S.; Stripay, J.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G. Improving Precision in Investigating Aging: Why Telomeres Can Cause Problems. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2010, 65A, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.C.; Matej, D.; Darren, J.B.; Jan, M.V.D. Cellular senescence in aging and age- related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424. [Google Scholar] [CrossRef]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867. [Google Scholar] [CrossRef]
- Gingell-Littlejohn, M.; McGuinness, D.; McGlynn, L.M.; Kingsmore, D.; Stevenson, K.S.; Koppelstaetter, C.; Clancy, M.J.; Shiels, P.G. Pre-Transplant CDKN2A Expression in Kidney Biopsies Predicts Renal Function and Is a Future Component of Donor Scoring Criteria.(Report). PLoS ONE 2013, 8, e68133. [Google Scholar] [CrossRef]
- Koppelstaetter, C.; Schratzberger, G.; Perco, P.; Hofer, J.; Mark, W.; Öllinger, R.; Oberbauer, R.; Schwarz, C.; Mitterbauer, C.; Kainz, A.; et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell 2008, 7, 491–497. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, D.; Camussi, G.; Leierer, J.; Shapter, O.; Mohammed, S.; Gingell-Littlejohn, M.; Kingsmore, D.B.; Little, A.-M.; Kerschbaum, J.; Schneeberger, S.; et al. Identification of Molecular Markers of Delayed Graft Function Based on the Regulation of Biological Ageing. PLoS ONE 2016, 11, e0146378. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Cai, G.-Y.; Chen, X.-M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520. [Google Scholar] [CrossRef] [PubMed]
- Joshua, N.F.; Ming, X.; Megan, M.W.; David, G.M.; Daniel, G.F.; Jennifer, L.O.; Brittany, A.N.; Jad, G.S.; Mikolaj, B.O.; Christine, M.H.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23. [Google Scholar] [CrossRef]
- Chen, Z.; Qureshi, A.R.; Ripsweden, J.; Wennberg, L.; Heimburger, O.; Lindholm, B.; Barany, P.; Haarhaus, M.; Brismar, T.B.; Stenvinkel, P. Vertebral bone density associates with coronary artery calcification and is an independent predictor of poor outcome in end-stage renal disease patients. Bone 2016, 92, 50–57. [Google Scholar] [CrossRef]
- Collins, J.A. Cardiovascular Mortality in End-Stage Renal Disease. Am. J. Med. Sci. 2003, 325, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Sharaf El Din, U.A.A.; Salem, M.M.; Abdulazim, D.O. Vascular calcification: When should we interfere in chronic kidney disease patients and how? World J. Nephrol. 2016, 5, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amann, K. Media calcification and intima calcification are distinct entities in chronic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 1599. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef]
- Fuery, M.A.; Liang, L.; Kaplan, F.S.; Mohler, E.R. Vascular ossification: Pathology, mechanisms, and clinical implications. Bone 2018, 109, 28–34. [Google Scholar] [CrossRef]
- Machowska, A.; Carrero, J.J.; Lindholm, B.; Stenvinkel, P. Therapeutics targeting persistent inflammation in chronic kidney disease. Transl. Res. 2016, 167, 204–213. [Google Scholar] [CrossRef]
- Pecoits Filho, R.; Barany, P.; Lindholm, B.; Heimbürger, O.; Stenvinkel, P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol. Dial. Transplant. 2002, 17, 1684–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iseki, K.; Tozawa, M.; Yoshi, S.; Fukiyama, K. Serum C-reactive protein (CRP) and risk of death in chronic dialysis patients. Nephrol. Dial. Transplant. 1999, 14, 1956–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benz, K.; Varga, I.; Neureiter, D.; Campean, V.; Daniel, C.; Heim, C.; Reimann, A.; Weyand, M.; Hilgers, K.F.; Amann, K. Vascular inflammation and media calcification are already present in early stages of chronic kidney disease. Cardiovasc. Pathol. 2017, 27, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.L.; Chonchol, M. Vascular calcification in end-stage renal disease. Hemodial. Int. 2013, 17, S17–S21. [Google Scholar] [CrossRef] [PubMed]
- Tonar, Z.; Kochova, P.; Cimrman, R.; Perktold, J.; Witter, K. Segmental differences in the orientation of smooth muscle cells in the tunica media of porcine aortae. Biomech. Model. Mechanobiol. 2015, 14, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular calcification: The killer of patients with chronic kidney disease. J. Am. Soc. Nephrol. JASN 2009, 20, 1453. [Google Scholar] [CrossRef]
- Chen, N.X.; O’neill, K.D.; Chen, X.; Kiattisunthorn, K.; Gattone, V.H.; Moe, S.M. Activation of Arterial Matrix Metalloproteinases Leads to Vascular Calcification in Chronic Kidney Disease. Am. J. Nephro. 2011, 34, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent cancer-associated fibroblasts secrete active MMP-2 that promotes keratinocyte dis-cohesion and invasion. Br. J. Cancer 2014, 111. [Google Scholar] [CrossRef]
- Li, M.X.; Yang, M.H.-Y.; Giachelli, M.C. Role of the Sodium-Dependent Phosphate Cotransporter, Pit-1, in Vascular Smooth Muscle Cell Calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a Candidate Gene for Cleidocranial Dysplasia Syndrome, Is Essential for Osteoblast Differentiation and Bone Development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Hruska, A.K.; Mathew, A.S.; Saab, A.G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shroff, C.R.; McNair, N.R.; Figg, E.N.; Skepper, M.J.; Schurgers, M.L.; Gupta, M.A.; Hiorns, M.M.; Donald, M.A.; Deanfield, M.J.; Rees, M.L.; et al. Dialysis Accelerates Medial Vascular Calcification in Part by Triggering Smooth Muscle Cell Apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, N.D.; Skepper, R.J.; Hegyi, M.L.; Bennett, L.M.; Shanahan, L.C.; Weissberg, L.P. Apoptosis Regulates Human Vascular Calcification In Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. J. Am. Heart Assoc. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Jing, W.; Jabbari, B.; Vaziri, N.D. Uremia induces upregulation of cerebral tissue oxidative/inflammatory cascade, downregulation of Nrf2 pathway and disruption of blood brain barrier. Am. J. Transl. Res. 2018, 10, 2137. [Google Scholar] [PubMed]
- Yao, L.; Wang, J.; Tian, B.Y.; Xu, T.H.; Sheng, Z.T. Activation of the Nrf2-ARE Signaling Pathway Prevents Hyperphosphatemia-Induced Vascular Calcification by Inducing Autophagy in Renal Vascular Smooth Muscle Cells. J. Cell. Biochem. 2017, 118, 4708–4715. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.M.; Park, S.; Choi, Y.K.; Jeong, J.Y.; Oh, C.J.; Bae, K.H.; Lee, S.J.; Kim, J.H.; Park, K.G.; Jun do, Y.; et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vasc. Pharmacol. 2014, 63, 29–36. [Google Scholar] [CrossRef]
- Zhang, P.; Li, Y.; Du, Y.; Li, G.; Wang, L.; Zhou, F. Resveratrol Ameliorated Vascular Calcification by Regulating Sirt-1 and Nrf2. Transplant. Proc. 2016, 48, 3378–3386. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Griffith, C.; Kuro-O, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 124. [Google Scholar] [CrossRef]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Maltese, G.; Psefteli, P.M.; Rizzo, B.; Srivastava, S.; Gnudi, L.; Mann, G.E.; Siow, R.C.M. The anti-ageing hormone klotho induces Nrf2-mediated antioxidant defences in human aortic smooth muscle cells. J. Cell. Mol. Med. 2017, 21, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Garcia-Cardena, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis.(Report)(Author abstract). J. Cell Biol. 2015, 209, 13. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, A.-K.; Lappalainen, J.P.; Heiskanen, E.; Merentie, M.; Sihvola, V.; Näpänkangas, J.; Lottonen-Raikaslehto, L.; Kansanen, E.; Adinolfi, S.; Kaarniranta, K.; et al. Nrf2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolemic mice. Cardiovasc. Res. 2018, 115, 243. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Souma, Y.; Akakabe, Y.; Kitamura, Y.; Matsuo, K.; Shimoda, Y.; Ueyama, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 39–44. [Google Scholar] [CrossRef]
- Dube, P.R.; Birnbaumer, L.; Vazquez, G. Evidence for constitutive bone morphogenetic protein-2 secretion by M1 macrophages: Constitutive auto/paracrine osteogenic signaling by BMP-2 in M1 macrophages. Biochem. Biophys. Res. Commun. 2017, 491, 154–158. [Google Scholar] [CrossRef]
- Kovacic, C.J.; Randolph, J.G. Vascular Calcification: Harder Than It Looks. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1249–1250. [Google Scholar] [CrossRef]
- Leszczynska, A.; Murphy, J.M. Vascular Calcification: Is it rather a Stem/Progenitor Cells Driven Phenomenon?(Report)(Author abstract). Front. Bioeng. Biotechnol. 2018, 6. [Google Scholar] [CrossRef]
- Huang, P.H.; Virmani, D.R.; Younis, T.H.; Burke, T.A.; Kamm, T.R.; Lee, T.R. The Impact of Calcification on the Biomechanical Stability of Atherosclerotic Plaques. Circ. J. Am. Heart Assoc. 2001, 103, 1051–1056. [Google Scholar] [CrossRef] [Green Version]
- Henein, M.; Granåsen, G.; Wiklund, U.; Schmermund, A.; Guerci, A.; Erbel, R.; Raggi, P. High dose and long-term statin therapy accelerate coronary artery calcification. Int. J. Cardiol. 2015, 184, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Puri, R.; Hammadah, M.; Duggal, B.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E.; Nicholls, S.J. Spotty calcification and plaque vulnerability in vivo: Frequency-domain optical coherence tomography analysis. Cardiovasc. Diagn. Ther. 2014, 4, 460. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2017, 25, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.J.; Uryga, K.A.; Reinhold, K.J.; Figg, K.N.; Baker, K.L.; Finigan, K.A.; Gray, K.K.; Kumar, K.S.; Clarke, K.M.; Bennett, K.M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-G.; Liu, L.; Zheng, X.-Y. Cyclin-dependent kinase inhibitor p16 INK4a and telomerase may co-modulate endothelial progenitor cells senescence. Ageing Res. Rev. 2008, 7, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras Induces Vascular Smooth Muscle Cell Senescence and Inflammation in Human Atherosclerosis. Circ. J. Am. Heart Assoc. 2003, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Giles, P.J.; Sheerin, A.N.P.; Smith, S.K.; Lawton, J.J.; Ostler, E.L.; Rhys-Williams, W.; Kipling, D.; Faragher, R.G.A. Microarray analysis of senescent vascular smooth muscle cells: A link to atherosclerosis and vascular calcification. Exp. Gerontol. 2009, 44, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Alique, M.; Ruíz-Torres, M.P.; Bodega, G.; Noci, M.V.; Troyano, N.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Carracedo, J.; et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017, 9, 778. [Google Scholar] [CrossRef]
- Joshi, F.R.; Rajani, N.K.; Abt, M.; Woodward, M.; Bucerius, J.; Mani, V.; Tawakol, A.; Kallend, D.; Fayad, Z.A.; Rudd, J.H.F. Does Vascular Calcification Accelerate Inflammation?: A Substudy of the dal-PLAQUE Trial: A Substudy of the dal-PLAQUE Trial. J. Am. Coll. Cardiol. 2016, 67, 69–78. [Google Scholar] [CrossRef]
- Glenn, M.C.; Steven, K.B.; Paolo, R. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002, 62, 245. [Google Scholar] [CrossRef]
- NIH. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02848131 (accessed on 22 January 2018).
- Hegner, B.; Schaub, T.; Janke, D.; Zickler, D.; Lange, C.; Girndt, M.; Jankowski, J.; Schindler, R.; Dragun, D. Targeting proinflammatory cytokines ameliorates calcifying phenotype conversion of vascular progenitors under uremic conditions in vitro. Sci. Rep. 2018, 8, 12087. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.G.; Cregor, M.; McAndrews, K.; Morales, C.C.; McCabe, L.D.; McCabe, G.P.; Peacock, M.; Burr, D.; Weaver, C.; Bellido, T. Nrf2 regulates mass accrual and the antioxidant endogenous response in bone differently depending on the sex and age.(Research Article). PLoS ONE 2017, 12, e0171161. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hobson, S.; Arefin, S.; Kublickiene, K.; Shiels, P.G.; Stenvinkel, P. Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment. Toxins 2019, 11, 82. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11020082
Hobson S, Arefin S, Kublickiene K, Shiels PG, Stenvinkel P. Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment. Toxins. 2019; 11(2):82. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11020082
Chicago/Turabian StyleHobson, Sam, Samsul Arefin, Karolina Kublickiene, Paul G. Shiels, and Peter Stenvinkel. 2019. "Senescent Cells in Early Vascular Ageing and Bone Disease of Chronic Kidney Disease—A Novel Target for Treatment" Toxins 11, no. 2: 82. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11020082