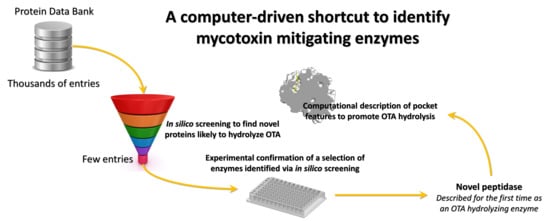
An In Silico Target Fishing Approach to Identify Novel Ochratoxin A Hydrolyzing Enzyme

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
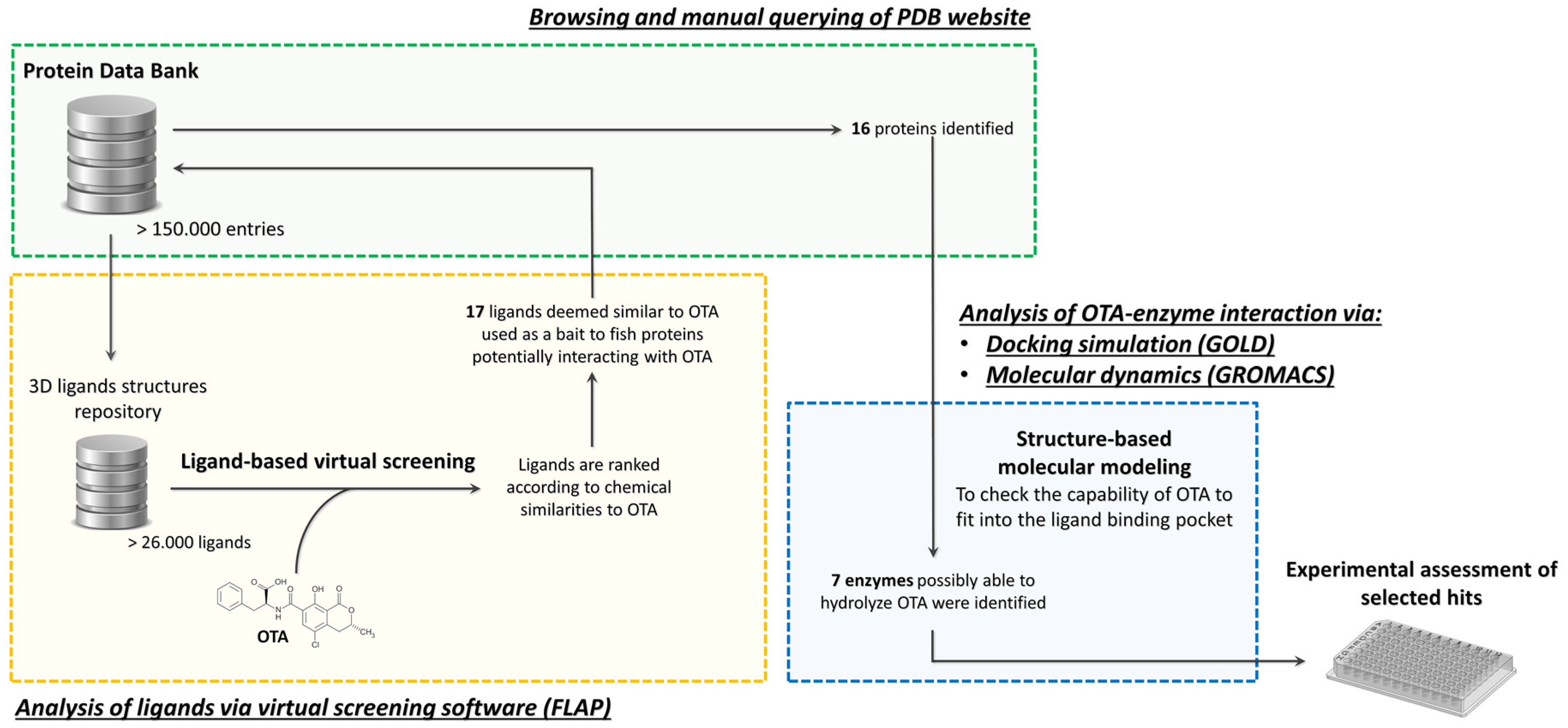
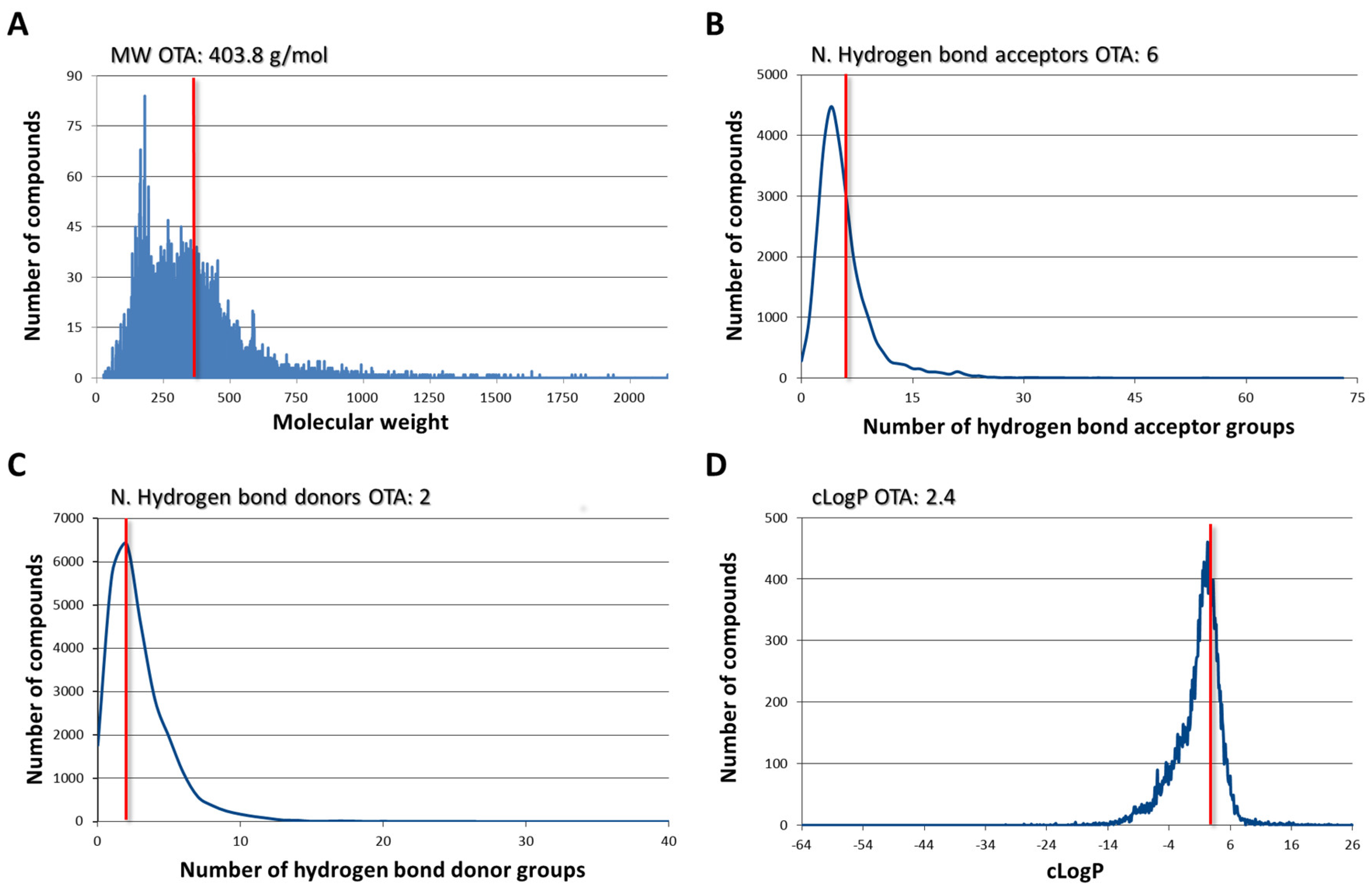
2.1. Ligand Dabase Anatomy
2.2. Ligand-Based Virtual Screening (VS)
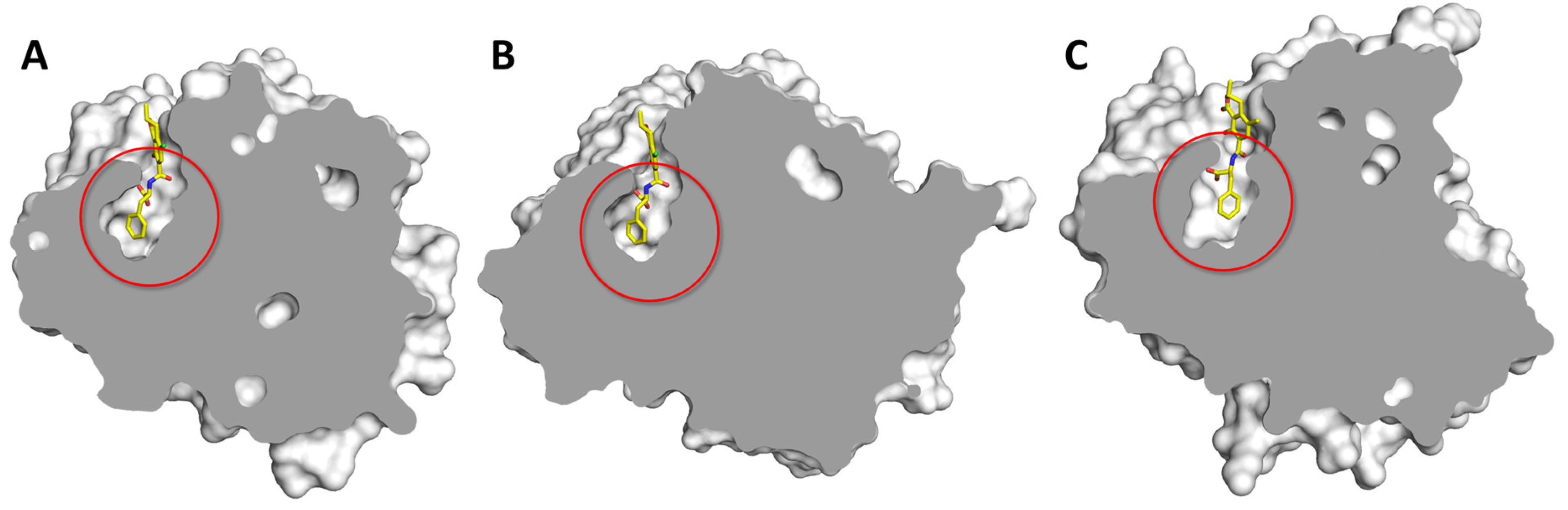
2.3. Structure-Based Molecular Modeling
2.3.1. Carboxypeptidase T (CPT)
2.3.2. Carboxypeptidase B (CPB)
2.3.3. Serine Carboxypeptidase II (Ser-CP II)
2.3.4. Neprilysin
2.3.5. Urokinase
2.3.6. Endothiapepsin
2.3.7. Cathepsin A
2.3.8. Matrix Metalloproteinase 12 (MMP-12)
2.3.9. Beta-Secretase 1 (BACE1)
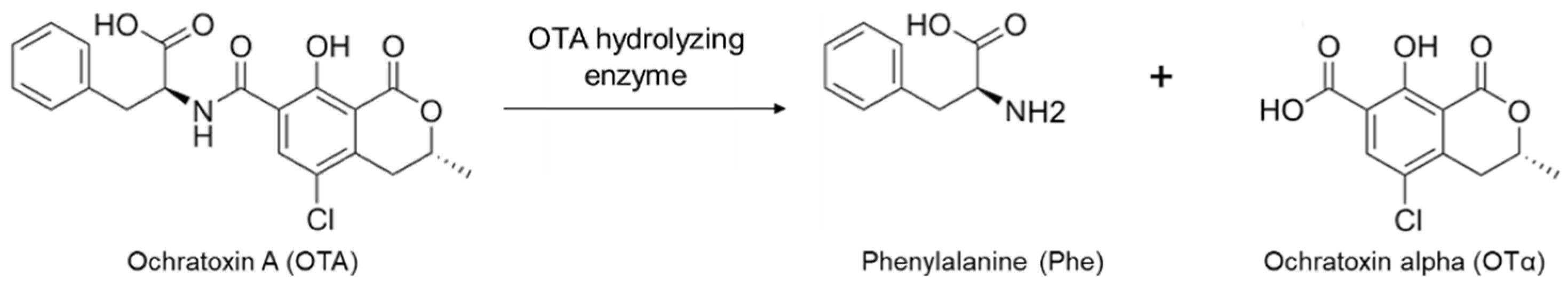
2.4. Confirmation of OTA Hydrolyzing Activity of Selected Enzymes
3. Conclusions
4. Materials and Methods
4.1. Assembly of the Ligands Database for Target Fishing
4.2. Ligand-Based Virtual Screening
- (i)
- VS-1, using the so defined “bit string” mode to speed up computation time. In addition, the “shape penalty” was allowed to promote the selection of molecules with shape analogies;
- (ii)
- VS-2, using the so defined “quadruplet” mode with a normal accuracy level;
- (iii)
- VS-3, using the “dissimilarity” penalty in addition to the setting of VS-2 to promote the selection of molecules with shape analogies;
- (iv)
- VS-4, using the so defined “quadruplet” mode with a maximum accuracy level; and,
- (v)
- VS-5, using the “dissimilarity” penalty in addition to the setting of VS-4 to promote the selection of molecules with shape analogies.
4.3. Structure-Based Molecular Modeling
4.4. Estimation of OTA Hydrolyzing Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, K.J.; Steyn, P.S.; Fourie, L.; Scott, D.B.; Theron, J.J. Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature 1965, 205, 1112–1113. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.H.; Dohnal, V.; Huang, L.L.; Kuca, K.; Wang, X.; Chen, G.Y.; Yuan, Z.H. Metabolic Pathways of Ochratoxin A. Curr. Drug Metab. 2011, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gan, F.; Zhou, Y.J.; Hou, L.L.; Qian, G.; Chen, X.X.; Huang, K.H. Ochratoxin A induces nephrotoxicity and immunotoxicity through different MAPK signaling pathways in PK15 cells and porcine primary splenocytes. Chemosphere 2017, 182, 630–637. [Google Scholar] [CrossRef]
- Wu, T.S.; Lin, Y.T.; Huang, Y.T.; Yu, F.Y.; Liu, B.H. Ochratoxin A triggered intracerebral hemorrhage in embryonic zebrafish: Involvement of microRNA-731 and prolactin receptor. Chemosphere 2020, 242, 125143. [Google Scholar] [CrossRef]
- IARC. Monographs on the Evaluation of Carcinogenic Risks to Humans: Chemical Agents and Related Occupations. A Review of Human Carcinogens. Lyon Fr. 2012, 100, 224. [Google Scholar]
- Denli, M.; Perez, J.F. Ochratoxins in Feed, a Risk for Animal and Human Health: Control Strategies. Toxins 2010, 2, 1065–1077. [Google Scholar] [CrossRef]
- Becker-Algeri, T.A.; Castagnaro, D.; de Bortoli, K.; de Souza, C.; Drunkler, D.A.; Badiale-Furlong, E. Mycotoxins in Bovine Milk and Dairy Products: A Review. J. Food Sci. 2016, 81, R544–R552. [Google Scholar] [CrossRef] [Green Version]
- Persi, N.; Pleadin, J.; Kovacevic, D.; Scortichini, G.; Milone, S. Ochratoxin A in raw materials and cooked meat products made from OTA-treated pigs. Meat Sci. 2014, 96, 203–210. [Google Scholar] [CrossRef]
- The European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuff. Off. J. Eur. Union 2006, L364, 5–8. [Google Scholar]
- The European Commission. Commission Recommendation (EU) 2016/1319 of 29 July 2016 amending recommendation 2006/576EC as regards deoxynivalenol, zearalenone and ochratoxin A in pet food. Off. J. Eur. Union 2006, L208, 58–60. [Google Scholar]
- Karlovsky, P.; Suman, M.; Berthiller, F.; De Meester, J.; Eisenbrand, G.; Perrin, I.; Oswald, I.P.; Speijers, G.; Chiodini, A.; Recker, T.; et al. Impact of food processing and detoxification treatments on mycotoxin contamination. Mycotoxin Res. 2016, 32, 179–205. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Y.I.; Zhou, T. Promising Detoxification Strategies to Mitigate Mycotoxins in Food and Feed. Toxins 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Varga, J.; Kocsube, S.; Peteri, Z.; Vagvolgyi, C.; Toth, B. Chemical, Physical and Biological Approaches to Prevent Ochratoxin Induced Toxicoses in Humans and Animals. Toxins 2010, 2, 1718–1750. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Li, C.; Zhang, B.Y.; Zhou, Z.; Shen, Y.B.; Liao, X.; Yang, J.Y.Q.; Wang, Y.; Li, X.H.; Li, Y.Z.; et al. Advances in Biodetoxification of Ochratoxin A-A Review of the Past Five Decades. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Galaverna, G.; Reverberi, M.; Dall’Asta, C. Degradation of Aflatoxins by Means of Laccases from Trametes versicolor: An In Silico Insight. Toxins (Basel) 2017, 9, E17. [Google Scholar] [CrossRef] [Green Version]
- Heinl, S.; Hartinger, D.; Thamhesl, M.; Vekiru, E.; Krska, R.; Schatzmayr, G.; Moll, W.D.; Grabherr, R. Degradation of fumonisin B1 by the consecutive action of two bacterial enzymes. J. Biotechnol. 2010, 145, 120–129. [Google Scholar] [CrossRef]
- Utermark, J.; Karlovsky, P. Role of zearalenone lactonase in protection of Gliocladium roseum from fungitoxic effects of the mycotoxin zearalenone. Appl. Environ. Microbiol. 2007, 73, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Grenier, B.; Schwartz-Zimmermann, H.E.; Gruber-Dorninger, C.; Dohnal, I.; Aleschko, M.; Schatzmayr, G.; Moll, W.D.; Applegate, T.J. Enzymatic hydrolysis of fumonisins in the gastrointestinal tract of broiler chickens. Poult. Sci. 2017, 96, 4342–4351. [Google Scholar] [CrossRef]
- Pitout, M.J. The hydrolysis of ochratoxin A by some proteolytic enzymes. Biochem. Pharmacol. 1969, 18, 485–491. [Google Scholar] [CrossRef]
- Abrunhosa, L.; Paterson, R.R.M.; Venancio, A. Biodegradation of Ochratoxin A for Food and Feed Decontamination. Toxins 2010, 2, 1078–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.J.; Wu, Z.D.; Wu, S.L.; Dai, Y.S.; Sun, C.P. Degradation of ochratoxin A by Bacillus amyloliquefaciens ASAG1. Food Addit. Contam. A 2015, 32, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Dobritzsch, D.; Wang, H.M.; Schneider, G.; Yu, S. Structural and functional characterization of ochratoxinase, a novel mycotoxin-degrading enzyme. Biochem. J. 2014, 462, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Haq, M.; Gonzalez, N.; Mintz, K.; Jaja-Chimedza, A.; De Jesus, C.L.; Lydon, C.; Welch, A.Z.; Berry, J.P. Teratogenicity of Ochratoxin A and the Degradation Product, Ochratoxin a, in the Zebrafish (Danio rerio) Embryo Model of Vertebrate Development. Toxins 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Dellafiora, L.; Aichinger, G.; Geib, E.; Sánchez-Barrionuevo, L.; Brock, M.; Cánovas, D.; Dall’Asta, C.; Marko, D. Hybrid in silico/in vitro target fishing to assign function to “orphan” compounds of food origin–The case of the fungal metabolite atromentin. Food Chem. 2019, 270, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Dellafiora, L.; Mena, P.; Del Rio, D.; Cozzini, P. Modeling the effect of phase II conjugations on topoisomerase I poisoning: Pilot study with luteolin and quercetin. J. Agr. Food Chem. 2014, 62, 5881–5886. [Google Scholar] [CrossRef]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The practice of structure activity relationships (SAR) in toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Dridi, F.; Marrakchi, M.; Gargouri, M.; Saulnier, J.; Jaffrezic-Renault, N.; Lagarde, F. Comparison of carboxypeptidase Y and thermolysin for ochratoxin A electrochemical biosensing. Anal. Methods 2015, 7, 8954–8960. [Google Scholar] [CrossRef]
- Uchiyama, S.; Saito, Y. Protein-binding potential of ochratoxin A in vitro and its fluorescence enhancement. J. Food Hyg. Soc. Japan 1987, 28, 453–460. [Google Scholar] [CrossRef]
- Koszegi, T.; Poor, M. Ochratoxin A: Molecular Interactions, Mechanisms of Toxicity and Prevention at the Molecular Level. Toxins 2016, 8. [Google Scholar] [CrossRef]
- Il’ichev, Y.V.; Perry, J.L.; Simon, J.D. Interaction of ochratoxin A with human serum albumin. Preferential bining of the dianion and pH effects. J. Phys. Chem. B 2002, 106, 452–459. [Google Scholar] [CrossRef]
- Dellafiora, L.; Galaverna, G.; Cruciani, G.; Dall’Asta, C. A computational study toward the “personalized” activity of alternariol - Does it matter for safe food at individual level? Food Chem. Toxicol. 2019, 130, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Abrunhosa, L.; Santos, L.; Venancio, A. Degradation of ochratoxin A by proteases and by a crude enzyme of Aspergillus niger. Food Biotechnol. 2006, 20, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Kolli, N.; Garman, S.C. Proteolytic Activation of Human Cathepsin A. J. Biol. Chem. 2014, 289, 11592–11600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, W. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acid Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Dellafiora, L.; Galaverna, G.; Dall’Asta, C.; Cozzini, P. Hazard identification of cis/trans-zearalenone through the looking-glass. Food Chem. Toxicol. 2015, 86, 65–71. [Google Scholar] [CrossRef]
- Maldonado-Rojas, W.; Olivero-Verbel, J. Potential interaction of natural dietary bioactive compounds with COX-2. J. Mol. Graph. Model. 2011, 30, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D.J. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinformatics 2016, 54, 5–6. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand PDB ID | Protein PDB ID | Protein Name | Source Organism | Note |
|---|---|---|---|---|
| TI2/TIO | 1QF0/1ZDP | Thermolysin * | Bacteria | a |
| CXA | 4DJL | Carboxypeptidase T (CPT) | Bacteria | a |
| CXA/ING | 1IY7 | Carboxypeptidase A (CPA) * | Bovine | a |
| CXA | 5J1Q | Carboxypeptidase B (CPB) | Pig | a |
| FC0 | 1BCR/1WHT | Serine Carboxypeptidase II (Ser-CP II) | Wheat | a |
| TIO | 5V48 | Neprilysin | Rabbit | a |
| 9UP | 4FUG | Urokinase | Human | a |
| 46L | 4Y3S | Endothiapepsin | Fungus | a |
| S61 | 4AZ0 | Cathepsin A | Human | a |
| DSV | 2K2G | Matrix metalloproteinase 12 (MMP-12) | Human | a |
| 1CH/ZY4/192 | 2WF4/1W51/4I11 | Beta-secretase 1 (BACE 1) | Human | a |
| BBL | 1ESB | Pancreatic elastase | Pig | a |
| P28 | 2ROY | Transthyretin | Human | b |
| 9NF | 2WX0 | Serum albumin ** | Human | b |
| 1HN | 3NKT | Salicylate 1,2-dioxygenase | Bacteria | c |
| NPQ | 5FJO | N-acyl amino acid racemase | Bacteria | c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dellafiora, L.; Gonaus, C.; Streit, B.; Galaverna, G.; Moll, W.-D.; Vogtentanz, G.; Schatzmayr, G.; Dall’Asta, C.; Prasad, S. An In Silico Target Fishing Approach to Identify Novel Ochratoxin A Hydrolyzing Enzyme. Toxins 2020, 12, 258. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040258
Dellafiora L, Gonaus C, Streit B, Galaverna G, Moll W-D, Vogtentanz G, Schatzmayr G, Dall’Asta C, Prasad S. An In Silico Target Fishing Approach to Identify Novel Ochratoxin A Hydrolyzing Enzyme. Toxins. 2020; 12(4):258. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040258
Chicago/Turabian StyleDellafiora, Luca, Christoph Gonaus, Barbara Streit, Gianni Galaverna, Wulf-Dieter Moll, Gudrun Vogtentanz, Gerd Schatzmayr, Chiara Dall’Asta, and Shreenath Prasad. 2020. "An In Silico Target Fishing Approach to Identify Novel Ochratoxin A Hydrolyzing Enzyme" Toxins 12, no. 4: 258. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040258