Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways

1
Department of Anesthesia and Perioperative Care, University of California at San Francisco, San Francisco, CA 94143-0542, USA
2
California Academy of Sciences, San Francisco, CA 94118, USA
Toxins 2020, 12(2), 68; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020068
Submission received: 18 November 2019
/
Revised: 12 January 2020
/
Accepted: 14 January 2020
/
Published: 22 January 2020
(This article belongs to the Special Issue Novel Strategies for the Diagnosis and Treatment of Snakebites)
Abstract
:The active components of snake venoms encompass a complex and variable mixture of proteins that produce a diverse, but largely stereotypical, range of pharmacologic effects and toxicities. Venom protein diversity and host susceptibilities determine the relative contributions of five main pathologies: neuromuscular dysfunction, inflammation, coagulopathy, cell/organ injury, and disruption of homeostatic mechanisms of normal physiology. In this review, we describe how snakebite is not only a condition mediated directly by venom, but by the amplification of signals dysregulating inflammation, coagulation, neurotransmission, and cell survival. Although venom proteins are diverse, the majority of important pathologic events following envenoming follow from a small group of enzyme-like activities and the actions of small toxic peptides. This review focuses on two of the most important enzymatic activities: snake venom phospholipases (svPLA2) and snake venom metalloproteases (svMP). These two enzyme classes are adept at enabling venom to recruit homologous endogenous signaling systems with sufficient magnitude and duration to produce and amplify cell injury beyond what would be expected from the direct impact of a whole venom dose. This magnification produces many of the most acutely important consequences of envenoming as well as chronic sequelae. Snake venom PLA2s and MPs enzymes recruit prey analogs of similar activity. The transduction mechanisms that recruit endogenous responses include arachidonic acid, intracellular calcium, cytokines, bioactive peptides, and possibly dimerization of venom and prey protein homologs. Despite years of investigation, the precise mechanism of svPLA2-induced neuromuscular paralysis remains incomplete. Based on recent studies, paralysis results from a self-amplifying cycle of endogenous PLA2 activation, arachidonic acid, increases in intracellular Ca2+ and nicotinic receptor deactivation. When prolonged, synaptic suppression supports the degeneration of the synapse. Interaction between endothelium-damaging MPs, sPLA2s and hyaluronidases enhance venom spread, accentuating venom-induced neurotoxicity, inflammation, coagulopathy and tissue injury. Improving snakebite treatment requires new tools to understand direct and indirect effects of envenoming. Homologous PLA2 and MP activities in both venoms and prey/snakebite victim provide molecular targets for non-antibody, small molecule agents for dissecting mechanisms of venom toxicity. Importantly, these tools enable the separation of venom-specific and prey-specific pathological responses to venom.
Keywords:
phospholipase A2; metalloprotease; snake venom; intracellular signaling; neuromuscular paralysis; intracellular calciumKey Contribution: Snakebite is not only a condition mediated directly by venom proteins but also by the reaction of the body to the venom. Venom phospholipase A2s and metalloproteases recruit endogenous homologs in prey; mediating paralysis; inflammation; and coagulopathy. Small molecule inhibitors of venom proteases now permit understanding of these mechanisms.
1. Introduction
The World Health Organization (WHO) estimates that snakes envenom about 400,000 people per year, causing more than 148,000 deaths. Permanent disabilities such as amputations, wound contractures, and functional loss of limbs are often the result of non-fatal envenoming [1,2]). In total, 5.8 billion people live directly in or within an hour of venomous snake habitats, putting nearly three-quarters of the world’s population at risk [3].
Current standard of care therapy for snakebite is focused on the use of antibody-based treatments (here termed “serotherapies”) intended to intercept, neutralize and remove venoms present in the circulation before they produce long-term effects [4]. The basic concepts and methodology underlying serotherapy were developed more than a century ago: stimulated by Pasteur’s and Behring’s work on rabies and diphtheria, Albert Calmette produced antisera effective against cobra venom [5]. Vital Brazil was the first to develop polyvalent antisera against South American snakes and described the chief component pathologies of envenoming: coagulopathy, hemolysis, cytotoxicity, and paralysis [6]. Attempts to understand the mechanisms of venom toxicity began even earlier, with Fayer’s seminal recognition in the 1860s of the physiological similarities between the actions of cobra venom and the plant toxin curare [7].
Proteomic science has revealed that snake venoms are a diverse and variable mixture of enzymatic and non-enzymatic proteins and peptides. Venom complexity presents several important challenges to the understanding of venom mechanisms. The most obvious challenge is dissecting the separate effects of venom components in an envenomed animal. A summary of the major known venom toxicities and mechanisms, classified by key effects, molecular nature (enzymatic or non-enzymatic), timing of action, and sites of effect, is presented in Table 1. A more detailed compendium of venom components was recently published by Waheed et al. [8]. Laustsen states that snake venom is the “most complex pharmaceutical target” known, composed not only of a multitude of toxin components but a multitude of biochemical interactions [9].
Snakebite is not only a condition mediated directly by venom proteins but also by the reaction of the body to the venom. Some of the ways that venom engages endogenous signaling systems in prey or victim are included in the far-right column of Table 1. Based on convergent biochemical and physiological information, it has been possible to resolve that the most important effects of venom are based on a handful of, rather than many, molecular effectors. This small number of endogenous processes have an outsized effect on the regulation of cell function and cell survival. Co-opting endogenous processes enables venoms to use the envenomed organisms’ own cellular machinery to disrupt neurotransmission, dysregulate coagulation, and produce mediators of inflammation.
The foci of this review are hypotheses related to the manner in which venoms enhance their lethality by co-opting prey signaling systems, disordering and amplifying the prey’s inflammation and cell survival machinery. Identifying key venom targets and their endogenous counterparts for inhibition should be applicable to most medically important snake species, and simultaneously address the fundamental matter of the recipient’s biological response to the venomous insult.
2. Enzymatic and Non-Enzymatic Components of Venom and Where They Act
Venom proteins can be broadly classified into those that contain intrinsic enzymatic activity and those that do not. The obvious significance of this distinction is the possible inhibition of the enzymatic venom components by small molecule therapeutic agents. Proteomic analysis reveals that snake venoms contain proteins from 26 protein families, with substantial species variation [28,29,30]. However, the most prevalent medically relevant components occur in just four families, in varying proportions [10,31]. These proteins are: (1) secreted phospholipase A2 (sPLA2); (2) metalloproteases (MPs); (3) serine-proteases (SPs); and (4) the non-enzymatic three-finger toxins (3-FTX, e.g., α-bungarotoxin, an antagonist of nicotinic receptors [28,31]). Not all snake venom toxins are so neatly classified, including dendrotoxins from mambas, which are competitive antagonists of voltage gated potassium channels, and myotoxins from Crotalids that create cation permeability channels in the sarcolemma [24,25,32]. The main actions of these groups of enzymatic and non-enzymatic venom components are shown in Figure 1. It is important to note that some quantitatively minor venom components can have outsized effects on venom toxicity; for example, hyaluronidase from Crotalus durissus terrificus represents only 0.23% of the total protein, but greatly potentiates crotoxin lethality [33].
Molecular understanding of venom toxicity, based on enzymatic and non-enzymatic actions, developed slowly. The first advance in the modern era was the recognition by Karl Slotta and Heinz Fraenkel-Conrat in 1938 that crotoxin crystalized from Crotalus durissus was a phospholipase [34]. Another leap occurred in the 1970s when it was established that α-neurotoxins are competitive nicotinic receptor blockers [21,35]. Gutierrez and Lomonte have published a valuable review of seminal developments in the field [18].
Venoms typically act quickly to immobilize prey, with non-lethal doses more slowly producing weakness and a dose-dependent range of tissue and organ toxicity. Few venoms cross the blood–brain barrier, or even gain access to the extravascular compartment environment without assisted vascular leakage. Instead, to exert biological effects, larger or enzymatic venom proteins either: (1) bind to other proteins in the body (e.g., α-toxins); or (2) enzymatically create small molecular mass signaling molecules that have spatially and pharmacologically broader effects. Based purely on molecular mass considerations, svMPs (we use the terms svMP and svPLA2 to designate snake venom metalloproteases and phospholipases A2 to distinguish those enzymes from the secreted or intracellular enzymes present in prey/victim.) are expected to have effects confined to the circulation. However, the actions of svMPs yield small molecular mass peptides that are both biologically active and spread quickly. Phospholipases are smaller proteins and gain earlier access to deeper compartments in the body, where they generate cell-specific signals. Excluding for the moment direct protein–protein interactions in the extracellular compartment (e.g., proteases that hydrolyze coagulation proteins), these biological effects include:
(1) Production of mediators that diffuse within or across cell membranes;
(2) Production of transmembrane signals by direct binding to cell surface receptors such as neurotransmitter receptors/ion channels or G-protein coupled receptors;
(3) Translocation into the cell via transporters, carriers or endocytosis.
Any molecular description of venom effects must also account for the variability and time-dependent pathology seen in both lethal and sub-lethal envenoming. These effects can be diverse, even when caused by envenoming by a single species or closely related group of snakes. For example, in a recent review, Frare and colleagues described the delayed and variable clinical manifestations of Crotalus durissus envenoming. The summarized clinical reports describe muscle and kidney damage, neurotoxicity and hemolytic/coagulopathic pathologies to varying degrees [36,37,38]. These variations in clinical presentation may be related to differences in venom composition and effects that vary with season, locality, and even with sub-populations of the same species of Crotalus [39].
2.1. Venom Translocation from Circulation to Interstitial Compartment
Based on current knowledge, venoms rely on penetration of barriers rather than specific transport mechanisms to leave the circulation. Relatively little is known about the rate of spread, extent of distribution, and persistence of venom proteins in the body. Knowledge concerning the distribution of venom in the circulation, based on pharmacokinetic models and published information, was summarized recently by Sanhajariya [40]. The conclusion of Sanhajariya’s review was that the limited knowledge about the pharmacokinetics and pharmacodynamics of venoms restricts our understanding of venom-venom and whole venom–host interactions. Since the effects of envenoming can be long-lived, it is of interest to know whether venom persistence or host-response persistence explains long-lasting toxicity.
One of the ubiquitous weapons in venom is metalloproteases [41]. These enzymes attack basement membranes and collagen in tissues to increase venom spread and disrupt blood vessels [14]. Importantly, the digestion products of metalloproteases can be biologically active: these bioactive peptides affect tissue growth, remodeling, repair and development [42]. In addition, these bioactive peptides increase venom toxicity by triggering and amplifying inflammation [15]. Further, they induce the transcription and translation of endogenous matrix metalloproteases that attack types of collagen that are outside the range of svMPs [41].
Hyaluronidases are also important in venom spread and represent a nearly ubiquitous venom component [12,27]. Hyaluronidase activity amplifies the toxicity of crotoxin by enhancing venom distribution. In one study, Crotalus durissus terrificus crotoxin injected into mice was only toxic when co-injected with purified hyaluronidase [33]. Other mechanisms of hyaluronidase toxicity involve bioactive products of the enzymatic digestion of hyaluronic acid [13].
2.2. Venom Binding or Interaction with Cell Surface Receptors
There are several lines of evidence that venoms bind or interact with cell surface proteins. However, clear demonstration of venom binding/incorporation into membranes or binding to a particular acceptor/receptor was a challenge for many years. While α-neurotoxins were known to bind with high affinity to nicotinic acetylcholine [21], there was no similar demonstration of a “receptor” for β-neurotoxins. The nature of weak membrane interactions of PLA2 was explored in the 1970s and 1980s, identifying only low affinity interactions [43]. Studies by Oberg and Kelly [44] used iodine-125 labeled-β-bungarotoxin to identify a class of membrane fragments associating with the toxin in rat brain. Binding sites were found in cell membrane and mitochondrial fractions but the studies did not characterize the affinity of binding or whether the binding sites were protein, carbohydrate or lipid. In the 1970s, McDermott et al. used [3H]-pyridoxylated β-bungarotoxin to identify binding in synaptosomes and synaptic vesicles from rat brain. Binding occurred at relatively low affinity to a protein “acceptor” that was distributed widely in several membrane fractions, including synaptic vesicles [45].
By the 1990s, it was becoming accepted that the neuro- and myotoxic sPLA2 interact with specific receptors or interacting proteins (reviewed by [46]). One of the clearest demonstrations of such specificity is the high-affinity cell surface binding of PLA2s from the venom of Oxyuranus scutellatus. Different components of Oxyuranus venom bind specifically to neuronal and muscle membranes [47]. Additional evidence for selectivity is the accelerated evolution of PLA2 genes, first clarified by the work of Nakashima and colleagues in the 1990s [48]. The model proposed by Kini and Evans [49] in 1989 explained the fundamental aspects, if not the molecular details, of this amazing diversity and specificity the of toxic activities of venom PLA2s.
As mentioned, it is clearly established that one class of venom proteins, the α-neurotoxins (3FTx), bind with high affinity to nicotinic acetylcholine receptors at the neuromuscular junction (NMJ) [50]. Nicotinic receptors rendered non-functional by α-bungarotoxins are eventually de-phosphorylated, a state that identifies them for internalization [50]. Internalized receptors are then degraded by the proteasome complex.
2.3. Venom Transport into the Intracellular Compartment
Evidence from several recent studies suggests that one of the ways venom proteins may cause intracellular effects is to physically translocate into the cytosolic compartment. For example, Lagonder and colleagues demonstrated the translocation of a β-neurotoxin (gold-labeled mutant ammodytoxin A) into the of terminal axon and terminal boutons of a mammalian motor neuron. Interestingly, the labelled toxin was not found in the muscle fibers themselves [51]. In contrast, myotoxic PLA2 was demonstrated to be exclusively found on the sarcolemma. Several additional demonstrations of internalization of β-neurotoxins into various neuronal cells have also been published. In one study, the fluorescent protein label Alexa was used to label notexin, β-bungarotoxin and taipotoxin. These labeled proteins were found to localize to mitochondria in rat cerebellar granular neurons and spinal cord motor neurons [52]. Vimentin, a ubiquitous filament protein that anchors organelles within the cytoplasm, has also been identified as associating with venom PLA2. Vimentin binding may both facilitate PLA2 hydrolytic activity and facilitate internalization of the catalytic subunit of PLA2 [46]. A recent report [53] found evidence of intracellular location of three types of snake venom PLA2s, based on elegant protein labeling studies in myotube cultures. Adding to this body of evidence, Sribar’s team suggested that venom proteins with PLA2 activity may be transported into the pre-synapse [54].
3. Molecular Effectors in Elapid and Viper Venoms
We will next review the main enzymatic activities of snake venom proteins. These primary enzymatic effects are frequently amplified within the prey, broadening and prolonging the pathology.
3.1. Snake Venom Phospholipases (svPLA2)
Phospholipase enzymes are found in nearly all forms of life including bacteria, plants, invertebrates and vertebrates. PLA2s have roles in the regulation of phospholipid turnover and membrane lipid content. Their most important physiological role is the production of arachidonic acid (AA). AA is the first step in the production of eicosanoids, leukotrienes and prostaglandins (Figure 2).
Vertebrate PLA2s comprise a large superfamily of enzymes composed of 16 recognized groups within six major types, as reviewed by Harris and Scott-Davey [46] (particularly see references 1–6 therein): These major types include the secreted PLA2s (sPLA2), the cytosolic PLA2s (cPLA2), the calcium independent PLA2s (iPLA2), the platelet activating factor (PAF) acetyl hydrolase/oxidized lipid lipoprotein associated PLA2s (LpPLA2s), the adipose PLA2 (AdPLA2s) and the lysosomal PLA2s (LPLA2s). Extracellular PLA2 requires millimolar to micromolar [Ca2+] for full activity, whereas the intracellular PLA2 is active in the nanomolar level Ca2+ range that characterizes the intracellular environment.
As is true of all PLA2s, svPLA2s catalyze specific hydrolysis of the ester linkage at the sn-2 position of glycerophospholipids. The catalytic site for the generation of AA lies in a grove accessible on the surface of the venom protein. However, PLA2-like proteins found in snake venoms may be devoid of catalytic activity. These non-catalytic subunits may exhibit myotoxic, neurotoxic, or pro-inflammatory effects [55,56]. The mechanisms of non-catalytically active PLA2 toxicity are poorly understood.
Snake venom PLA2s are probably the most pharmacologically active, multi-effect venom components [46]. Phospholipases are found to varying degrees in virtually all snake venoms and in the saliva of non-venomous and minimally venomous snakes as well, with a notable preponderance in the venoms of Elapid snakes. The catalytic components of svPLA2s principally act by hydrolyzing the glycerol ester of membrane bound arachidonic acid, liberating free arachidonic acid (AA). Arachidonic acid is highly reactive and stimulates important pathways that govern a myriad of biological processes. Arachidonic acid stimulates the creation of a family of biologically active signals, including: (1) members of the cyclooxygenase pathway, involving prostaglandins and thromboxane; (2) members of the lipoxygenase pathway, forming leukotrienes; and (3) regulation of the Cytochrome P450 group of enzymes, including lipoxygenases that form hydroperoxy-eicosatetraenoic acids (HPETEs) and hydro-eicosatetraenoic acids (HETEs). A summary of the role of these signaling molecules following snake envenoming with notations as to how they contribute to venom pathology (light blue boxes) is shown in Figure 2.
svPLA2s exist as monomers, dimers, heterodimers, trimers and hexamers composed of varying combinations of catalytic and non-catalytic subunits. The detailed functions of most non-catalytic units are still poorly defined, but some are thought to be involved in trafficking the catalytic unit to specific tissues, and in some cases direct cytotoxicity. This basic concept of PLA2 toxicity was first incorporated into a model by Kini and Evans about 30 years ago [49], although the enzymatic and non-enzymatic potential of PLA2 heterodimer components was not known at the time. It is now clear that formation of multi-unit complexes of catalytic and non-catalytic PLA2s can dramatically alter toxicity. In a remarkable demonstration, in vitro dimerization of catalytic and non-catalytic subunits from different families of snakes can produce enhanced toxicity: dimerization of crotoxin-A and a single β-chain of agkistrodotoxin increased toxicity over monomers [58]. For a review of the importance of venom component complexes to toxicity, see Doley and Kini [59].
svPLA2s have, by far, the broadest pathologic effects of any snake venom proteins. An excellent historical perspective and review of the development of knowledge about the role of PLA2s in venom toxicity was written by Gutierrez and Lomonte, recounting progress from the first crystallization of venom PLA2 to recent molecular understanding of PLA2 action [18]. Snake venom PLA2s are directly responsible for early- and late-onset symptomology, as well as synergistic and regulatory roles for other snake venom components [60]. sPLA2s are intimately involved in the peripheral neuro-myotoxicity caused by envenoming bites of many dangerous snakes and because both s- and cPLA2 are implicated in inflammatory and degenerative disease of the nervous system, roles that are discussed below. PLA2 can also mediate cell-based toxicity, for example in Bothrops PLA2 myotoxicity [61]. PLA2s can be the dominant venom component in some species. A recent paper from Calvete’s group showed that 60% of the proteins in Russell’s viper belong to the PLA2 family [62]. The widespread distribution of PLA2 in snake venoms suggests a universal potential for toxicity involving the dysregulation of processes involving arachidonic acid.
Intracellular PLA2 activity must liberate arachidonic acid from membranes inside the cell, having powerful actions. Importantly, this would include release of Ca2+ from intracellular sites of sequestration, mediated by arachidonic acid and activation of ryanodine receptors [63]. Because membrane bound PLA2 generates intracellular arachidonic acid, and intracellular action of PLA2 does not, in principle, require translocation of sPLA2 into the intracellular compartment.
One of the most interesting and important features of svPLA2s is their capacity to activate PLA2 homologs in the tissues of prey, in a process of homologous protein activation. This results in an amplification of the effects of svPLA2 beyond that of the venom alone. This mechanism also provides a relay of toxicity by non-catalytic svPLA2s to endogenous catalytic PLA2 proteins. A transcellular relay of phospholipase activity was first described by Shier who reported experiments in 1979 showing activation of endogenous PLA2 by a fraction of cobra venom [20]. This action, mediated by cobra “Lytic factor”, seems to be specific to activation of endogenous intracellular PLA2. Further, purified mellitin, which contains no intrinsic PLA2 activity, greatly increases intracellular PLA2 activity in cultured cells. These key results were among the first to show that venom can relay protease activity to the intracellular compartment, co-opting the cells own machinery. This relay and amplification indicate that venom PLA2s can have effects that are larger than would be guessed based on their percentage distribution in venoms.
Because of its near ubiquity in snake venoms and clinically significant effects, sPLA2 is an appealing candidate for inhibition by small molecule therapeutics [64] and recent studies have demonstrated that PLA2 activity of a diverse group of venoms can be inhibited by the agent varespladib [65,66,67,68]. Varespladib was recently shown to bind to both catalytic and non-catalytic PLA2s, reducing myotoxicity mediated solely by a non-catalytic PLA2 [69].
3.2. Snake Venom Metalloprotease (svMP)
Metalloproteases are large molecular mass proteins, >50 kDa. Metalloproteases are a family of proteases that were originally grouped by their requirement for divalent cations (zinc and cobalt) for full activity. Subsequent studies revealed important diversity and subgroups based on structure, substrate and regulatory control [70]. Simply by virtue of their size, these enzymes act predominately in the circulatory compartment, playing roles to facilitate the dispersion of smaller molecular weight venom components (e.g., phospholipases) and in signaling and amplifying the toxicity of other venom components. It is important to note that metalloproteases are distinct from matrix metalloproteases (MMPs), although there is a key interaction, via inflammation, and induced gene expression by the digestion products of both classes of proteases [71,72].
It is generally accepted that metalloproteases in snake venoms play central roles in hemorrhage, by loosening the connective tissue components responsible for blood vessel structural integrity [41]. The pioneering histologic studies of McKay and Owenby [73,74] described destruction of basement membranes of capillaries in tissues exposed to hemorrhagic venom components, actions now thought primarily to be mediated by these metalloprotease enzymes. The ability of svMPs to degrade various types of extracellular matrix proteins has been demonstrated in vitro by protein electrophoresis and immunoblot techniques, allowing visualization of digestion fragments. svMPs hydrolyze laminin, nidogen, enactin, type IV collagen, fibronectin, and proteoglycans (for review see [41]). A recent proteomic analysis revealed an even greater range of protein targets [75]. Although metalloproteases have numerous effects in vitro that support a causative role in bleeding, the relative contribution of svMP to hemorrhage in vivo has been difficult to clearly ascertain for several reasons. First, other components of snake venoms, such as phospholipases and serine proteases, inhibit or alter coagulation proteins [8]. Second, tools such as selective small molecule inhibitors to isolate svMP effects from matrix metalloproteases have only recently been available. Examples of metalloprotease inhibitors include prinomastat and marimastat, developed to inhibit cancer metastasis [76].
The mechanism of action of hemorrhagic svMPs involves cleavage of structurally important basement membrane components. This includes type IV collagen, followed by the mechanical disruption of vessels due to hemodynamic biophysical forces (i.e., a “two-step” process, see [41]). The identification of the regions in the molecular structure of svMPs that determine their ability to bind to the cleavage sites of basement membrane proteins are incompletely known. Studies by Gutierrez and Fox indicate that fragments of extracellular matrix proteins and other types of proteins released in the tissues as a consequence of svMP action may be normally involved in the processes of tissue repair and regeneration [41]. svMPs have a synergistic effect with PLA2 activity, related to these protein fragments. The complexity of potential interactions between PLA2s and MPs in producing tissue damage has recently been reviewed [77].
A number of observations suggest that endogenous proteases contribute importantly to the structural damage caused by snake venom structural proteases (for review [41]). Matrix metalloproteases are synthesized and secreted by resident and infiltrating cells in the course of the inflammation that follows tissue damage by venom. A further important effect of metalloproteases is that their action initiates and supports ongoing inflammation. Evidence for this action derives from studies in which relatively pure venom metalloprotease preparations have been injected into rodents and immune responses quantified. A number of such studies have reported increases in interleukins, PGE2, TNF-α, as well as changes in leukocyte populations and migration from the action of svMPs [78,79,80]. For a recent review of svMPs and immune modulation, see the review of Burin et al. [17]. Additional studies are needed to clarify the mechanism of immune modulation by metalloproteases, whether it is direct signaling by cleavage products or simply exposure of basement membranes and tissue factors during the digestion of the extracellular protein matrix.
3.3. Snake Venon Hyaluronidases
Although not present in large quantitites in the venom of any known snakes, hyuronidases apparently act to amplify the toxicity of other venom components by increasing the rate and spread of the injected toxins [12]. The products of proteoglycan hydrolysis produced by hyaluronidases have biological activity as well, with hyaluronan fragments participating in the acute pharmacological effects of envenoming, including the inflammatory response, by upregulating matrix metalloproteases [81]. This interesting body of work is reviewed by Kemparaju et al. [13]. Inhibition of venom hyaluronidase activity with natural or synthetic compounds has been a subject of several recent studies [12,82].
3.4. Other Directly Cytotoxic Proteins
A number of snake venom proteins have been reported to be directly cytotoxic in cell culture. Some venom proteins create cation channels in cell membranes, flooding cells with sodium and calcium and producing cell death by calcium intoxication, ionic imbalance and gross water movement/swelling/cell rupture. For review, see Waheed et al. 2017 [8]. Some of these toxicities include non-catalytic components of PLA2 heteromers.
Death receptors DR4 and DR5 are cell membrane proteins that trigger apoptosis. It is unclear if this is a direct binding or the consequence of paracellular or intracellular signals generated that activate these receptors via phospholipase cascades. Death receptor DR4 and DR5 may be activated by some types of venom, e.g., Vipera lebetina [26], but the interaction of venom and these receptors is not yet well characterized. Apoptosis is an actively mediated form of cell death and would seem not to be part of the immediate threat of envenoming. Apoptosis involves complex intracellular machinery and is an example of how a venom co-opts the prey’s own signaling apparatus to kill cells.
3.5. Non-Enzymatic 3-Finger Toxins (3FTx, -Neurotoxins)
3FTx proteins are found predominately in elapid venoms. In king cobras (Ophiophagus hannah) and Eastern green mambas (Dendroaspis angusticeps), 3FTx proteins make up about 70% of the proteins in venom [83]. In desert coral snakes (Micrurus tschudii), the proportion is reported as high as 95% [84]. This group of snake venom toxins are generally high affinity competitive antagonists of nicotinic acetylcholine receptors, making them one of the few groups of venom proteins whose action is well characterized. The diversity of 3FTs and the genomics of these proteins were recently reviewed [85]. High affinity binding of α-bungarotoxin to nAChRs is widely employed in receptor biology to label and study nAChRs.
4. Prey Response to Venoms: How Venoms Co-Opt Signaling Pathways to Produce Toxicity
Snakebite is not only a condition mediated directly by venom proteins but by the reaction of the envenomed animal to the venom. Understanding secondary causes of venom toxicity is a challenge to designing effective therapies to combat the serious delayed effects of envenoming in initial snake bite survivors.
4.1. Inflammation and Inflammation-Mediated Cytotoxicity
4.1.1. General Processes Involved in Activating Inflammation: Importance of PLA2
In view of the diversity of venom proteins, it is perhaps not surprising that snake venoms are capable of activating all pathways of the immune response. A novel proposal relating to immune reactions to snakebite is that type II immune responses evolved in vertebrates to protect against venom. This hypothesis was advanced by Galli and colleagues [86]. Type II immune responses usually involve acquired immunity, mediated by IgE antibodies and mast cells, producing seemingly inappropriate immune responses like anaphylaxis to foods and other common antigens. Mast cells can degranulate following envenoming even with no prior specific immunization or sensitization. The resulting intense humoral immune response protects against otherwise fatal venom exposures [86,87]. However, the interaction of the immune system and snake venoms is more complex than just type II responses. As highlighted by Burin and colleagues [17], snake venom accomplishes both immune suppression and immune stimulation, with the pattern of reaction to venom snake species-specific. The potential of using these actions of venom for immune therapy and for understanding immune responses has been much discussed [88].
A particularly broad and complex venom-induced inflammatory response is seen after cobra envenoming, with both pro- and anti-inflammatory components [89]. Naja annulifera venom triggers acute systemic inflammation, including PLA2-dependent neutrophilia and increased plasma levels of IL-6 and MCP-1. In mice, 2LD50 dose of Naja venom caused both neutrophilia and monocytosis. In an in vivo experimental model in mice, Silva-de-Franca et al. [90] found that Naja annulifera venom induced swelling and several histopathologic changes in the hind paws of the animals. In addition, myonecrosis associated with inflammation was observed, an event that is commonly found in experimental models of elapid envenoming. This was attributed to a cytotoxic action of some PLA2 component.
Nearly all snake venoms initiate and sustain pathological inflammation in the body [91,92]. The consequences of this pathological inflammation include both short- and long-term effects of tissue damage and organ system failure. The first demonstration of an inflammatory reaction to snake venom seems to be that of Damerau et al. who in 1975 showed that cobra venom lytic factor caused mast cell degranulation, histamine release and cytokine production [93]. Brain and Whittle in 1977 reported that the inflammatory actions of Russell’s viper PLA2 generates a dose-dependent release of histamine. Brain and Whittle also believed that the venom released or enhanced endogenous PLA2 [94]. Recent studies have coalesced on the concept that svPLA2 and svMP appear to be the most important venom components in producing inflammation [16,95]. svPLA2 and svMP activities sidestep the usual foreign protein recognition aspect of immune activation to directly stimulate immune cells, via arachidonic acid and pro-inflammatory digestion products [17]. This creates a response disproportionate to the immediate direct immunogenicity of the venom proteins. PLA2 activity in venom produces high levels of arachidonic acid and related inflammatory cytokines both in serum and inside cells. Activation of innate immune cells by venoms triggers a self-amplifying cascade of pro-inflammatory cytokine production, including IL-6, TNF- and IL-1, chemokines, and lipid mediators, which produce a positive feedback loop of leukocyte migration and activation. Lipid mediators include eicosanoids derived from the arachidonic acid metabolism, such as prostaglandins, leukotrienes, and thromboxanes. These mediators, in combination with cytokines and chemokines, trigger various clinical and pathologic features of inflammation, including edema, pain, chemotaxis, cytokine release, and leukocyte activation. In addition, hyaluronidase, glycosylated proteins and svMPs promote different aspects of inflammation, which includes a range of effects, including complement activation [15].
We speculate that endogenous intracellular PLA2 recruitment in immune cells increases the venom’s ability to create a robust humoral immune response. The proposed mechanism involves arachidonic acid, ryanodine receptors, intracellular calcium, protein kinase C, endogenous PLA2 and calcium release from intracellular reservoirs, a universal mechanism involved in PLA2 signaling [63]. Activation of endogenous PLA2 is also probably involved. An example is in streptococcal bacterial infections in the lungs. PLA2-like proteins from these bacterial recruit and activate endogenous PLA2s in pulmonary macrophages and lung parenchyma [96]. A similar pattern of injury mediated by PLA2 in muscle tissue has been described [16]. Mediation of inflammatory pathways by metalloproteases is also likely to contribute to these effects [90].
In the above examples, the primary toxicity caused by svPLA2 is amplified and prolonged by the recruitment of endogenous PLA2. As with other venom effects, the contributions of primary and prey-enhanced inflammatory responses remain to be fully defined. Modern techniques to track systemic mediators of inflammation (cytokine arrays) could be employed to assess these events, as could cytokine mRNA arrays and the use of small molecule inhibitors.
4.1.2. Inflammation Underlies Increased Vascular Permeability and Tissue Edema
Inflammation produced in response to envenoming has the potential to affect every organ in the body. Inflammation-mediated fluid extravasation into tissue, when profound, can lead to tissue edema and systemic hypovolemia and circulatory shock. Longer-term effects of inflammation can lead to cell death and organ failure.
Inflammation-induced pulmonary edema is a common pulmonary complication of snakebite. Other impacts on the respiratory system include pulmonary hemorrhage [97] and, obviously, neuromuscular paralysis and respiratory failure [98]. Pulmonary edema following envenomation is most likely related to capillary leak mediated by inflammatory mediators [99], although cardiogenic pulmonary edema can occur as well [100].
Systemic inflammation and circulating cytokines such as TNF-α following envenoming may be associated with venom-induced lung injury in humans. In different models of hemorrhagic shock, plasma, pulmonary and hepatic increases in IL-6 and MCP-1 are observed with inflammation and lung injury, which may culminate in acute respiratory distress syndrome [101]. It is important to emphasize that in addition to cytokines, some other venom proteins such as enzymes that attack basement membranes (metalloproteases and hyaluronidases) may produce pulmonary hemorrhage [8].
4.2. Coagulation Disorders
Bleeding and disordered coagulation is seen with many types of envenoming, suggesting strong natural selection for venom proteins that produce coagulopathy. Coagulopathy following envenoming is complex, with contributions from svPLA2s, svMPs, hyaluronidases, serine proteases, and others [8]. Because phospholipids serve as potent co-factors for numerous enzymatic conversions in the intrinsic and extrinsic clotting cascades, it is not surprising that phospholipases play a potent role in regulating or disrupting coagulation. Venoms, through their PLA2 activity, achieve potent, multi-site anticoagulation by co-opting just a few key processes that regulate coagulation. Accordingly, venom PLA2 is a prime target for therapeutic intervention in the coagulopathy of snakebite [2,8,90,102].
4.2.1. Specific Role of svPLA2 in Disordered Coagulation
Dysregulated platelet adhesion, mediated by svPLA2, is a significant component of the pathology of snake-venom-induced coagulation disorder. This was apparently first suggested by Boffa and Boffa in the 1970s, and was presented model form by Kini and Evans [49]. PLA2-dependent effects include inhibition of platelet adhesion, release of thromboxane, serotonin and adenosine diphosphate. Each of these platelet-derived factors contribute importantly to disordered coagulation. The initiating event in producing all these mediators is the PLA2-dependent production of arachidonic acid from phospholipids in the platelet membrane or in circulating lipoprotein particles. The importance of PLA2 in coagulopathy was neatly demonstrated by the neutralization of the coagulotoxic effects of Naja venom with the specific PLA2 inhibitor varespladib [102]. Similar PLA2-dependent coagulation disruption was demonstrated by Anilkumar et al. [103] with a series of imidazopyridines that inhibit PLA2 activity in Russell’s viper venom.
4.2.2. Role of svMPs in Hemorrhage
Venom metalloproteases and venom-activated prey matrix metalloproteases are important in hemorrhage. It is not clear whether svMP activity is entirely direct, or whether its action also sets in motion other signaling molecules such as bioactive protein digestion products, expression of tissue factor and coagulation and expression of endogenous metalloproteases, in an amplification cascade. Current evidence reveals a very complex interrelationship between svMPs and endogenous matrix metalloproteases (MMPs). Gutierrez and colleagues [41] note that the degradation of some types of fibrillar collagen after envenoming depends upon the action of endogenous matrix metalloproteases (MMPs) in prey. Endogenous MMPs are rapidly expressed in prey as a result of the induction of inflammation mediated by svPLA2 and activation of endogenous PLA2. svMPs generate biologically active proteins from the hydrolysis of proteins in the extracellular matrix. For example, hydrolysis of types XV and XVIII collagen generates endostatin, an angiogenesis inhibitor. The cleavage of the α-3 chain of type IV collagen by matrix MPs releases tumustatin, another antiangiogenic molecule. This complex mixture of biologically active fragments of extracellular matrix degradation serves to amplify and broaden the initial effects of the svMPs. Therefore, the interaction of inflammation and protease action on the extracellular matrix and blood vessel integrity is crucial in determining the pathologic effects of venoms containing svMPs. These topics have been extensively explored because of the implications for inhibition of angiogenesis in cancer treatment [76]. Only recently has research moved more broadly to consider the effects of svMPs other than effects mediating structural damage to basement membranes including those of blood vessels. Disruption of microvessel integrity is accepted as the most important cause of hemorrhage caused by viperid venoms [14]. The expression of matrix MPs and tissue inhibitors of MPs by cells is regulated by numerous cytokines (particularly interleukin-1, IL-1), growth factors and hormones, some of which are specific to cell type and others that are ubiquitous (e.g., transforming growth factor beta, TGF-beta) [76]. Many of these factors are products of monocytes/macrophages and their production in inflammatory situations is therefore part of the chain of events leading to tissue degradation. Tissue destruction, both physiological and pathological, is correlated with an imbalance of inhibitors and activators [42]; snake venoms would likely tip the scales.
4.3. Paralysis
Paralysis is one of the most dramatic and consequential effects of envenoming. However, other than the well-defined actions of 3FTxs (e.g., α-bungarotoxin) as competitive antagonists of nicotinic acetylcholine receptors, one of the least understood. Paralysis of skeletal muscles, including respiratory muscles, is the cardinal effect of elapid venoms but also a clinical feature of many types of viper envenoming. A very strong body of evidence points to PLA2 activity in venom (i.e., β-neurotoxins) as the main cause of paralysis produced by bites from some of the most medically important snakes in the world. Among the clinically relevant effects of β-neurotoxins are depletion of pre-synaptic neurotransmitter vesicles, inactivation of nicotinic acetylcholine receptors and eventual physical degeneration of the neuromuscular endplate. Ranawaka and colleagues recently reviewed some of the controversies related to PLA2 paralysis, from the perspective of the actions of krait β-neurotoxin [104]. An integrated two-part model for how we believe PLA2 mediates a multi-target attack on neuromuscular transmission is shown in Figure 3. The initial effects of facilitation, then rundown of acetylcholine release is depicted in the upper panel, and inactivation of transmission at the post-junction are shown in the lower panel. Together, these effects explain both the rapid and sustained effects of PLA2 at the neuromuscular junction.
4.3.1. Clinical Features of Paralysis from β-Neurotoxins
There are several features of the paralysis produced by PLA2 venoms that must be accounted for in any mechanistic explanation. The first is that fasciculations (myokymia) frequently precede or accompany the onset of clinical weakness. In contrast, fasciculations are never seen in neuromuscular block produced by competitive antagonists of nicotinic acetylcholine receptors, e.g., the curare-like non-depolarizing agents used to produce muscle relaxation for surgery or critical care [105]. Fasciculations also are never seen even during subclinical block or during block recovery from non-depolarizing agents. However, fasciculations are a clear feature of drugs that produce depolarizing block, such as succinylcholine or decamethonium, or presynaptic toxins such as botulinum or nerve agents. As a result of facilitated acetylcholine release, these agents cause both depletion of pre-junctional neurotransmitter and depolarization of the post-junctional membrane, at least for a period of time. Fasciculations are a clinical sign of disordered release and accumulation acetylcholine, inhibited disposal of acetylcholine in the neuromuscular junction, or activation of extra-junctional receptors. Fasciculations have been clearly described in bites involving PLA2 venoms, in both elapids [106] and vipers [107,108].
Fasciculations precede and cause a second and very important (although short-lived) phenomenon at the neuromuscular junction: desensitization of post-junctional nicotinic acetylcholine receptors. Acute desensitization was one of the first autoregulatory aspects of nicotinic transmission at the neuromuscular junction described by Sir Bernard Katz in seminal studies of neuromuscular junction function [109]. The details of this have been revealed in the frog neuromuscular junction to involve the action of calcium-dependent negative feedback inhibition of neurotransmission mediated by pre-synaptic nicotinic and muscarinic autoreceptors [110]. At the same time that desensitization of post-junctional nicotinic receptors is occurring, depletion of synaptic vesicles develops, such that nerve depolarization does not accomplish neurotransmission. This pre-synaptic effect is based on the antecedent enhanced vesicle fusion and release, Ca2+ entry, and Ca2+ facilitation of phospho-activation of the vesicle release protein system. Basically, the neurotransmitter system is depleted and not replenished (see Figure 3).
The third effect is seen at the post-junctional component of the NMJ. When desensitization is accompanied by post-synaptic increases in [Ca2+], calcium-sensitive phosphatases are activated and nicotinic receptors undergo the process of inactivation. As this persists, receptors are removed from the synapse in a process of cytoskeleton-mediated internalization. This is a long-lasting effect. The pharmacology of receptor desensitization and inactivation is detailed in a review by Albuquerque [50]. This action by β-neurotoxins toxins is more speculative, although it is a mechanism based on many studies of nicotinic receptor regulation in various preparations [111]. It is important to note that there is some evidence that PLA2s can act as competitive nAChR antagonists, similar to the better defined α-neurotoxins [112]. The quantitative importance of this α-effect is unresolved.
The final observation that must be accounted for in a mechanistic explanation is that, following very prolonged paralysis mediated by β-neurotoxins, physical damage to the synaptic structure may occur. A model of PLA2 toxicity must also account for the observation that relatively long periods of neuromuscular block (4–6 h) can be reversed by small molecule PLA2 inhibitors. Such a rescue was in swine envenomed with Micrurus venom was observed with the PLA2 inhibitor varespladib [67].
4.3.2. Is PLA2 and Arachidonic Acid Sufficient to Cause Synaptic Failure
Not excluding other mechanisms, we believe that arachidonic acid generated by svPLA2 is sufficiently potent to account for the clinical and pathologic features outlined above. A key mechanism is that AA generated by catalytic svPLA2s or endogenous PLA2s potently stimulates Ca2+ release from the endoplasmic reticulum [54], accounting for the initial surge of acetylcholine release and fasciculations. Increased Ca2+ in the pre- and post-junctional compartments are amplified by other mechanisms as well, including Ca2+ entry through voltage-gated calcium channels. Observed electrophysiological effects of crotoxin are consistent with these actions. For example, both crotoxin dimer (one catalytically active and one catalytically inactive PLA2 subunit) and the basic PLA2 subunit monomer, have a biphasic and calcium-dependent effect on nerve-evoked transmitter exocytosis. A transient initial facilitation followed by a sustained decay of transmitter release, is observed. Monomer and dimer reduce nerve-evoked radiolabeled-ACh release by 60% and 69%, respectively, but only the crotoxin heterodimer decreased the amplitude of nerve-evoked muscle twitches [19]. This model is also consistent with in vitro studies of C. durissus crotoxin [113]. Experiments of neuromuscular junction electrophysiology and intracellular Ca2+ measurements and small molecule inhibitors of secreted PLA2s would be very helpful in confirming or refuting the validity of the role of Ca2+ in these interpretations.
Augmenting pre-junctional failure, AA inhibits the choline reuptake transporter, contributing to the depletion of pre-synaptic terminals of releasable acetylcholine [114]. Combined with the other presynaptic actions of arachidonic acid, blockade of choline re-uptake is a significant contributor to paralysis. These actions comprise pre-synaptic focus of PLA2 toxin effects, consistent with electrophysiology done in the seminal work of Chang et al. [115]. However, technically speaking the work of Cheng did not rule out some blockade at the post-synapse, just not a block of the muscle itself, such as a prolonged muscle depolarization or toxicity to myofibrils.
Arachidonic acid also interacts with SNARE proteins that regulate neurotransmitter release, contributing to the biphasic effects of β-neurotoxins on neuromuscular transmission. This produces an initial facilitation followed by a long-lasting depression of neuromuscular function. This effect most likely involves both vesicle depletion and receptor inactivation as discussed above; i.e., both pre- and post-synaptic inhibition of NMJ function [116]. Consistent with our model, the initial effects of svPLA2 increase transmitter release in an exuberant and uncontrolled manner, and then produce deactivation of post-junctional receptors.
Arachidonic acid mediates release of Ca2+ from intracellular stores such as the endoplasmic reticulum; these alterations can be cytotoxic, particularly in the context of other co-occurring cellular stress [117].
4.3.3. Post-Junctional Effects of PLA2 Venoms
In the post-synapse, AA activates protein kinase C (PKC) [118]. Activated PKC phosphorylates nicotinic receptors, increasing their activity but contributing rapidly to desensitization, which is a conformational state of the receptor protein which renders it less capable of activation by acetylcholine [112]. A second and key stage of receptor inactivation occurs with receptor de-phosphorylation by calcium-sensitive phosphatases, resulting in prolonged internalization of these now inactivated nicotinic receptors. Inactivated receptors can remain intact in the intracellular compartment of the cell for 5 or more hours, before being re-inserted into in the post-synaptic membrane as functional receptors. This is a period of profound synaptic inactivation [119]. In addition to functional changes in the synapse, internalization of nicotinic receptors causes cytoskeletal-dependent structural changes in the synaptic structure. Thus, PLA2, via AA, co-opts machinery crucial to the function of both the pre- and post-synapse in the neuromuscular junction, producing a multidimensional, profound and persistent blockade of neuromuscular function.
PLA2 or closely associated proteins have been proposed to directly antagonize nAchRs [113], similar to a-toxins. This does not seem to us to be a complete explanation for several reasons. First, the initial clinical presentation of PLA2-induced paralysis does not fit that of a competitive antagonist (see above) or explain the initial facilitation of neurotransmitter release seen in electrophysiology studies. Also, there is no demonstrated high-affinity of -neurotoxins for nicotinic receptors.
4.3.4. Role of Calcium in PLA2 Mediated Pre- and Post-Junctional Block of Neurotransmission
The cycle of effects initiated by PLA2 forms a positive feedback loop involving plasma membrane PLA2, endogenous PLA2, Ca2+ release, kinase activation and activation of Ca2+ release/influx by multiple mechanisms (upper panel in Figure 3). This process is not unique to snake venoms, but a widespread mechanism clearly described in other cells [63]. One of the most important effects of the AA-Ca2+ amplification cycle is grossly elevated intracellular Ca2+. A study by Tedesco neatly demonstrated this action: the large and sustained increases in interterminal [Ca2+] produced by a snake PLA2 neurotoxin (β-bungarotoxin, taipaitoxin) was similar to that of the well-characterized black widow venom α-latrotoxin [120].
Bothrops asper myotoxins type I and II induce Ca2+ release from inside the cell, most likely from the endoplasmic reticulum, which contains by far the largest amount of Ca2+ inside cells [121,122,123]. This is also observed in human brain endothelial cells, where arachidonic acid releases intracellular Ca2+ by inositol triphosphate and ryanodine receptors. This work is convergent with our earlier work showing that release of calcium from the ER is a key player in adaptation to hypoxia and in ischemic preconditioning in neurons [124]. In addition, triggered release of Ca2+ by venom is a plausible mechanism of cellular toxicity, and would involve mitochondrial dysfunction caused by mitochondrial Ca2+ overload, as in neurodegenerative diseases [125]. The neurotoxic secreted phospholipase A2 from the Vipera a. ammodytes venom targets cytochrome-c oxidase in neuronal mitochondria [54].
4.3.5. PLA2 and Rapid Degeneration of the Synapse
A key question is whether venoms permanently destroy neuromuscular junctions or cause potentially reversible physical changes in the structure and function of the junction. Current dogma posits rapid destruction of the pre-synapse [126]. It has been known for some time that -neurotoxins such as bungarotoxin produce physical changes in the structure of motor nerve terminals that precede axonal degeneration of motor nerves [127]. Indeed, rat muscles paralyzed with β-bungarotoxin show loss of synaptic vesicles, changes in mitochondria, loss of boutons and other changes and ultrastructural changes observed between 3 and 6 h. However, the hypothesis that the morphological changes observed within 3–6 h represent degeneration (and presumed loss of rescue potential) do not fit with the observation that PLA2 inhibitors can reverse induced antivenom resistant paralysis in swine caused by Micrurus venom some 4 h after envenoming [67]. Thus, the hypothesis that β-neurotoxins cause irreversible damage to the NMJ soon after envenoming may need modification.
Structure and function of the neuromuscular junction are intimately linked. The neuromuscular junction is highly plastic, changing anatomical form with function, and requiring constant input from neurotransmission and trophic factors for its health and permanence. Nicotinic receptor activation state is a key part of this regulation, because receptors are connected to the cytoskeleton. When receptors are inactivated, they are internalized, and the cytoskeleton complex that determines the shape of the NMJ is altered at the same time. Neuromuscular junction function and morphology both can change over the course of minutes [128]. These cytoskeletal features are highly conserved across different animals, with similar mechanisms in mammals, insects and mollusks [129]. Accordingly, neuromuscular block, regardless of mechanism, leads to reversible changes in synaptic structure. Some of the changes in synaptic morphology seen in ER studies could represent structural changes due to receptor internalization, without actual cell degeneration. This possibility widens the therapeutic window for reversing neuromuscular block induced by venoms, consistent with late rescue of neuromuscular dysfunction by varespladib [67]. Thus, β-neurotoxins may be more like nerve agent toxins than botulinum toxin in their effects on the neuromuscular junction, at least in terms of reversibility, if not mechanism. Certainly, persistent paralysis can cause atrophic degeneration; this is also seen in the curare-like drugs used for long periods (even in humans with botulism or prolonged neuromuscular block in the intensive care unit [130]).
5. Interaction and Amplification of Venom MPs and PLA2s in Inflammation and Coagulation
We have described some ways that svMPs and svPLA2s and probably other venom components act synergistically, mediated by their biologically active products. Teixeira and colleagues have written an excellent recent review of the convergence and interaction of inflammation and coagulation disorders in snake envenoming [88]. More work is needed to uncover these important interacting mechanisms and to discover therapeutic strategies based on them. Figure 4 is a synthesis of known and speculative pathway convergence.
6. Tools Needed to Dissect the Multi-Compartment Action of Snake Venoms
The hypothesis that venom mechanisms involve a substantial contribution from prey response can be tested with a number of tools and approaches. These tools include: (1) small molecule selective inhibitors of specific signaling components; (2) genetic models that have been engineered for deletion or amplification of specific signal elements; (3) assay of prey gene expression or cytokine expression profiles, either alone or in combination with #1 and #2. None of these approaches have been extensively used to understand reaction to snake venoms.
In contrast, antivenom serotherapy has a limited capacity to dissect venom mechanisms. Serotherapy is a treatment that predominately intercepts venom molecules in the plasma or lymph. Whether polyvalent or monoclonal, antibodies are mostly restricted to the blood and perhaps to a limited degree to the interstitial compartment (i.e., antiserum has a small volume of distribution) and this is also a limitation of their utility as research tools for dissection of venom mechanisms.
7. Areas Where New Knowledge Concerning the Cellular Effects of Venom Is Needed
The complexity of snake venoms has clearly deterred efforts to formulate molecular understandings of venom action. However, we are optimistic that progress towards such an understanding is possible. This optimism is supported by evidence that small molecule drugs, acting at specific enzyme sites, can rescue late toxicity, such as in the case of the PLA2 inhibitor varespladib [67,68,69]. Other highly selective small molecule inhibitors have the potential to reveal previously unknown, or underappreciated, components of venom toxicity, and venom-initiated endogenous responses.
Detailed understanding of the processes involving co-option of endogenous pathways and processes will contribute to new therapeutic directions for snakebite treatment. Several of the outstanding questions related to these endogenous components are presented in Table 2. Addressing the hypotheses in the Table will uncover host-based pathological responses to venom and suggest the advantages and disadvantages to different therapeutic approaches. Ultimately, whether the dissection approach is genetic, proteomic, or biochemical pathway specific, systems need to be understood in their functional totality. The World Health Organization’s recent attention to priority actions on snakebite therapeutics has directed attention to some of the newer approaches.
8. Conclusions
Snake venoms rely on proteases and small toxic peptides to elaborate signals that co-opt universal vertebrate signaling pathways that regulate coagulation, inflammation, neuromuscular function, and cell survival. Venoms act in the circulation, in the intracellular compartment, within cell membranes, and inside cells. Just a few types of protease activities underlie the capabilities of venom to derail regulation in the PLA2 and MP pathways, making it likely that small molecule inhibitors of PLA2s and MPs will provide synergistic therapeutic benefit. In addition, because these small molecules are not restricted in distribution as is current serotherapy, they have a potentially much wider therapeutic window for late effects of venom proteins. A major gap in our understanding is whether the intracellular effects of venom are due to intracellular translocation of venom protein including non-catalytic subunits of svPLA2s, or second messenger effects from arachidonic acid, Ca2+, etc. A further gap in our knowledge is whether the benefits of drugs like varespladib are due to inhibition of venom protease activity, inhibition of prey protease activity, or a mixture of the two effects.
Recent advances in testing and development of small molecule inhibitors of snake toxins and snake toxin effects take us into new territory in several important ways. First, they provide rescue therapy beyond what antivenoms can do. Second, they provide insight into the physiology and pathology of snakebite in ways that antivenoms never could. Because of the exquisite selectivity of new agents against venom protease activities, we can begin to unravel how snake venoms not only exert direct toxicity, but co-opt reactive inflammation in the prey/victim to produce toxicity to organs, cause paralysis, and other short and long-term effects.
Funding
This research received no external funding
Conflicts of Interest
The author owns equity in Ophirex, Inc., a public benefit corporation.
References
- Longbottom, J.; Shearer, F.M.; Devine, M.; Alcoba, G.; Chappuis, F.; Weiss, D.J.; Ray, S.E.; Ray, N.; Warrell, D.A.; Ruiz de Castaneda, R.; et al. Vulnerability to snakebite envenoming: A global mapping of hotspots. Lancet 2018, 392, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primers 2017, 3, 17079. [Google Scholar] [CrossRef]
- Isbister, G.K.; Silva, A. Addressing the global problem of snake envenoming. Lancet 2018, 392, 619–620. [Google Scholar] [CrossRef] [Green Version]
- Waiddyanatha, S.; Silva, A.; Siribaddana, S.; Isbister, G.K. Long-term Effects of Snake Envenoming. Toxins 2019, 11, 193. [Google Scholar] [CrossRef] [Green Version]
- Calmette, A. The treatment of anomals poisoned with snakevenom by the injection of antivenomous serum. Br. Med. J. 1896, 2, 399–400. [Google Scholar] [CrossRef] [Green Version]
- Hawgood, B.J. Pioneers of anti-venomous serotherapy: Dr. Vital Brazil. Toxicon 1992, 30, 573–579. [Google Scholar] [CrossRef]
- Lauder Brunton, T.; Fayrer, T. On the nature and physiological action of the poison of Naja tripudians and other Indian venomous snakes. Part I. Proc. R. Soc. 1873, 145, 357–374. [Google Scholar]
- Waheed, H.; Moin, S.F.; Choudhary, M.I. Snake Venom: From Deadly Toxins to Life-saving Therapeutics. Curr. Med. Chem. 2017, 24, 1874–1891. [Google Scholar] [CrossRef]
- Laustsen, A.H. Toxin-centric development approach for next-generation antivenoms. Toxicon 2018, 150, 195–197. [Google Scholar] [CrossRef] [Green Version]
- Tasoulis, T.; Isbister, G.K. A Review and Database of Snake Venom Proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef] [Green Version]
- Cesar, P.H.S.; Braga, M.A.; Trento, M.V.C.; Menaldo, D.L.; Marcussi, S. Snake Venom Disintegrins: An Overview of their Interaction with Integrins. Curr. Drug Targets 2019, 20, 465–477. [Google Scholar] [CrossRef]
- Oliveira-Mendes, B.B.R.; Miranda, S.E.M.; Sales-Medina, D.F.; Magalhaes, B.F.; Kalapothakis, Y.; Souza, R.P.; Cardoso, V.N.; De Barros, A.L.B.; Guerra-Duarte, C.; Kalapothakis, E.; et al. Inhibition of Tityus serrulatus venom hyaluronidase affects venom biodistribution. PLoS Negl. Trop. Dis. 2019, 13, e0007048. [Google Scholar] [CrossRef] [PubMed]
- Kemparaju, K.; Girish, K.S.; Nagaraju, S. Hyaluronidases, a neglected class of glycosidases from snake venom: Beyond a spreading factor. In Handbook of Venoms and Toxins of Reptiles; Mackessey, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2016; Volume 1, pp. 237–261. [Google Scholar]
- Escalante, T.; Rucavado, A.; Fox, J.W.; Gutierrez, J.M. Key events in microvascular damage induced by snake venom hemorrhagic metalloproteinases. J. Proteom. 2011, 74, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Luchini, L.S.G.; Pidde, G.; Squaiella-Baptistao, C.C.; Tambourgi, D.V. Corrigendum: Complement System Inhibition Modulates the Pro-Inflammatory Effects of a Snake Venom Metalloproteinase. Front. Immunol. 2019, 10, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, C.F.; Landucci, E.C.; Antunes, E.; Chacur, M.; Cury, Y. Inflammatory effects of snake venom myotoxic phospholipases A2. Toxicon 2003, 42, 947–962. [Google Scholar] [CrossRef]
- Burin, S.M.; Menaldo, D.L.; Sampaio, S.V.; Frantz, F.G.; Castro, F.A. An overview of the immune modulating effects of enzymatic toxins from snake venom. Int J. Biol. Macromol. 2018, 109, 664–671. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Lomonte, B. Phospholipases A2: Unveiling the secrets of a functionally versatile group of snake venom toxins. Toxicon 2013, 62, 27–39. [Google Scholar] [CrossRef]
- Cavalcante, W.L.G.; Noronha-Matos, J.B.; Timoteo, M.A.; Fontes, M.R.M.; Gallacci, M.; Correia-de-Sa, P. Neuromuscular paralysis by the basic phospholipase A2 subunit of crotoxin from Crotalus durissus terrificus snake venom needs its acid chaperone to concurrently inhibit acetylcholine release and produce muscle blockage. Toxicol. Appl. Pharmacol. 2017, 334, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Shier, W.T. Activation of high levels of endogenous phospholipase A2 in cultured cells. Proc. Natl. Acad. Sci. USA 1979, 76, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Katz, B.; Miledi, R. The effect of alpha-bungarotoxin on acetylcholine receptors. Br. J. Pharmacol. 1973, 49, 138–139. [Google Scholar] [CrossRef]
- Lodovicho, M.E.; Costa, T.R.; Bernardes, C.P.; Menaldo, D.L.; Zoccal, K.F.; Carone, S.E.; Rosa, J.C.; Pucca, M.B.; Cerni, F.A.; Arantes, E.C.; et al. Investigating possible biological targets of Bj-CRP, the first cysteine-rich secretory protein (CRISP) isolated from Bothrops jararaca snake venom. Toxicol. Lett. 2017, 265, 156–169. [Google Scholar] [CrossRef]
- Burgers, P.M.; Eckstein, F.; Hunneman, D.H. Stereochemistry of hydrolysis by snake venom phosphodiesterase. J. Biol. Chem. 1979, 254, 7476–7478. [Google Scholar]
- Oguiura, N.; Boni-Mitake, M.; Affonso, R.; Zhang, G. In vitro antibacterial and hemolytic activities of crotamine, a small basic myotoxin from rattlesnake Crotalus durissus. J. Antibiot. 2011, 64, 327–331. [Google Scholar] [CrossRef]
- Ownby, C.L.; Aird, S.D.; Kaiser, I.I. Physiological and immunological properties of small myotoxins from the venom of the midget faded rattlesnake (Crotalus viridis concolor). Toxicon 1988, 26, 319–332. [Google Scholar] [CrossRef]
- Park, M.H.; Jo, M.; Won, D.; Song, H.S.; Han, S.B.; Song, M.J.; Hong, J.T. Snake venom toxin from Vipera lebetina turanica induces apoptosis of colon cancer cells via upregulation of ROS- and JNK-mediated death receptor expression. BMC Cancer 2012, 12, 228. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.W. A brief review of the scientific history of several lesser-known snake venom proteins: L-amino acid oxidases, hyaluronidases and phosphodiesterases. Toxicon 2013, 62, 75–82. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Lohse, B.; Lomonte, B.; Engmark, M.; Gutiérrez, J.M. Selecting key toxins for focused development of elapid snake antivenoms and inhibitors guided by a Toxicity Score. Toxicon. 2015, 104, 43–45. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.J.; Gutiérrez, J.M.; Calvete, J.J.; Wüster, W.; Ratanabanangkoon, K.; Paiva, O.; Brown, N.I.; Casewell, N.R.; Harrison, R.A.; Rowley, P.D.; et al. Ending the drought: New strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteom. 2011, 74, 1735–1767. [Google Scholar] [CrossRef]
- Calvete, J.J.; Lomonte, B. A bright future for integrative venomics. Toxicon 2015, 107, 159–162. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Wüster, W.; Cook, D.A.N.; Bolton, F.M.S.; King, S.I.; Pla, D.; Sanz, L.; Calvete, J.J.; Harrison, R.A. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 9205–9210. [Google Scholar] [CrossRef] [Green Version]
- Laustsen, A.H.; Lomonte, B.; Lohse, B.; Fernández, J.; Gutiérrez, J.M. Unveiling the nature of black mamba (Dendroaspis polylepis) venom through venomics and antivenom immunoprofiling: Identification of key toxin targets for antivenom development. J. Proteom. 2015, 119, 126–142. [Google Scholar] [CrossRef]
- Bordon, K.C.; Perino, M.G.; Giglio, J.R.; Arantes, E.C. Isolation, enzymatic characterization and antiedematogenic activity of the first reported rattlesnake hyaluronidase from Crotalus durissus terrificus venom. Biochimie 2012, 94, 2740–2748. [Google Scholar] [CrossRef]
- Slotta, K.H.; Fraenkel-Conrat, H. Two active proteins from rattlesnake venom. Nature 1938, 142, 213. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Barnard, E.A.; Chiu, T.H.; Lapa, A.J.; Dolly, J.O.; Jansson, S.E.; Daly, J.; Witkop, B. Acetylcholine receptor and ion conductance modulator sites at the murine neuromuscular junction: Evidence from specific toxin reactions. Proc. Natl. Acad. Sci. USA 1973, 70, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Sousa, I.D.L.; Barbosa, A.R.; Salvador, G.H.M.; Frihling, B.E.F.; Santa-Rita, P.H.; Soares, A.M.; Pessoa, H.L.F.; Marchi-Salvador, D.P. Secondary hemostasis studies of crude venom and isolated proteins from the snake Crotalus durissus terrificus. Int. J. Biol. Macromol. 2019, 131, 127–133. [Google Scholar] [CrossRef]
- Jorge, M.T.; Ribeiro, L.A. Epidemiology and clinical features of South American rattlesnakes (Crotalus durissus) envenomation. Rev. Do Inst. Demedicina Trop. De São Paulo 1992, 34, 347–354. [Google Scholar] [CrossRef]
- Silveira, P.V.; Nishioka Sde, A. South American rattlesnake bite in a Brazilian teaching hospital. Clinical and epidemiological study of 87 cases, with analysis of factors predictive of renal failure. Trans. R Soc. Trop. Med. Hyg. 1992, 86, 562–564. [Google Scholar] [CrossRef]
- Oliveira, I.S.; Cardoso, I.A.; Bordon, K.D. Global proteomic and functional analysis of Crotalus durissus collilineatus individual venom variation and its impact on envenoming. J. Proteom. 2019, 191, 153–165. [Google Scholar] [CrossRef]
- Sanhajariya, S.; Duffull, S.B.; Isbister, G.K. Pharmacokinetics of snake venom. Toxins 2018, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.M.; Escalante, T.; Rucavado, A.; Herrera, C.; Fox, J.W. A Comprehensive View of the Structural and Functional Alterations of Extracellular Matrix by Snake Venom Metalloproteinases (SVMPs): Novel Perspectives on the Pathophysiology of Envenoming. Toxins 2016, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, J.J. Collagenases and tissue inhibitors of metalloproteinases: A functional balance in tissue degradation. Oral Dis. 1996, 2, 70–76. [Google Scholar] [CrossRef]
- Diraviyam, K.; Murray, D. Computational analysis of the membrane association of group IIA secreted phospholipases A2: A differential role for electrostatics. Biochemistry 2006, 45, 2584–2598. [Google Scholar] [CrossRef]
- Oberg, S.G.; Kelly, R.B. Saturable binding to cell membranes of the presynaptic neurotoxin β-bungarotoxin. Biochem. Biophys. Acta 1976, 433, 662–673. [Google Scholar] [CrossRef]
- MacDermot, J.W.; Westgaard, R.H.; Thompson, E.J. The binding of [3H]-pyridoxylated Beta-bungarotoxin to a high molecular weight protein receptor. Biochem. J. 1978, 175, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.B.; Scott-Davey, T. Secreted Phospholipases A2 of Snake Venoms: Effects on the Peripheral Neuromuscular System with Comments on the Role of Phospholipases A2 in Disorders of the CNS and Their Uses in Industry. Toxins 2013, 5, 2533–2571. [Google Scholar] [CrossRef] [Green Version]
- Lambeau, G.; Schmid-Alliana, A.; Lazdunski, M.; Barhanin, J. Identification and purification of a very high affinity binding protein for toxic phospholipases A2 in skeletal muscle. J. Biol. Chem. 1990, 265, 9526–9532. [Google Scholar]
- Nakashima, K.; Ogawa, T.; Oda, N.; Hattori, M.; Sakaki, Y.; Kihara, H.; Ohno, M. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc. Natl. Acad. Sci. USA 1993, 90, 5964–5968. [Google Scholar] [CrossRef] [Green Version]
- Kini, R.M.; Evans, H.J. A model to explain the pharmacological effects of snake venom phospholipases A2. Toxicon 1989, 27, 613–635. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [Green Version]
- Logonder, U.; Jenko-Praznikar, Z.; Scott-Davey, T.; Pungercar, J.; Krizaj, I.; Harris, J.B. Ultrastructural evidence for the uptake of a neurotoxic snake venom phospholipase A2 into mammalian motor nerve terminals. Exp. Neurol. 2009, 219, 591–594. [Google Scholar] [CrossRef]
- Rigoni, M.; Paoli, M.; Milanesi, E.; Caccin, P.; Rasola, A.; Bernardi, P.; Montecucco, C. Snake phospholipase A2 neurotoxins enter neurons, bind specifically to mitochondria, and open their transition pores. J. Biol. Chem. 2008, 283, 34013–34020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spolaore, B.; Fernandez, J.; Lomonte, B.; Massimino, M.L.; Tonello, F. Enzymatic labelling of snake venom phospholipase A2 toxins. Toxicon 2019, 170, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Sribar, J.; Oberckal, J.; Krizaj, I. Understanding the molecular mechanism underlying the presynaptic toxicity of secreted phospholipases A(2): An update. Toxicon 2014, 89, 9–16. [Google Scholar] [CrossRef]
- Borges, R.J.; Lemke, N.; Fontes, M.R.M. PLA2-like proteins myotoxic mechanism: A dynamic model description. Sci. Rep. 2017, 7, 15514. [Google Scholar] [CrossRef] [Green Version]
- Leiguez, E.; Giannotti, K.C.; Moreira, V.; Matsubara, M.H.; Gutiérrez, J.M.; Lomonte, B.; Rodríguez, J.P.; Balsinde, J.; Teixeira, C. Critical role of TLR2 and MyD88 for functional response of macrophages to a group IIA-secreted phospholipase A2 from snake venom. PLoS ONE 2014, 9, e93741. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.A.; Brash, A.R.; Murphy, R.C. The discovery and early structural studies of arachidonic acid. J. Lipid Res. 2016, 57, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Choumet, V.; Saliou, B.; Fideler, L.; Chen, Y.C.; Gubensek, F.; Bon, C.; Delot, E. Snake-venom phospholipase A2 neurotoxins. Potentiation of a single-chain neurotoxin by the chaperon subunit of a two-component neurotoxin. Eur. J. Biochem. 1993, 211, 57–62. [Google Scholar] [CrossRef]
- Doley, R.; Kini, R.M. Protein complexes in snake venom. Cell. Mol. Life Sci. 2009, 66, 2851–2871. [Google Scholar] [CrossRef]
- Laustsen, A.H. Toxin synergism in snake venoms. Toxin Rev. 2016, 35, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Salvador, G.H.M.; Cardoso, F.F.; Gomes, A.A.; Cavalcante, W.L.G.; Gallacci, M.; Fontes, M.R.M. Search for efficient inhibitors of myotoxic activity induced by ophidian phospholipase A2-like proteins using functional, structural and bioinformatics approaches. Sci. Rep. 2019, 9, 510. [Google Scholar] [CrossRef]
- Pla, D.; Sanz, L.; Quesada-Bernat, S.; Villalta, M.; Baal, J.; Chowdhury, M.A.W.; Leon, G.; Gutierrez, J.M.; Kuch, U.; Calvete, J.J. Phylovenomics of Daboia russelii across the Indian subcontinent. Bioactivities and comparative in vivo neutralization and in vitro third-generation antivenomics of antivenoms against venoms from India, Bangladesh and Sri Lanka. J. Proteom. 2019, 207, 103443. [Google Scholar] [CrossRef]
- Paillamanque, J.; Sanchez-Tusie, A.; Carmona, E.M.; Trevino, C.L.; Sandoval, C.; Nualart, F.; Osses, N.; Reyes, J.G. Arachidonic acid triggers [Ca2+] i increases in rat round spermatids by a likely GPR activation, ERK signalling and ER/acidic compartments Ca2+ release. PLoS ONE 2017, 12, e0172128. [Google Scholar] [CrossRef]
- Williams, D.J.; Faiz, M.A.; Abela-Ridder, B.; Ainsworth, S.; Bulfone, T.C.; Nickerson, A.D.; Habib, A.G.; Junghanss, T.; Fan, H.W.; Turner, M.; et al. Strategy for a globally coordinated response to a priority neglected tropical disease: Snakebite envenoming. PLoS Negl. Trop. Dis. 2019, 13, e0007059. [Google Scholar] [CrossRef] [Green Version]
- Bulfone, T.C.; Samuel, S.P.; Bickler, P.E.; Lewin, M.R. Developing Small Molecule Therapeutics for the Initial and Adjunctive Treatment of Snakebite. J. Trop. Med. 2018, 2018, 4320175. [Google Scholar] [CrossRef] [Green Version]
- Herzel, B.J.; Samuel, S.P.; Bulfone, T.C.; Raj, C.S.; Lewin, M.; Kahn, J.G. Snakebite: An Exploratory Cost-Effectiveness Analysis of Adjunct Treatment Strategies. Am. J. Trop. Med. Hyg. 2018, 99, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Lewin, M.R.; Gilliam, L.L.; Gilliam, J.; Samuel, S.P.; Bulfone, T.C.; Bickler, P.E.; Gutierrez, J.M. Delayed LY333013 (Oral) and LY315920 (Intravenous) Reverse Severe Neurotoxicity and Rescue Juvenile Pigs from Lethal Doses of Micrurus fulvius (Eastern Coral Snake) Venom. Toxins 2018, 10, 479. [Google Scholar] [CrossRef] [Green Version]
- Lewin, M.R.; Gutierrez, J.M.; Samuel, S.P.; Herrera, M.; Bryan-Quiros, W.; Lomonte, B.; Bickler, P.E.; Bulfone, T.C.; Williams, D.J. Delayed Oral LY333013 Rescues Mice from Highly Neurotoxic, Lethal Doses of Papuan Taipan (Oxyuranus scutellatus) Venom. Toxins 2018, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- Salvador, G.; Gomes, A.A.S.; Bryan-Quirós, W.; Fernández, J.; Lewin, M.R.; Gutiérrez, J.M.; Lomonte, B.; Fonte, M.R.M. Structural basis for phospholipase A2-like toxin inhibition by the synthetic compound Varespladib (LY315920). Nat. Sci. Rep. 2019, in press. [Google Scholar] [CrossRef]
- Nagase, H. Metalloproteases. Curr. Protoc. Protein Sci. 2001, 24, 21.4.1–21.4.13. [Google Scholar] [CrossRef]
- Saklatvala, J.; Dean, J.; Clark, A. Control of the expression of inflammatory response genes. Biochem. Soc. Symp. 2003, 70, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperi, C.; Papavassiliou, A.G. Molecular mechanisms regulating matrix metalloproteinases. Curr. Top. Med. Chem. 2012, 12, 1095–1112. [Google Scholar] [CrossRef] [PubMed]
- Ownby, C.L.; Bjarnason, J.; Tu, A.T. Hemorrhagic toxins from rattlesnake (Crotalus atrox) venom. Pathogenesis of hemorrhage induced by three purified toxins. Am. J. Pathol. 1978, 93, 201–218. [Google Scholar] [PubMed]
- McKay, D.G.; Moroz, C.; DeVries, A.; Csavossy, I.; Cruse, V. The action of hemorrhagin and phospholipase derived from Vipera palestinae venom on the microcirculation. Lab. Intestig. 1970, 22, 387–399. [Google Scholar]
- Zelanis, A.; Oliveira, A.K.; Prudova, A.; Huesgen, P.F.; Tashima, A.K.; Kizhakkedathu, J.; Overall, C.M.; Serrano, S.M.T. Deep Profiling of the Cleavage Specificity and Human Substrates of Snake Venom Metalloprotease HF3 by Proteomic Identification of Cleavage Site Specificity (PICS) Using Proteome Derived Peptide Libraries and Terminal Amine Isotopic Labeling of Substrates (TAILS) N-Terminomics. J. Proteome Res. 2019, 18, 3419–3428. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.M.; Rucavado, A.; Escalante, T.; Herrera, C.; Fernandez, J.; Lomonte, B.; Fox, J.W. Unresolved issues in the understanding of the pathogenesis of local tissue damage induced by snake venoms. Toxicon 2018, 148, 123–131. [Google Scholar] [CrossRef]
- Rucavado, A.; Lomonte, B.; Ovadia, M.; Gutierrez, J.M. Local tissue damage induced by BaP1, a metalloproteinase isolated from Bothrops asper (Terciopelo) snake venom. Exp. Mol. Pathol. 1995, 63, 186–199. [Google Scholar] [CrossRef]
- Fernandes, C.M.; Pereira Teixeira Cde, F.; Leite, A.C.; Gutiérrez, J.M.; Rocha, F.A. The snake venom metalloproteinase BaP1 induces joint hypernociception through TNF-alpha and PGE2-dependent mechanisms. Br. J. Pharmacol. 2007, 151, 1254–1261. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.M.; Zamuner, S.R.; Zuliani, J.P.; Rucavado, A.; Gutierrez, F.M.; Teixeira, F. Inflammatory effects of BaP1 a metalloproteinase isolated from Bothrops asper snake venom: Leukocyte recruitment and release of cytokines. Toxicon 2006, 47, 549–559. [Google Scholar] [CrossRef]
- Fieber, C.; Baumann, P.; Vallon, R.; Termeer, C.; Simon, J.C.; Hofmann, M.; Angel, P.; Herrlich, P.; Sleeman, J.P. Hyaluronan-oligosaccharide-induced transcription of metalloproteases. J. Cell Sci. 2004, 117, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Sivaramakrishnan, V.; Ilamathi, M.; Girish, K.S.; Kemparaju, K.; Rangappa, K.S.; Dhananjaya, B.L. Viper venom hyaluronidase and its potential inhibitor analysis: A multipronged computational investigation. J. Biomol. Struct. Dyn. 2017, 35, 1979–1989. [Google Scholar] [CrossRef]
- Lauridsen, L.P.; Laustsen, A.H.; Lomonte, B.; Gutiérrez, J.M. Toxicovenomics and antivenom profiling of the Eastern green mamba snake (Dendroaspis angusticeps). J. Proteom. 2016, 136, 248–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, L.; Pla, D.; Pérez, A.; Rodríguez, Y.; Zavaleta, A.; Salas, M.; Lomonte, B.; Calvete, J.J. Venomic Analysis of the Poorly Studied Desert Coral Snake, Micrurus tschudii tschudii, Supports the 3FTx/PLA₂ Dichotomy across Micrurus Venoms. Toxins 2016, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albulescu, L.O.; Kazandjian, T.; Slagboom, J.; Bruyneel, B.; Ainsworth, S.; Alsolaiss, J.; Wagstaff, S.C.; Whiteley, G.; Harrison, R.A.; Ulens, C.; et al. A Decoy-Receptor Approach Using Nicotinic Acetylcholine Receptor Mimics Reveals Their Potential as Novel Therapeutics Against Neurotoxic Snakebite. Front. Pharmacol. 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, S.J.; Starkl, P.; Marichal, T.; Tsai, M. Mast cells and IgE can enhance survival during innate and acquired host responses to venoms. Trans. Am. Clin. Clim. Assoc. 2017, 128–221, 193–221. [Google Scholar]
- Stone, S.F.; Isbister, G.K.; Shahmy, S.; Mohamed, F.; Abeysinghe, C.; Karunathilake, H.; Ariaratnam, A.; Jacoby-Alner, T.E.; Cotterell, C.L.; Brown, S.G. Immune response to snake envenoming and treatment with antivenom; complement activation, cytokine production and mast cell degranulation. PLoS Negl. Trop. Dis. 2013, 7, e2326. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Fernandes, C.M.; Leiguez, E.; Chudzinski-Tavassi, A.M. Inflammation Induced by Platelet-Activating Viperid Snake Venoms: Perspectives on Thromboinflammation. Front. Immunol. 2019, 10, 2082. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Z.; Qin, Z.H. Anti-Inflammatory and Immune Regulatory Actions of Naja naja atra Venom. Toxins 2018, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Silva-de-Franca, F.; Villas-Boas, I.M.; Serrano, S.M.T.; Cogliati, B.; Chudzinski, S.A.A.; Lopes, P.H.; Kitano, E.S.; Okamoto, C.K.; Tambourgi, D.V. Naja annulifera Snake: New insights into the venom components and pathogenesis of envenomation. PLoS Negl. Trop. Dis. 2019, 13, e0007017. [Google Scholar] [CrossRef]
- Gutierrez, J.M.; Rucavado, A.; Chaves, F.; Diaz, C.; Escalante, T. Experimental pathology of local tissue damage induced by Bothrops asper snake venom. Toxicon 2009, 54, 958–975. [Google Scholar] [CrossRef]
- Teixeira, C.; Cury, Y.; Moreira, V.; Picolo, G.; Chaves, F. Inflammation induced by Bothrops asper venom. Toxicon 2009, 54, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Damerau, B.; Lege, L.; Oldigs, H.D.; Vogt, W. Histamine release, formation of prostaglandin-like activity (SRS-C) and mast cell degranulation by the direct lytic factor (DLF) and phospholipase A of cobra venom. Naunyn Schmiedebergs Arch. Pharmacol. 1975, 287, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Brain, S.; Lewis, G.P.; Whittle, B.J. Actions of phospholipase-A on mast-cell histamine release and paw oedema in the rat [proceedings]. Br. J. Pharmacol. 1977, 59, 440P–441P. [Google Scholar] [PubMed]
- Boeno, C.N.; Paloschi, M.V.; Lopes, J.A.; Pires, W.L.; Setubal, S.D.S.; Evangelista, J.R.; Soares, A.M.; Zuliani, J.P. Inflammasome Activation Induced by a Snake Venom Lys49-Phospholipase A2 Homologue. Toxins 2019, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Clark, S.; Bonventre, J.V.; Leong, J.M.; McCormick, B.A. Cytosolic Phospholipase A2α Promotes Pulmonary Inflammation and Systemic Disease during Streptococcus pneumoniae Infection. Infect. Immun. 2017, 85, e00280-17. [Google Scholar] [CrossRef] [Green Version]
- Palangasinghe, D.R.; Weerakkody, R.M.; Dalpatadu, C.G.; Gnanathasan, C.A. A fatal outcome due to pulmonary hemorrhage following Russell’s viper bite. Saudi Med. J. 2015, 36, 634–637. [Google Scholar] [CrossRef]
- Gnanathasan, A.; Rodrigo, C. Pulmonary effects and complications of snakebites. Chest 2014, 146, 1403–1412. [Google Scholar] [CrossRef]
- Siddall, E.; Khatri, M.; Radhakrishnan, J. Capillary leak syndrome: Etiologies, pathophysiology, and management. Kidney Int. 2017, 92, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Singh, A.P.; Aggarwal, A.N. Pulmonary oedema complicating snake bite due to Bungarus caeruleus. Singapore Med. J. 2007, 48, e227–e230. [Google Scholar] [PubMed]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittenbinder, M.A.; Zdenek, C.N.; Op den Brouw, B.; Youngman, N.J.; Dobson, J.S.; Naude, A.; Vonk, F.J.; Fry, B.G. Coagulotoxic Cobras: Clinical Implications of Strong Anticoagulant Actions of African Spitting Naja Venoms That Are Not Neutralised by Antivenom but Are by LY315920 (Varespladib). Toxins 2018, 10, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anilkumar, N.C.; Sundaram, M.S.; Mohan, C.D.; Rangappa, S.; Bulusu, K.C.; Fuchs, J.E.; Girish, K.S.; Bender, A.; Basappa; Rangappa, K.S. A One Pot Synthesis of Novel Bioactive Tri-Substitute-Condensed-Imidazopyridines that Targets Snake Venom Phospholipase A2. PLoS ONE 2015, 10, e0131896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranawaka, U.K.; Lalloo, D.G.; De Silva, H. Neurotoxicity in snakebite—The limits of our knowledge. PLoS Negl. Trop. Dis. 2013, 7, e2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, W.C. Neuromuscular block. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S277–S286. [Google Scholar] [CrossRef]
- Bamford, N.J.; Sprinkle, S.B.; Cudmore, L.A.; Cullimore, A.M.; Van Eps, A.W.; Verdegaal, E.; Tennent-Brown, B.S. Elapid snake envenomation in horses: 52 cases (2006-2016). Equine Vet. J. 2018, 50, 196–201. [Google Scholar] [CrossRef]
- Bosak, A.R.; Ruha, A.M.; Graeme, K.A. A case of neurotoxicity following envenomation by the Sidewinder rattlesnake, Crotalus cerastes. J. Med. Toxicol. 2014, 10, 229–231. [Google Scholar] [CrossRef] [Green Version]
- Richardson, W.H.; Goto, C.S.; Gutglass, D.J.; Williams, S.R.; Clark, R.F. Rattlesnake envenomation with neurotoxicity refractory to treatment with crotaline Fab antivenom. Clin. Toxicol. (Phila.) 2007, 45, 472–475. [Google Scholar] [CrossRef]
- Katz, B.; Thesleff, S. A study of the desensitization produced by acetylcholine at the motor end-plate. J. Physiol. 1957, 29, 63–80. [Google Scholar] [CrossRef]
- Khaziev, E.; Samigullin, D.; Zhilyakov, N.; Fatikhov, N.; Bukharaeva, E.; Verkhratsky, A.; Nikolsky, E. Acetylcholine-Induced Inhibition of Presynaptic Calcium Signals and Transmitter Release in the Frog Neuromuscular Junction. Front. Physiol. 2016, 7, 621. [Google Scholar] [CrossRef] [Green Version]
- Eilers, H.; Schaeffer, E.; Bickler, P.E.; Forsayeth, J.R. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase C-dependent mechanism. Mol. Pharmacol. 1997, 52, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Vulfius, C.A.; Kasheverov, I.E.; Kryukova, E.V.; Spirova, E.N.; Shelukhina, I.V.; Starkov, V.G.; Andreeva, T.V.; Faure, G.; Zouridakis, M.; Tsetlin, V.I.; et al. Pancreatic and snake venom presynaptically active phospholipases A2 inhibit nicotinic acetylcholine receptors. PLoS ONE 2017, 12, e0186206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bon, C. Synergism of the two subunits of crotoxin. Toxicon 1982, 20, 105–109. [Google Scholar] [CrossRef]
- Boksa, P.; Mykita, S.; Collier, B. Arachidonic acid inhibits choline uptake and depletes acetylcholine content in rat cerebral cortical synaptosomes. J. Neurochem. 1988, 50, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chen, T.F.; Lee, C.Y. Studies of the presynaptic effect of -bungarotoxin on neuromuscular transmission. J. Pharmacol. Exp. 1973, 184, 339–345. [Google Scholar]
- Garcia-Martinez, V.; Gimenez-Molina, Y.; Villanueva, J.; Darios, F.D.; Davletov, B.; Gutierrez, L.M. Emerging evidence for the modulation of exocytosis by signalling lipids. FEBS Lett. 2018, 592, 3493–3503. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca(2+) Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [Green Version]
- Schaechter, J.D.; Benowitz, L.I. Activation of protein kinase C by arachidonic acid selectively enhances the phosphorylation of GAP-43 in nerve terminal membranes. J. Neurosci. 1993, 13, 4361–4371. [Google Scholar] [CrossRef] [Green Version]
- St John, P.A.; Gordon, H. Agonists cause endocytosis of nicotinic acetylcholine receptors on cultured myotubes. J. Neurobiol. 2001, 49, 212–223. [Google Scholar] [CrossRef]
- Tedesco, E.; Rigoni, M.; Caccin, P.; Grishin, E.; Rossetto, O.; Montecucco, C. Calcium overload in nerve terminals of cultured neurons intoxicated by alpha-latrotoxin and snake PLA2 neurotoxins. Toxicon 2009, 54, 138–144. [Google Scholar] [CrossRef]
- Cintra-Francischinelli, M.; Pizzo, P.; Angulo, Y.; Gutierrez, J.M.; Montecucco, C.; Lomonte, B. The C-terminal region of a Lys49 myotoxin mediates Ca2+ influx in C2C12 myotubes. Toxicon 2010, 55, 590–596. [Google Scholar] [CrossRef]
- Angulo, Y.; Lomonte, B. Differential susceptibility of C2C12 myoblasts and myotubes to group II phospholipase A2 myotoxins from crotalid snake venoms. Cell Biochem. Funct. 2005, 23, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Angulo, Y.; Rufini, S.; Cho, W.; Giglio, J.R.; Ohno, M.; Daniele, J.J.; Geoghegan, P.; Gutierrez, J.M. Comparative study of the cytolytic activity of myotoxic phospholipases A2 on mouse endothelial (tEnd) and skeletal muscle (C2C12) cells in vitro. Toxicon 1999, 37, 145–158. [Google Scholar] [CrossRef]
- Bickler, P.E.; Fahlman, C.S.; Gray, J.; McKleroy, W. Inositol 1,4,5-triphosphate receptors and NAD(P)H mediate Ca2+ signaling required for hypoxic preconditioning of hippocampal neurons. Neuroscience 2009, 160, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Wills, Z.; Chu, C.T. Excitatory Dendritic Mitochondrial Calcium Toxicity: Implications for Parkinson’s and Other Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 523. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.B.; Grubb, B.D.; Maltin, C.A.; Dixon, R. The neurotoxicity of the venom phospholipases A(2), notexin and taipoxin. Exp. Neurol. 2000, 161, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.W.; Harris, J.B. Nerve terminal damage by b-bungarotoxin: it’s clinical significance. Am. J. Pathol. 1999, 154, 447–455. [Google Scholar] [CrossRef]
- Chibalin, A.V.; Benziane, B.; Zakyrjanova, G.F.; Kravtsova, V.V.; Krivoi, I.I. Early endplate remodeling and skeletal muscle signaling events following rat hindlimb suspension. J. Cell Physiol. 2018, 233, 6329–6336. [Google Scholar] [CrossRef]
- Bohme, M.A.; McCarthy, A.W.; Grasskamp, A.T.; Beuschel, C.B.; Goel, P.; Jusyte, M.; Laber, D.; Huang, S.; Rey, U.; Petzoldt, A.G.; et al. Rapid active zone remodeling consolidates presynaptic potentiation. Nat. Commun. 2019, 10, 1085. [Google Scholar] [CrossRef] [Green Version]
- Deem, S. Intensive-care-unit-acquired muscle weakness. Respir. Care 2006, 51, 1042–1052. [Google Scholar]
Figure 1.
General targets of major snake venom proteins divided into venoms that have intrinsic enzymatic activity and those that are non-enzymatic. Enzymatic venom proteins are typically hydrolases such as PLA2, serine proteases, metalloproteases, or hyaluronidases, releasing biologically active products that act on the extracellular matrix, on membrane proteins, on membrane-based signaling molecules or inside cells. Examples of non-enzymatic venom components include the curare-like 3-finger toxins from kraits, potassium channel blocking dendrotoxins and pore-forming myotoxins. Enzymatic destruction of the extracellular matrix by metalloproteases and hyaluronidases enhance venom spread and amplify toxicity. Other, direct acting, non-enzymatic protein toxins no doubt exist in yet to be characterized venoms. Further, venom proteins may simultaneously have enzyme-based and non-enzyme-based toxicities, such as components of PLA2 heterodimers, blurring these distinctions. Considerable cross-talk between enzymatic and non-enzymatic venom components may exist, for example non-enzymatic svPLA2s may dimerize and activate endogenous catalytic PLA2 proteins [18].
Figure 1.
General targets of major snake venom proteins divided into venoms that have intrinsic enzymatic activity and those that are non-enzymatic. Enzymatic venom proteins are typically hydrolases such as PLA2, serine proteases, metalloproteases, or hyaluronidases, releasing biologically active products that act on the extracellular matrix, on membrane proteins, on membrane-based signaling molecules or inside cells. Examples of non-enzymatic venom components include the curare-like 3-finger toxins from kraits, potassium channel blocking dendrotoxins and pore-forming myotoxins. Enzymatic destruction of the extracellular matrix by metalloproteases and hyaluronidases enhance venom spread and amplify toxicity. Other, direct acting, non-enzymatic protein toxins no doubt exist in yet to be characterized venoms. Further, venom proteins may simultaneously have enzyme-based and non-enzyme-based toxicities, such as components of PLA2 heterodimers, blurring these distinctions. Considerable cross-talk between enzymatic and non-enzymatic venom components may exist, for example non-enzymatic svPLA2s may dimerize and activate endogenous catalytic PLA2 proteins [18].

Figure 2.
Arachidonic acid metabolism stimulated by snake venom phospholipases. The primary effect of svPLA2 is production of arachidonic acid. Direct effects of arachidonic acid include activation of the transcription factor NFκΒ, responsible for the transcription of numerous genes encoding cytokines, release of intracellular Ca2+ from the endoplasmic reticulum, and phosphorylation of intracellular kinases. Arachidonic acid is also metabolized by cyclooxygenases and lipoxygenases, producing prostaglandins and leukotrienes [57]. Once set in motion, the inflammatory cascade is thus diversified and amplified by additional signaling molecules.
Figure 2.
Arachidonic acid metabolism stimulated by snake venom phospholipases. The primary effect of svPLA2 is production of arachidonic acid. Direct effects of arachidonic acid include activation of the transcription factor NFκΒ, responsible for the transcription of numerous genes encoding cytokines, release of intracellular Ca2+ from the endoplasmic reticulum, and phosphorylation of intracellular kinases. Arachidonic acid is also metabolized by cyclooxygenases and lipoxygenases, producing prostaglandins and leukotrienes [57]. Once set in motion, the inflammatory cascade is thus diversified and amplified by additional signaling molecules.

Figure 3.
Multi-site failure of synaptic transmission mediated by svPLA2s. Upper panel shows cycle of amplification of arachidonic acid and calcium signaling causing rapid depletion of pre-synaptic acetylcholine vesicles, increases in intracellular Ca2+ [Ca2+]i and acute desensitization of post-synaptic nicotinic acetylcholine receptors. Key events include snake venom (svPLA2)-mediated increase in pre-synaptic arachidonic acid (AA), and increases in pre-synaptic [Ca2+]i from release from intra-neuronal stores in the endoplasmic reticulum and augmented by voltage-gated calcium channels (not depicted). These actions are amplified by direct AA activation of protein kinase C, which facilitates activation of the vesicle fusion protein complex. Both catalytic and non-catalytic PLA2 subunits (shaded and cross-hatched circles, respectively) are potentially able to co-activate endogenous PLA2. Activation of intracellular, endogenous, PLA2 is part of the amplification cycle. The net effect is depletion of pre-synaptic transmitter vesicles and mitochondrial Ca2+ uptake. AA inhibition of the choline re-uptake transporter amplifies the decrease of releasable acetylcholine. Lower panel depicts the short and longer-term effects of PLA2 at the neuromuscular junction. Following the initial burst of acetylcholine release, post-junctional acetylcholine receptors are desensitized and then inactivated (dephosphorylated, internalized) analogous to their state in a phase II neuromuscular block produced by large/repeated doses of succinylcholine. As in the pre-synapse, PLA2 mediates a self-amplifying cycle of increase in arachidonic acid, intracellular calcium, and calcium-sensitive phosphatase activation. The process is augmented both by internalization of svPLA2 and/or activation of endogenous PLA2. The post-synaptic membrane is now depolarized and unexcitable for a prolonged period.
Figure 3.
Multi-site failure of synaptic transmission mediated by svPLA2s. Upper panel shows cycle of amplification of arachidonic acid and calcium signaling causing rapid depletion of pre-synaptic acetylcholine vesicles, increases in intracellular Ca2+ [Ca2+]i and acute desensitization of post-synaptic nicotinic acetylcholine receptors. Key events include snake venom (svPLA2)-mediated increase in pre-synaptic arachidonic acid (AA), and increases in pre-synaptic [Ca2+]i from release from intra-neuronal stores in the endoplasmic reticulum and augmented by voltage-gated calcium channels (not depicted). These actions are amplified by direct AA activation of protein kinase C, which facilitates activation of the vesicle fusion protein complex. Both catalytic and non-catalytic PLA2 subunits (shaded and cross-hatched circles, respectively) are potentially able to co-activate endogenous PLA2. Activation of intracellular, endogenous, PLA2 is part of the amplification cycle. The net effect is depletion of pre-synaptic transmitter vesicles and mitochondrial Ca2+ uptake. AA inhibition of the choline re-uptake transporter amplifies the decrease of releasable acetylcholine. Lower panel depicts the short and longer-term effects of PLA2 at the neuromuscular junction. Following the initial burst of acetylcholine release, post-junctional acetylcholine receptors are desensitized and then inactivated (dephosphorylated, internalized) analogous to their state in a phase II neuromuscular block produced by large/repeated doses of succinylcholine. As in the pre-synapse, PLA2 mediates a self-amplifying cycle of increase in arachidonic acid, intracellular calcium, and calcium-sensitive phosphatase activation. The process is augmented both by internalization of svPLA2 and/or activation of endogenous PLA2. The post-synaptic membrane is now depolarized and unexcitable for a prolonged period.


Figure 4.
Interaction of the effects of enzymatic venom components to produce and amplify immediate and long-term toxicity for immobilizing prey and deterring predators. cPLA2, cytosolic/endogenous PLA2s. cPLA2 = cytosolic/endogenous PLA2. MMP = endogenous, inducible matrix metalloproteases, DIC = disseminated intravascular coagulation.
Figure 4.
Interaction of the effects of enzymatic venom components to produce and amplify immediate and long-term toxicity for immobilizing prey and deterring predators. cPLA2, cytosolic/endogenous PLA2s. cPLA2 = cytosolic/endogenous PLA2. MMP = endogenous, inducible matrix metalloproteases, DIC = disseminated intravascular coagulation.


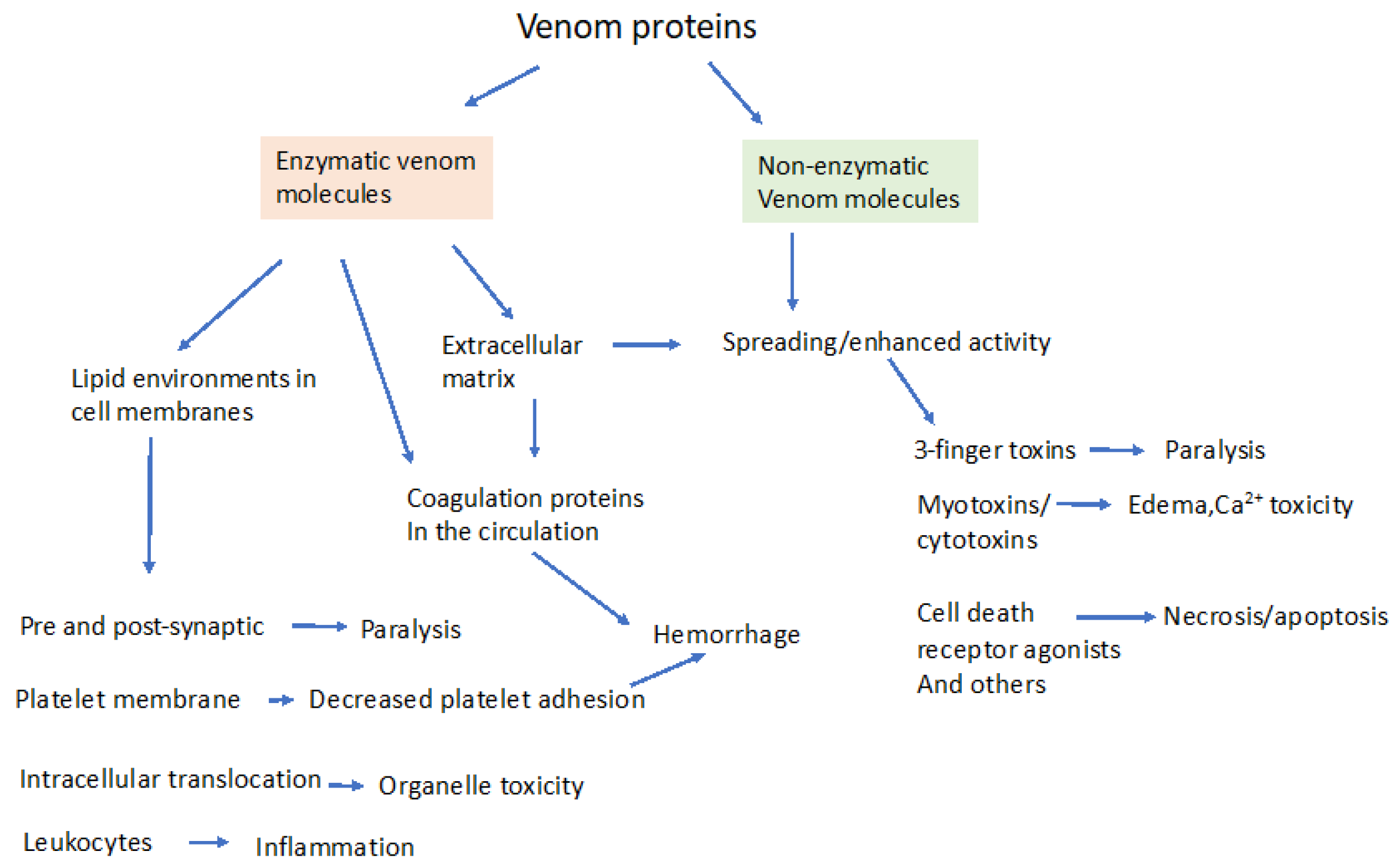{kind=link}
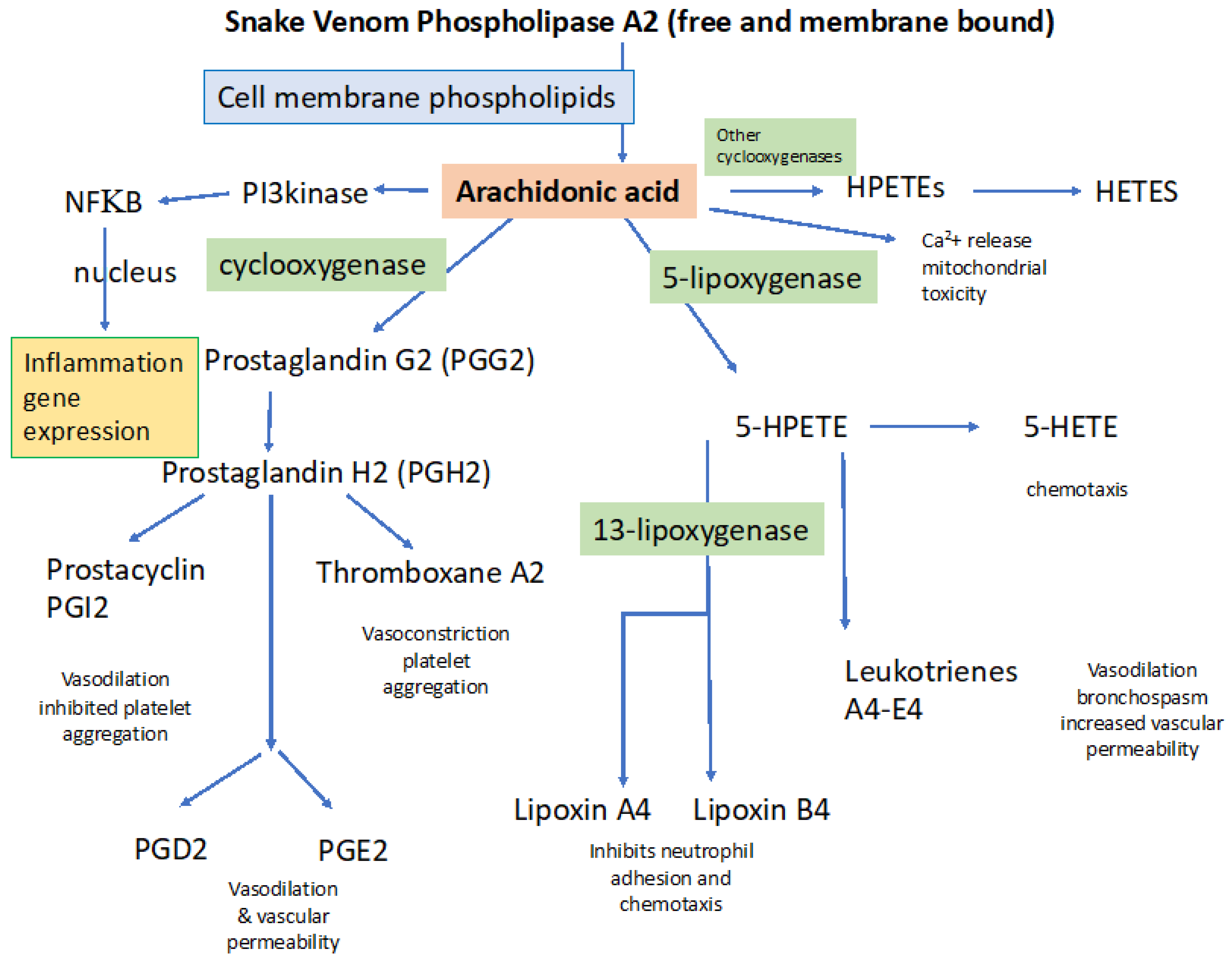{kind=link}
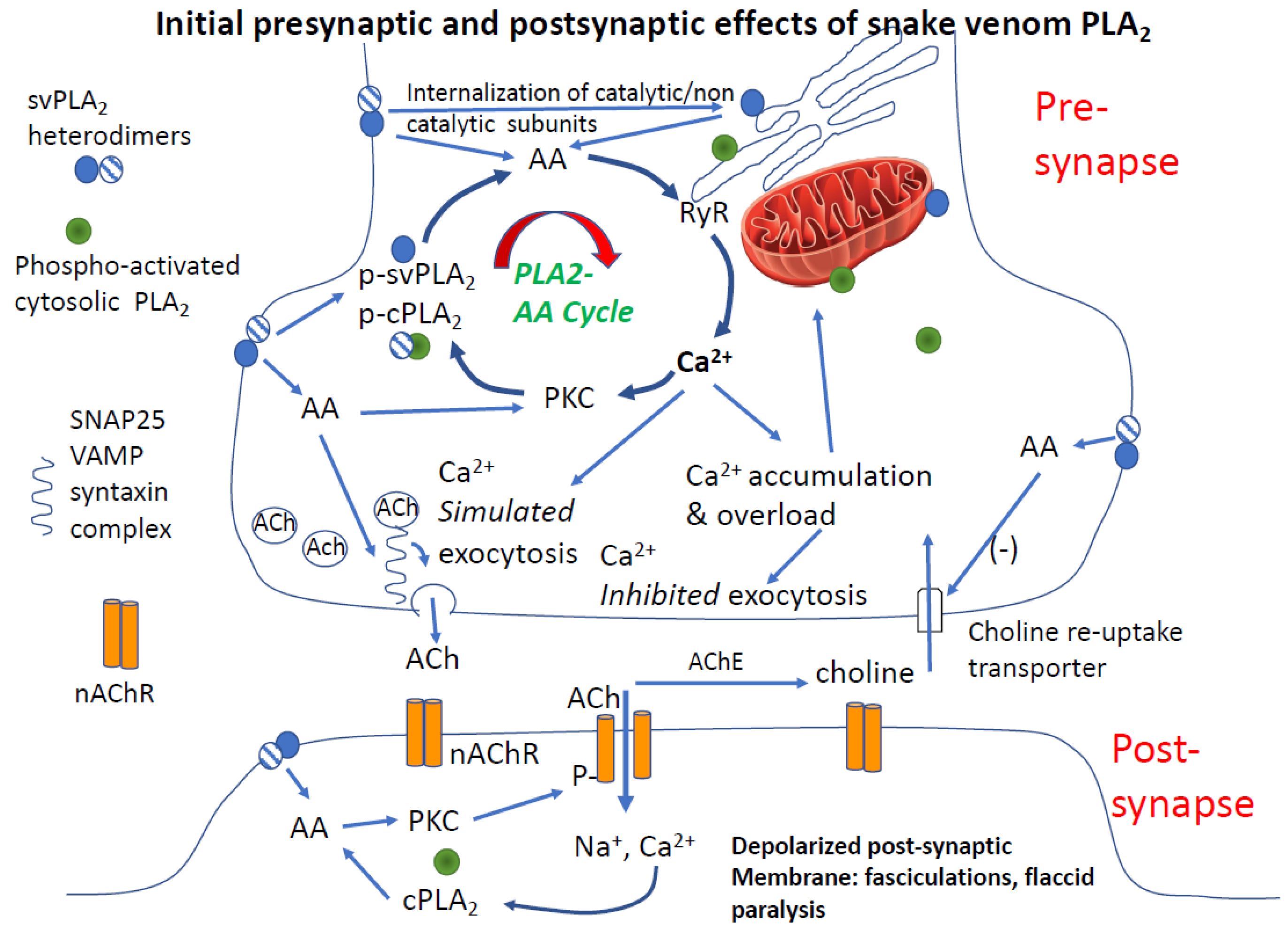{kind=link}
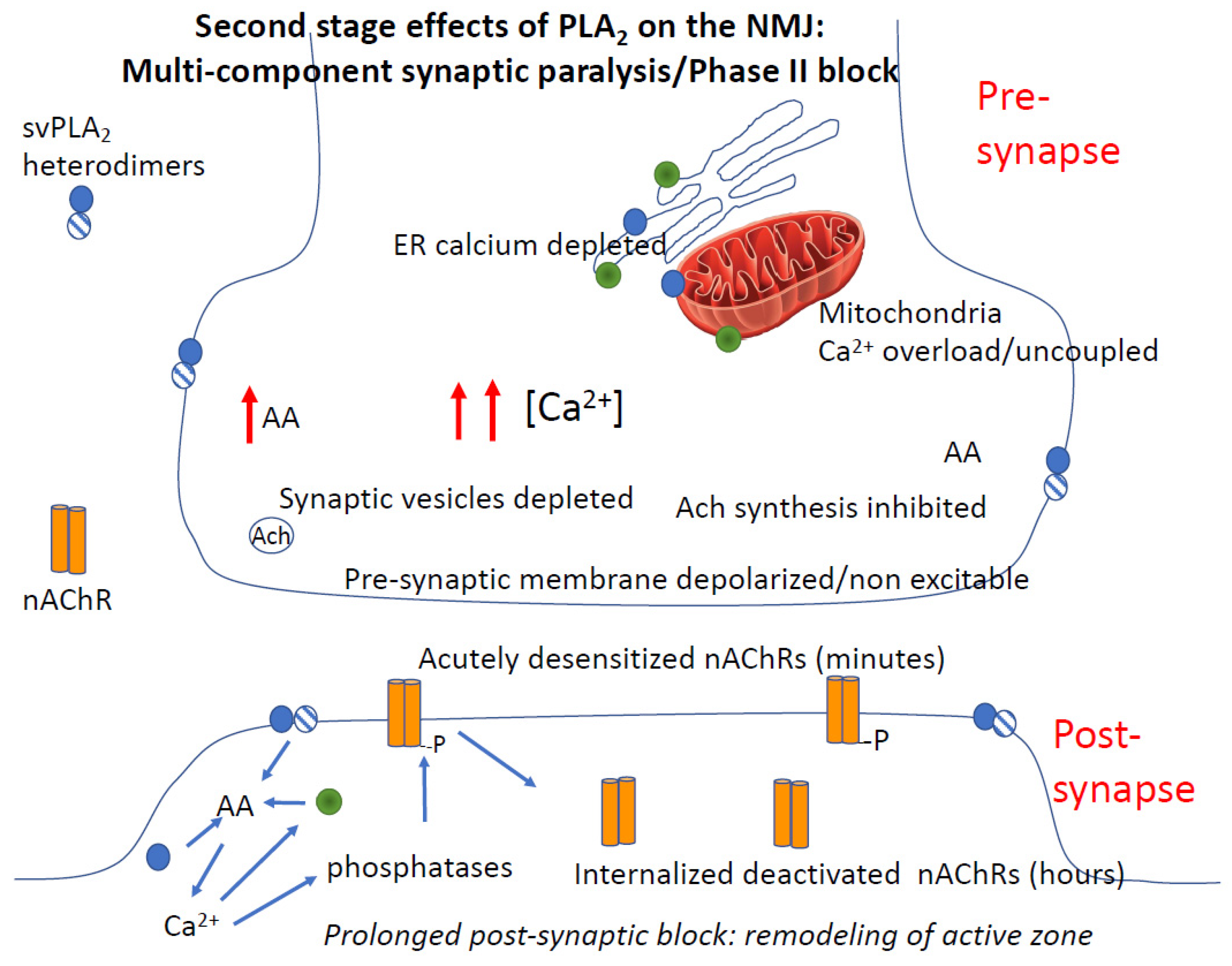{kind=link}
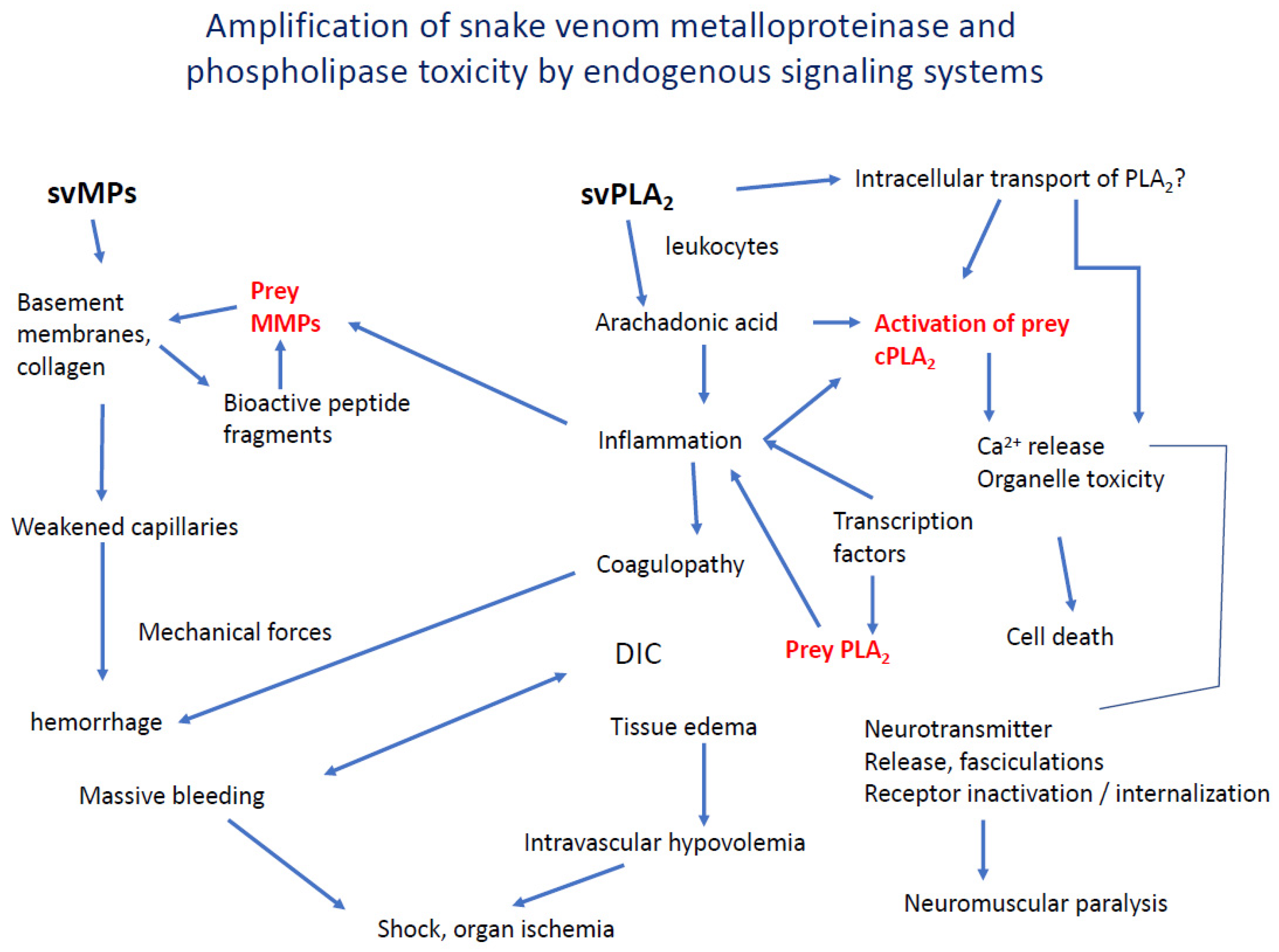{kind=link}
Table 1.
Main snake venom components, grouped by broad effects (structural mechanisms light green, coagulation yellow, paralysis grey, cardiovascular/cell signaling orange, and cell toxicity light blue). The percent contribution of each venom component varies; PLA2 and MP components predominate in many venoms. Also presented are the chief mechanism of effect and time course, and whether toxicities involve subversion of the envenomed animal’s homeostasis regulating machinery. Classification timing of action reflects: (1) Rapid; immediate (less than a few minutes) in blood compartment: does not require translocation or second messengers; (2) Intermediate (minutes to an hour): requires generation of second messenger signals and translocation outside circulation; and (3) Delayed (initiated or persistent for hours to days): requires extensive translocation and slower acting/regulated events such as apoptosis. Abbreviations: ECM extracellular matrix.
Table 1.
Main snake venom components, grouped by broad effects (structural mechanisms light green, coagulation yellow, paralysis grey, cardiovascular/cell signaling orange, and cell toxicity light blue). The percent contribution of each venom component varies; PLA2 and MP components predominate in many venoms. Also presented are the chief mechanism of effect and time course, and whether toxicities involve subversion of the envenomed animal’s homeostasis regulating machinery. Classification timing of action reflects: (1) Rapid; immediate (less than a few minutes) in blood compartment: does not require translocation or second messengers; (2) Intermediate (minutes to an hour): requires generation of second messenger signals and translocation outside circulation; and (3) Delayed (initiated or persistent for hours to days): requires extensive translocation and slower acting/regulated events such as apoptosis. Abbreviations: ECM extracellular matrix.
| Venom Component | Primary Pathologic Effect | Site of Action | Timing of Effect | Enzymatic or Non-Enzymatic | Venom Action Amplified by Prey |
|---|---|---|---|---|---|
| Disintegrins | Inhibit cell-ECM, loosen anchoring tissue [10] | Interstitial spaces | Intermediate and late? | Non | Yes, augments inflammation [11] |
| Hyaluronidases | Loosens tissue, enhances venom spread [12], exposes tissue factor | Capillaries and Interstitial spaces | Intermediate | Enzymatic | Yes, augments inflammation, coagulopathy [13] |
| Metalloproteases | Loosens/digests basal lamina [14] | Capillaries connective tissue | Intermediate | Enzymatic | Yes, bioactive peptides [15], gene expression |
| Serine Proteases | Inhibit coagulation, anti-thrombin effect [8] | Blood | Rapid | Enzymatic | Yes, signal cascades |
| Antithrombins | Hydrolysis of thrombin, clot destabilizer [8] | Blood | Rapid | Enzymatic | Unknown |
| PLA2s (inflammation, coagulation) | Production of arachidonic acid, mediators of inflammation [16,17] | leukocytes, platelets, endothelial cells | Rapid | Enzymatic and non-enzymatic subunits [18] | Yes, Ca2+, arachidonate, phosphorylation, gene expr. [17] |
| PLA2s (-neurotoxin) | Paralysis | Neuromuscular junction [19] | Usually rapid but may evolve slowly | Enzymatic and non-enzymatic | Yes, homologous protein activation [20] |
| 3FTx (-neurotoxins) | Paralysis/anticholinergic | Antagonists of nicotinic/muscarinic receptors [21] | Rapid | Direct | No |
| Cysteine-rich secretory proteins (CRISPS) | Target ion channels, Ca2+ release | Endothelium, leukocytes [22] | Delayed | unknown | Unknown |
| Kallikrein-like proteins | Shock, physiological disturbance [8] | Vasodilator | Rapid | Direct | Yes, amplifies inflammation |
| Phosphodiesterases | Hydrolysis of cyclic nucleotides/cell signaling/vasodilation [23] | Cell membrane, intracellular | Intermediate | Enzymatic | Yes, cell signaling pathways |
| Myotoxins | Cell damage [24,25] | Sarcolemma | Intermediate Delayed | Direct | Possible, overlap/identity to some PLA2s |
| Activators of cell death receptors DR4 and DR5 | Programmed cell death (apoptosis) [26] | Liver, kidneys, muscle | Delayed | Direct | Yes, cell apoptosis machinery |
| L-Amino Acid Oxidases | Free radicals tissue damage, immune activation [27] | Blood, extracellular fluid | Intermediate | Enzymatic | Yes, cytokine gene expression |
Table 2.
Important questions and hypotheses concerning the relative roles of direct or recipient-endogenous components in snake venom toxicity. Included is evidence supporting or in opposition to each question or hypothesis. The column on the right lists some newer models or experimental tools that could be used to test these hypotheses or generate new questions. MMP = endogenous matrix metalloproteases, SMT = small molecular therapeutics, EM = electron microscopy, NMJ = neuromuscular junction, ER = endoplasmic reticulum.
Table 2.
Important questions and hypotheses concerning the relative roles of direct or recipient-endogenous components in snake venom toxicity. Included is evidence supporting or in opposition to each question or hypothesis. The column on the right lists some newer models or experimental tools that could be used to test these hypotheses or generate new questions. MMP = endogenous matrix metalloproteases, SMT = small molecular therapeutics, EM = electron microscopy, NMJ = neuromuscular junction, ER = endoplasmic reticulum.
| Key Question/Hypothesis | Best Evidence for | Evidence Against | What Is Needed? |
|---|---|---|---|
| Is the catalytic action of svPLA2 responsible for paralysis? | Fasciculations [107], Pre-junctional effects, Ca2+ measurements [54], arachidonic acid | EM of altered NMJ morphology Post junctional effects [127] | Electrophysiology calcium imaging knockout mice. Small molecule inhibitor studies |
| Is endogenous PLA2 activation required for paralysis by β-toxins? | Small molecule inhibitors of endogenous PLA2 reverse paralysis [67,68] | None, excluding possible α-toxin effects of some PLA2s [113] | Ca2+-chelators, ER calcium imaging/release inhibitors should block synaptic failure |
| Do β-toxins cause reversible changes in the NMJ? | Delayed rescue by SMTs possible [67,68] | Ultrastructural images of damage [127] | Small molecule inhibitor studies, longer term assessment of NMJ structure |
| Is recruitment of prey PLA2 required to initiate or sustain inflammation? | Inflammation is sustained for long duration, failure of serotherapy to address [4] | No specific evidence against | Small molecule inhibitors selective for endogenous PLA2, cytokine gene arrays, genetic models |
| Can inhibition of endogenous PLA2 prevent organ toxicity? | Not studied | Organ damage caused by non PLA2 venom components [8] | Assessment of renal, hepatic, pulmonary function. Small molecule inhibitor studies |
| Are svPLA2s and svMPs synergistic in producing inflammation? | Elevations of cytokines, expression of MMPs [17] | Evidence lacking | Cytokine gene arrays with and without SMTs Cytokine levels |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bickler, P.E. Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways. Toxins 2020, 12, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020068
AMA Style
Bickler PE. Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways. Toxins. 2020; 12(2):68. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020068
Chicago/Turabian StyleBickler, Philip E. 2020. "Amplification of Snake Venom Toxicity by Endogenous Signaling Pathways" Toxins 12, no. 2: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12020068
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.