Cnf1 Variants Endowed with the Ability to Cross the Blood–Brain Barrier: A New Potential Therapeutic Strategy for Glioblastoma

 , ,
, ,  and
and 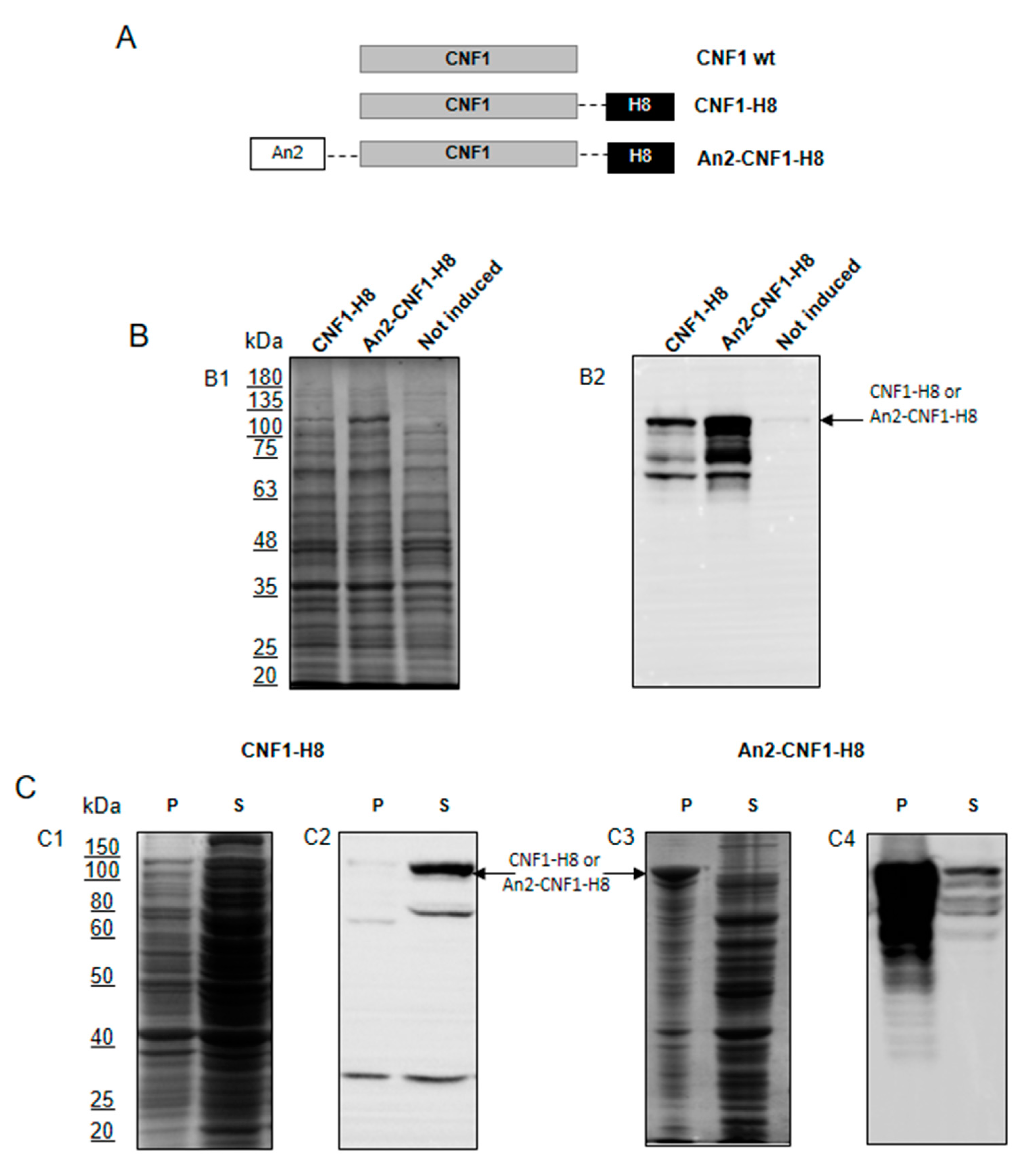{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Design and Production of a CNF1 Variant Bearing the Potentiality to Cross the BBB When Peripherally Injected
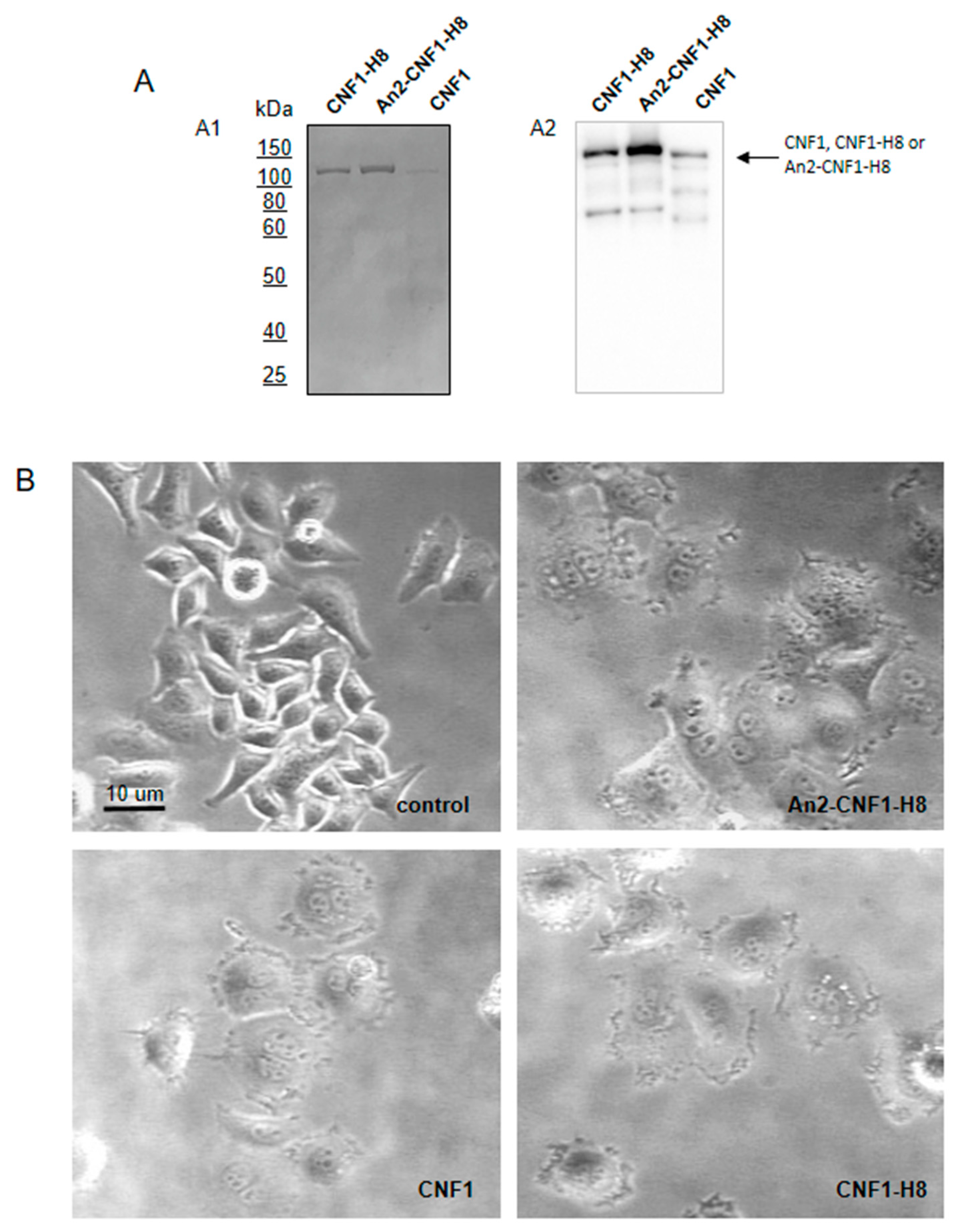
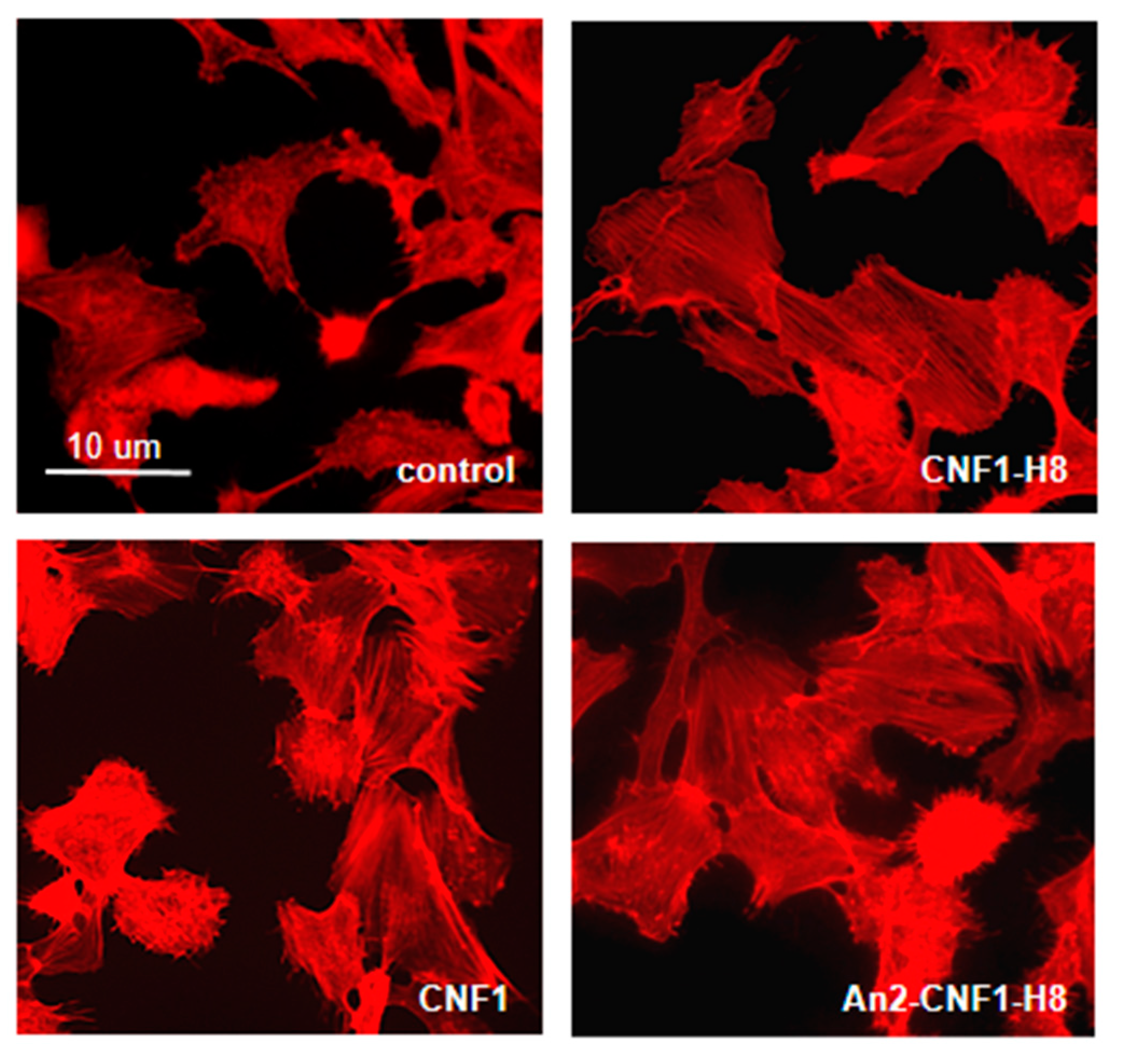
2.2. The An2-CNF1-H8 Variant Preserves the Original Activity Profile of wt CNF1 in HEp-2 Cells
2.3. The An2-CNF1-H8 Variant Enters the Cells in the Same Vein of wt CNF1
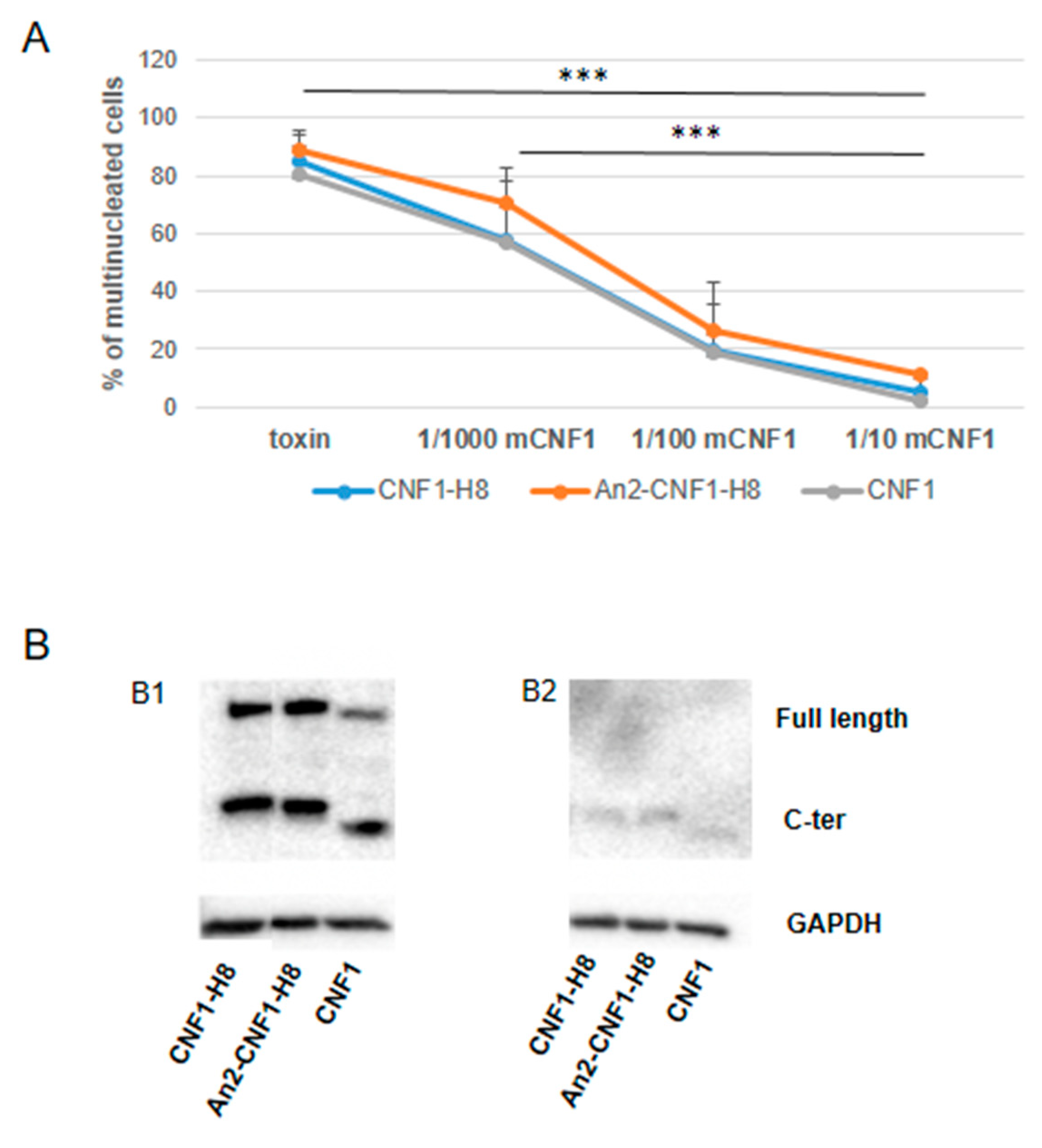
2.4. Activities of the An2-CNF1-H8 Variant on the Human Brain Endothelial Cell Line HBEC-5i
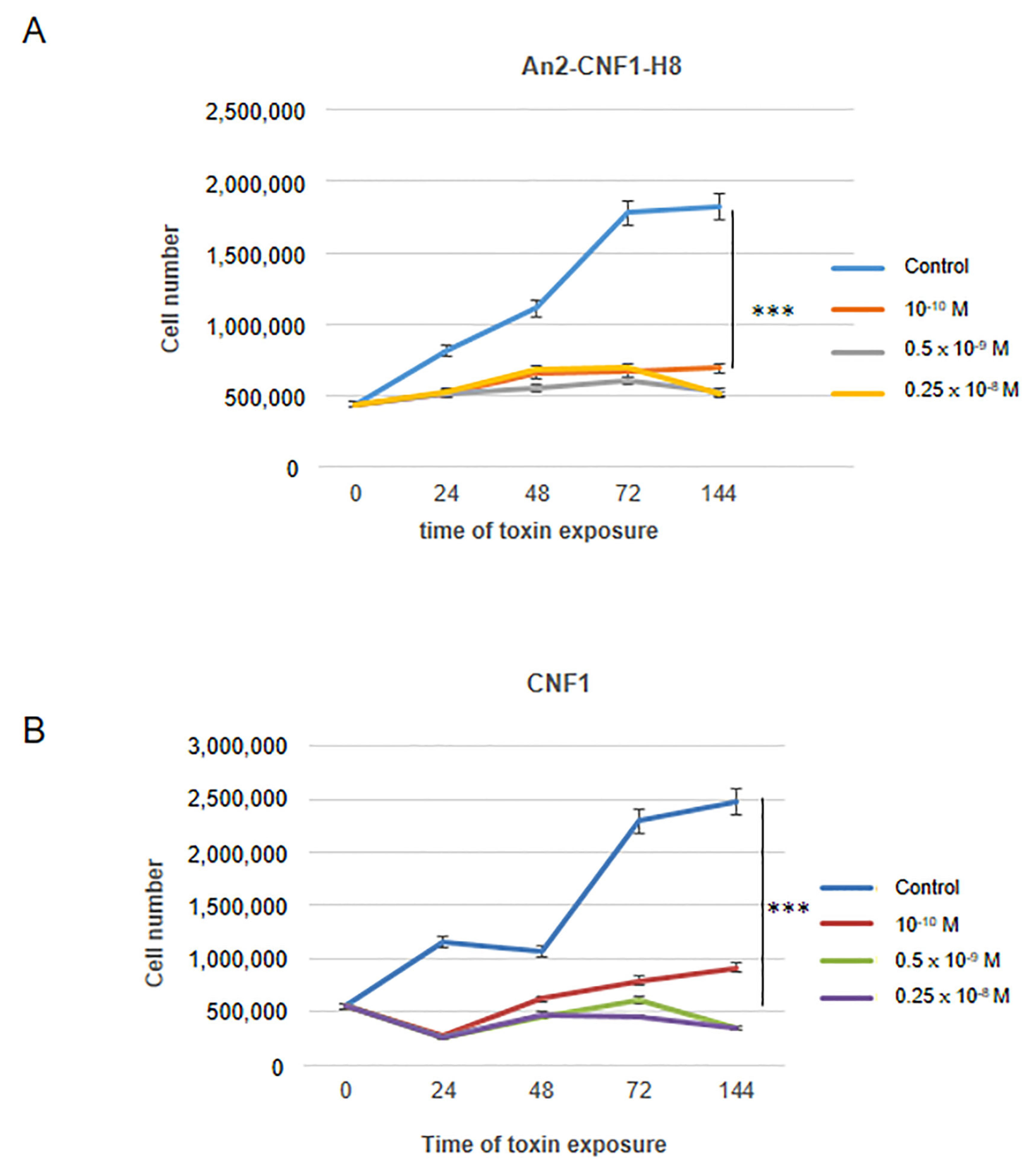
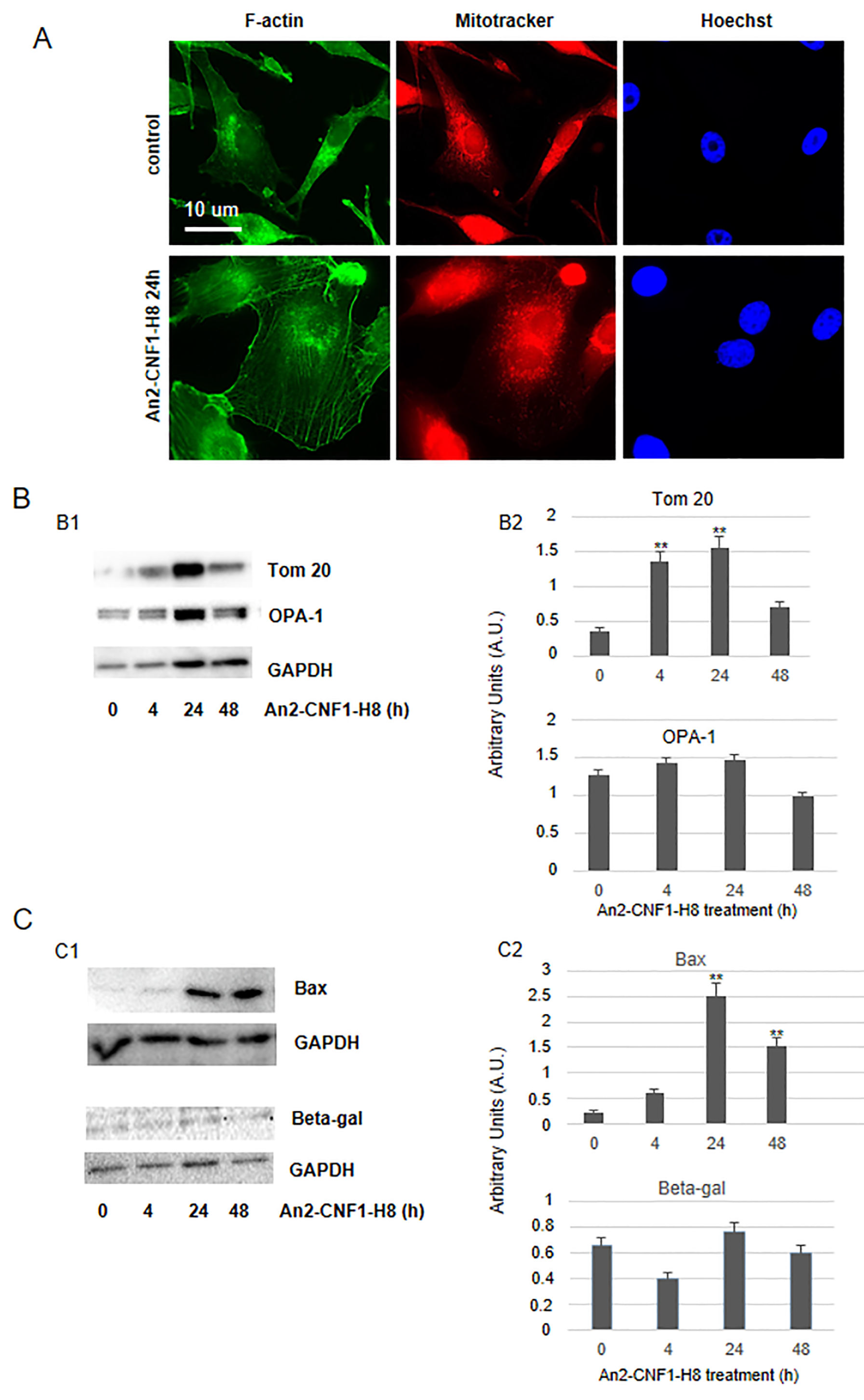
2.5. Activities of the An2-CNF1-H8 Variant on the Human GBM Cells
3. Discussion
4. Materials and Methods
4.1. Production and Purification of CNF1 and CNF1 Variants
4.2. Cell Cultures
4.3. CNF1 Variant Titration
4.4. Competition Experiments with CNF1 C866S
4.5. Protein Extraction of Polyacrylamide and Western Blot Gel
4.6. Cell Growth Experiments
4.7. Mouse Studies
4.8. Fluorescence Microscopy
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Niu, L.; Bai, Y.; Le, W. Glioblastoma: Targeting the autophagy in tumorigenesis. Brain Res. Bull. 2019, 153, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Rosadi, F.; Ballan, G.; Maroccia, Z.; Giambenedetti, M.; Guidotti, M.; Ødum, N.; Krejsgaard, T.; Fiorentini, C. The Escherichia coli protein toxin cytotoxic necrotizing factor 1 induces epithelial mesenchymal transition. Cell. Microbiol. 2019, 22, e13138. [Google Scholar] [CrossRef] [PubMed]
- Vannini, E.; Olimpico, F.; Middei, S.; Ammassari-Teule, M.; de Graaf, E.L.; McDonnell, L.; Schmidt, G.; Fabbri, A.; Fiorentini, C.; Baroncelli, L.; et al. Electrophysiology of glioma: A Rho GTPase-activating protein reduces tumor growth and spares neuron structure and function. Neuro. Oncol. 2016, 18, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases Improves the Behavioral Phenotype and Reverses Astrocytic Deficits in a Mouse Model of Rett Syndrome. Neuropsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 Increases Brain Energy Level, Counteracts Neuroinflammatory Markers and Rescues Cognitive Deficits in a Murine Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e65898. [Google Scholar] [CrossRef]
- Travaglione, S.; Ballan, G.; Fortuna, A.; Ferri, A.; Guidotti, M.; Campana, G.; Fiorentini, C.; Loizzo, S. CNF1 enhances brain energy content and counteracts spontaneous epileptiform phenomena in aged DBA/2J mice. PLoS ONE 2015, 10, e0140495. [Google Scholar] [CrossRef] [Green Version]
- Vannini, E.; Panighini, A.; Cerri, C.; Fabbri, A.; Lisi, S.; Pracucci, E.; Benedetto, N.; Vannozzi, R.; Fiorentini, C.; Caleo, M.; et al. The bacterial protein toxin, cytotoxic necrotizing factor 1 (CNF1) provides long-term survival in a murine glioma model. BMC Cancer 2014, 14, 449. [Google Scholar] [CrossRef]
- Vannini, E.; Maltese, F.; Olimpico, F.; Fabbri, A.; Costa, M.; Caleo, M.; Baroncelli, L. Progression of motor deficits in glioma-bearing mice: Impact of CNF1 therapy at symptomatic stages. Oncotarget 2017, 8, 23539–23550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeule, M.; Currie, J.C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, A.; Caterino, M.; Fabbri, A.; Fiorentini, C.; Vergara, A.; Sica, F.; Parrilli, E.; Tutino, M.L. High yield purification and first structural characterization of the full-length bacterial toxin CNF1. Biotechnol. Prog. 2018, 34, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Travaglione, S.; Loizzo, S.; Rizza, T.; Del Brocco, A.; Ballan, G.; Guidotti, M.; Vona, R.; Di Nottia, M.; Torraco, A.; Carrozzo, R.; et al. Enhancement of mitochondrial ATP production by the Escherichia coli cytotoxic necrotizing factor 1. FEBS J. 2014, 281, 3473–3488. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [CrossRef] [Green Version]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 2000, 11, 1775–1787. [Google Scholar] [CrossRef] [Green Version]
- Knust, Z.; Blumenthal, B.; Aktories, K.; Schmidt, G. Cleavage of escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect. Immun. 2009, 77, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Lerm, M.; Schmidt, G.; Goehring, U.M.; Schirmer, J.; Aktories, K. Identification of the region of Rho involved in substrate recognition by Escherichia coli cytotoxic necrotizing factor 1 (CNF1). J. Biol. Chem. 1999, 274, 28999–29004. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 (CNF1): Toxin biology, in vivo applications and therapeutic potential. Toxins (Basel) 2010, 2, 283–296. [Google Scholar] [CrossRef]
- Giamboi-Miraglia, A.; Travaglione, S.; Filippini, P.; Fabbri, A.; Fiorentini, C.; Falzano, L. A multinucleating Escherichia coli cytotoxin perturbs cell cycle in cultured epithelial cells. Toxicol. In Vitro 2007, 21, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Maroccia, Z.; Guidotti, M.; Pierri, C.; Primiano, G.; Servidei, S.; Loizzo, S.; Fiorentini, C. The Bacterial Protein CNF1 as a Potential Therapeutic Strategy against Mitochondrial Diseases: A Pilot Study. Int. J. Mol. Sci. 2018, 19, 1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.E.; Jensen, R.E. Barreling through the membrane. Nat. Struct. Mol. Biol. 2004, 11, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High Prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [Green Version]
- Keener, A.B. Host with the most: Targeting host cells instead of pathogens to fight infectious disease. Nat. Med. 2017, 23, 528–531. [Google Scholar] [CrossRef]
- Serna, N.; Céspedes, M.V.; Saccardo, P.; Xu, Z.; Unzueta, U.; Álamo, P.; Pesarrodona, M.; Sánchez-Chardi, A.; Roldán, M.; Mangues, R.; et al. Rational engineering of single-chain polypeptides into protein-only, BBB-targeted nanoparticles. Nanomedicine 2016, 12, 1241–1251. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, G.; Selzer, J.; Lerm, M.; Aktories, K. The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J. Biol. Chem. 1998, 273, 13669–13674. [Google Scholar] [CrossRef] [Green Version]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colarusso, A.; Maroccia, Z.; Parrilli, E.; Germinario, E.A.P.; Fortuna, A.; Loizzo, S.; Ricceri, L.; Tutino, M.L.; Fiorentini, C.; Fabbri, A. Cnf1 Variants Endowed with the Ability to Cross the Blood–Brain Barrier: A New Potential Therapeutic Strategy for Glioblastoma. Toxins 2020, 12, 291. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050291
Colarusso A, Maroccia Z, Parrilli E, Germinario EAP, Fortuna A, Loizzo S, Ricceri L, Tutino ML, Fiorentini C, Fabbri A. Cnf1 Variants Endowed with the Ability to Cross the Blood–Brain Barrier: A New Potential Therapeutic Strategy for Glioblastoma. Toxins. 2020; 12(5):291. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050291
Chicago/Turabian StyleColarusso, Andrea, Zaira Maroccia, Ermenegilda Parrilli, Elena Angela Pia Germinario, Andrea Fortuna, Stefano Loizzo, Laura Ricceri, Maria Luisa Tutino, Carla Fiorentini, and Alessia Fabbri. 2020. "Cnf1 Variants Endowed with the Ability to Cross the Blood–Brain Barrier: A New Potential Therapeutic Strategy for Glioblastoma" Toxins 12, no. 5: 291. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12050291