Signatures of TRI5, TRI8 and TRI11 Protein Sequences of Fusarium incarnatum-equiseti Species Complex (FIESC) Indicate Differential Trichothecene Analogue Production


Abstract
:
1. Introduction
2. Results
2.1. Comparative DNA Polymorphism
2.1.1. TRI5
2.1.2. TRI8
2.1.3. TRI11
2.2. Phylogeny
2.2.1. TRI5 Gene Tree
2.2.2. TRI5 Protein Tree
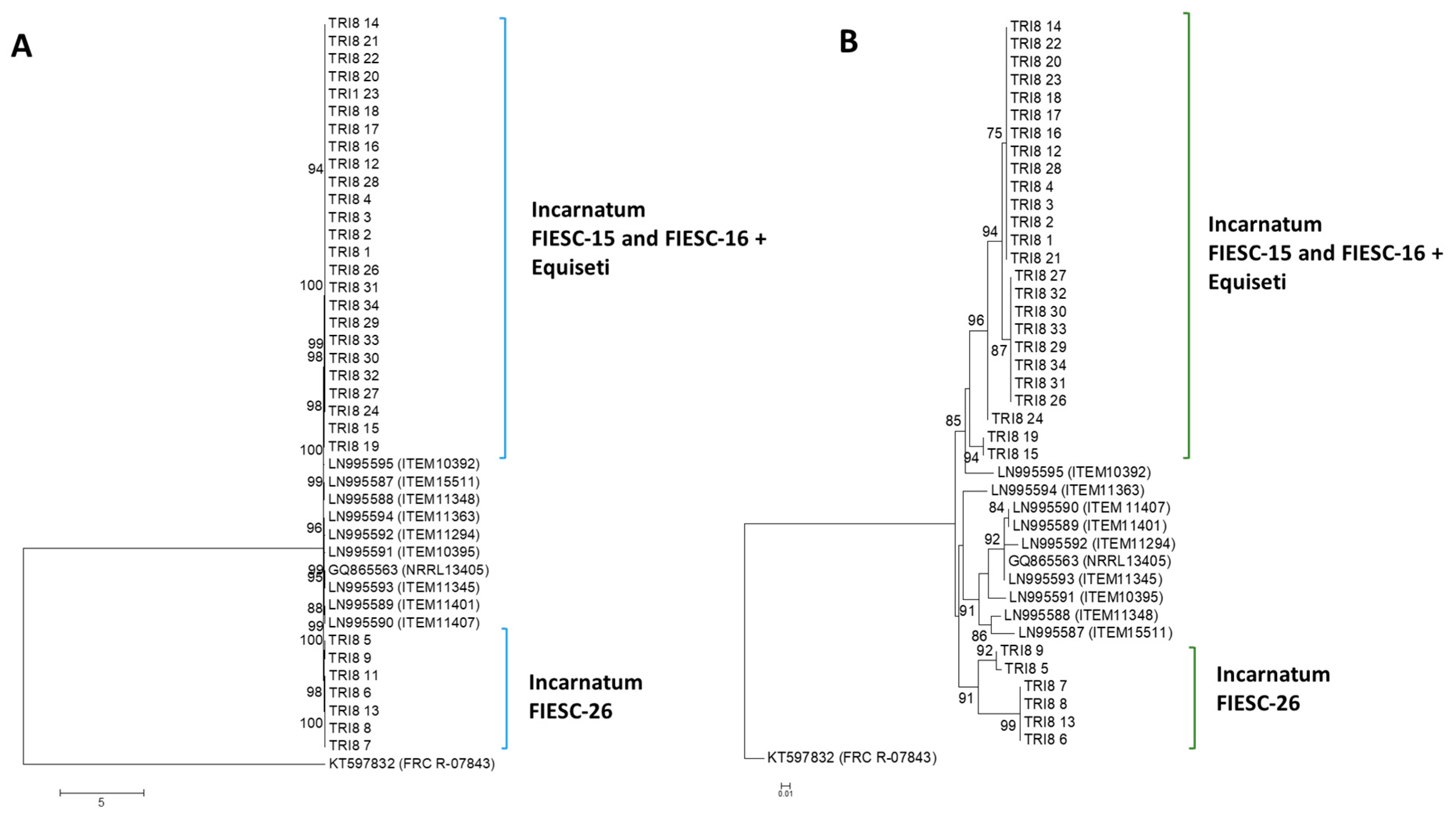
2.2.3. TRI8 Gene Tree
2.2.4. TRI8 Protein Tree
2.2.5. TRI11 Gene Tree
2.2.6. TRI11 Protein Tree
2.3. Mutation Effect Prediction
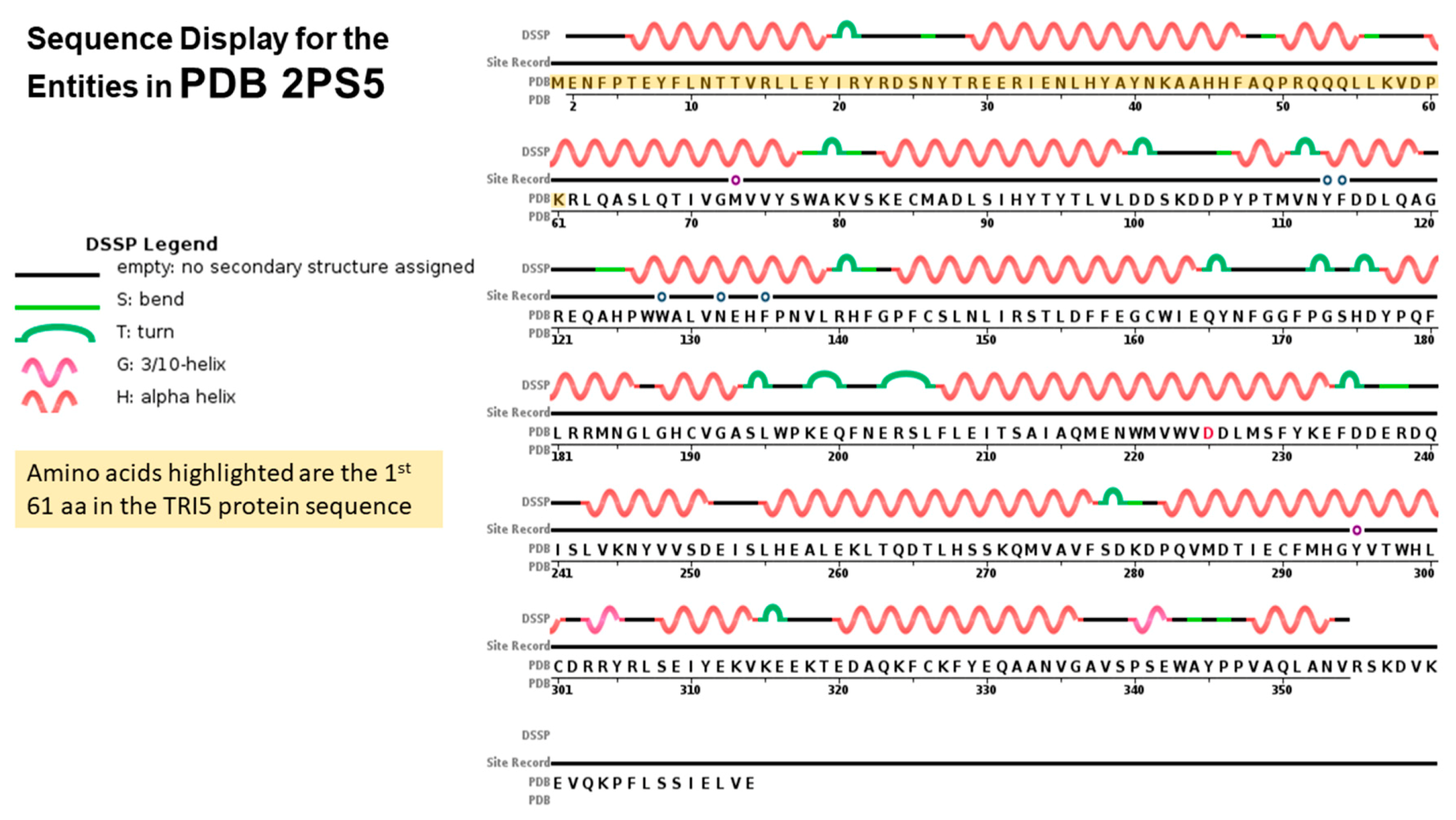
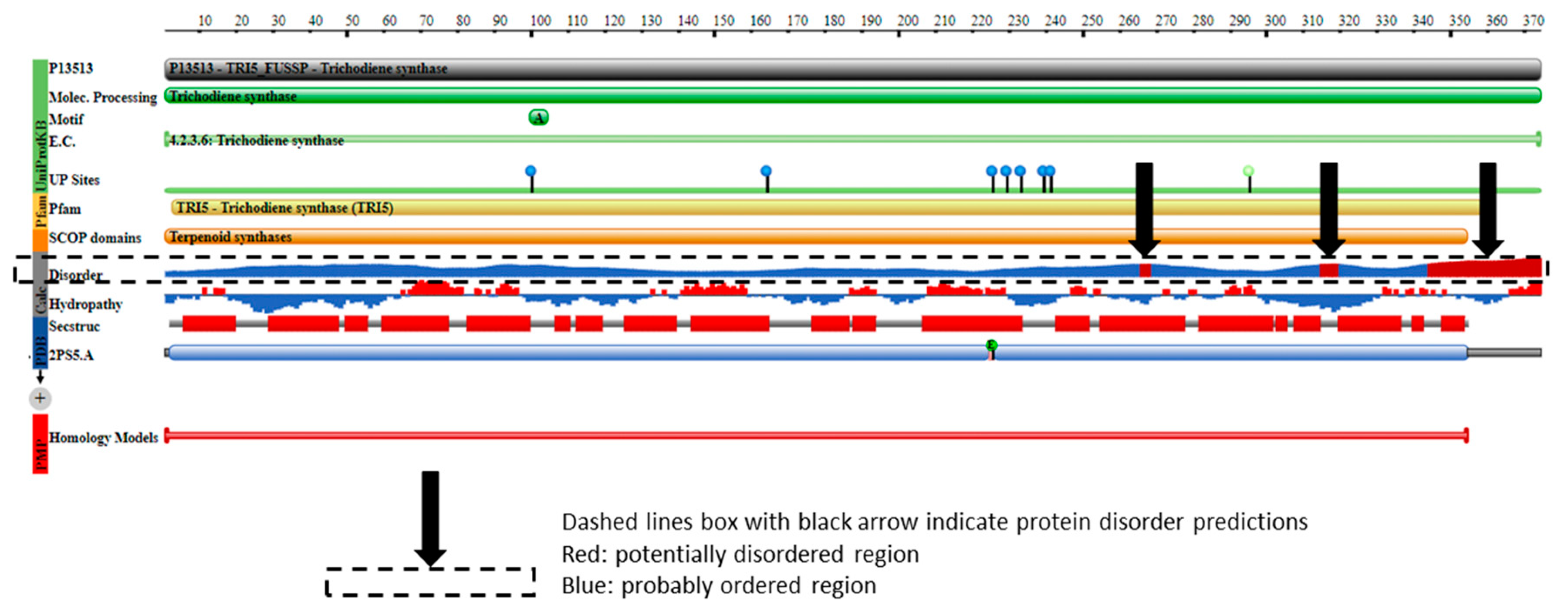
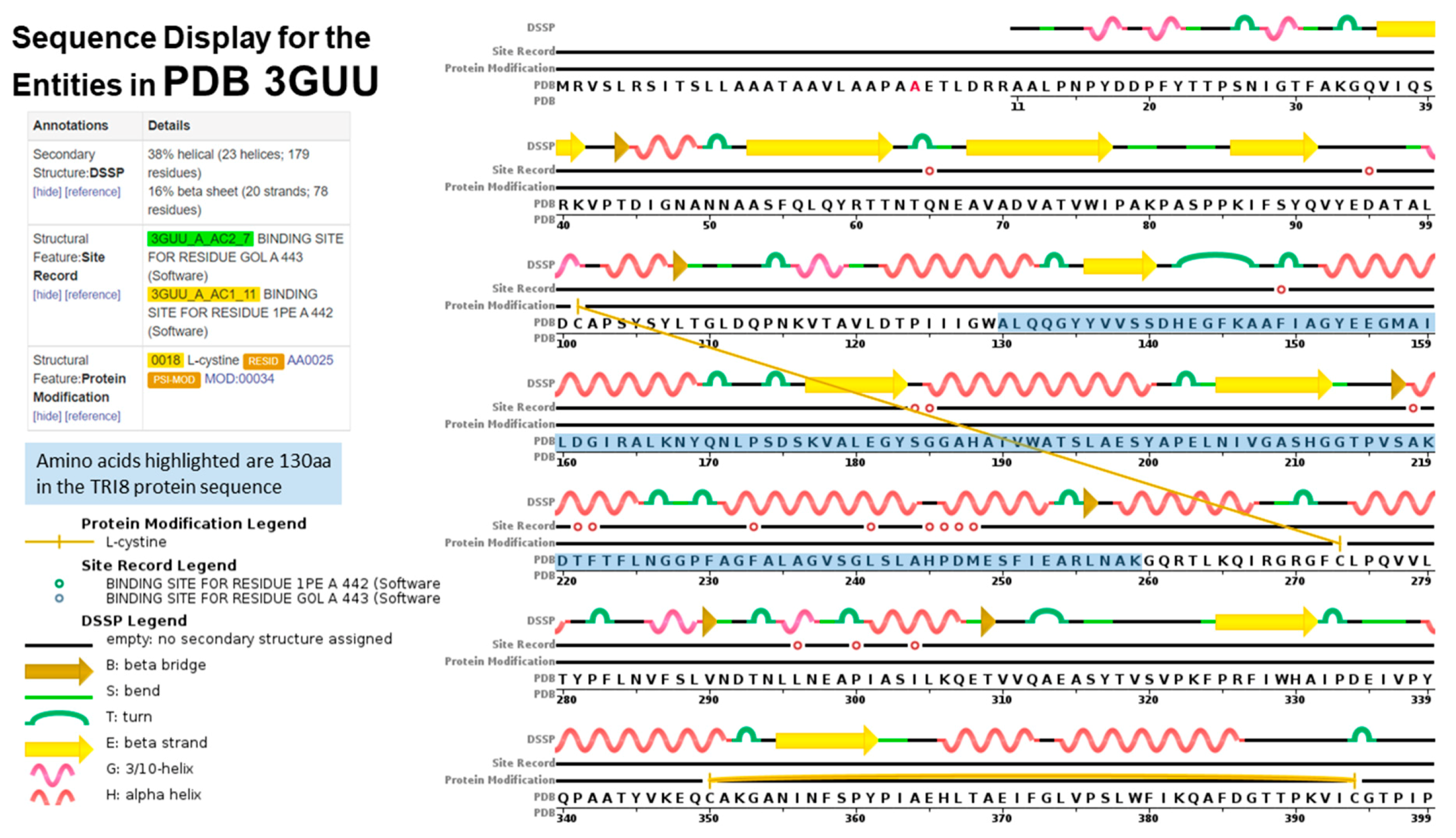
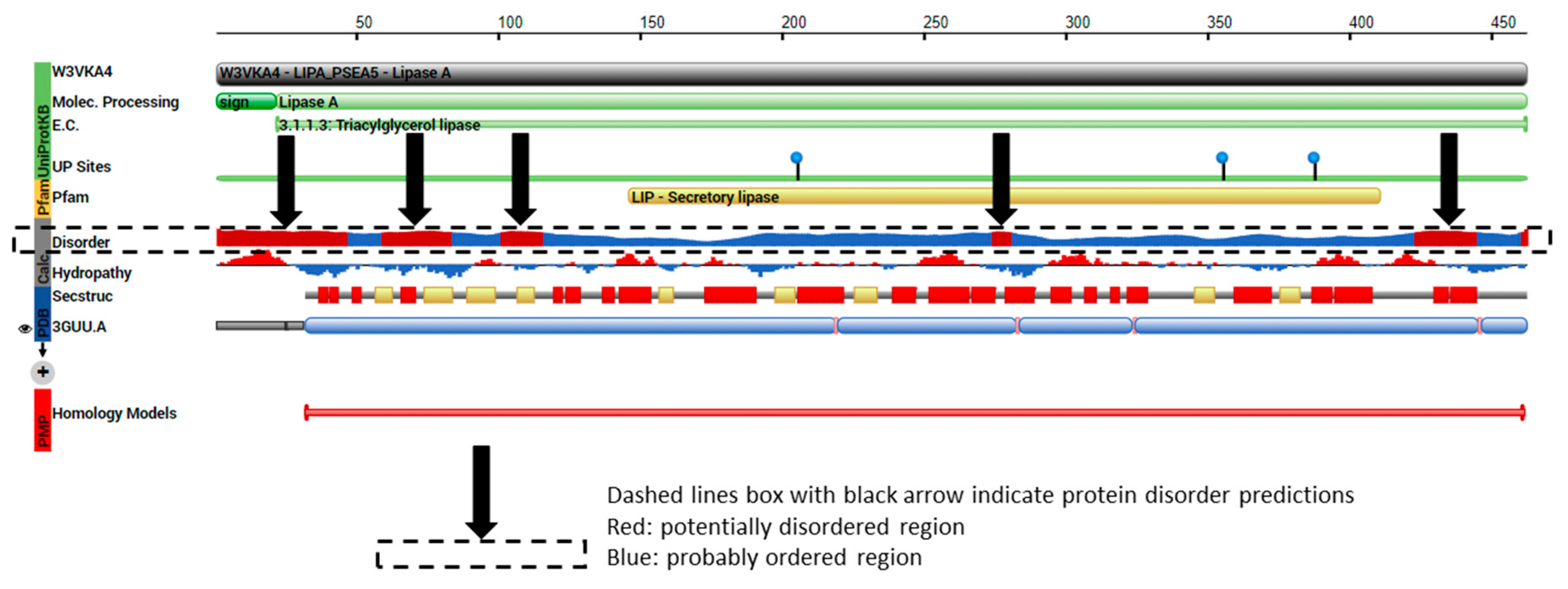
2.4. TRI5p and PDB Mapping
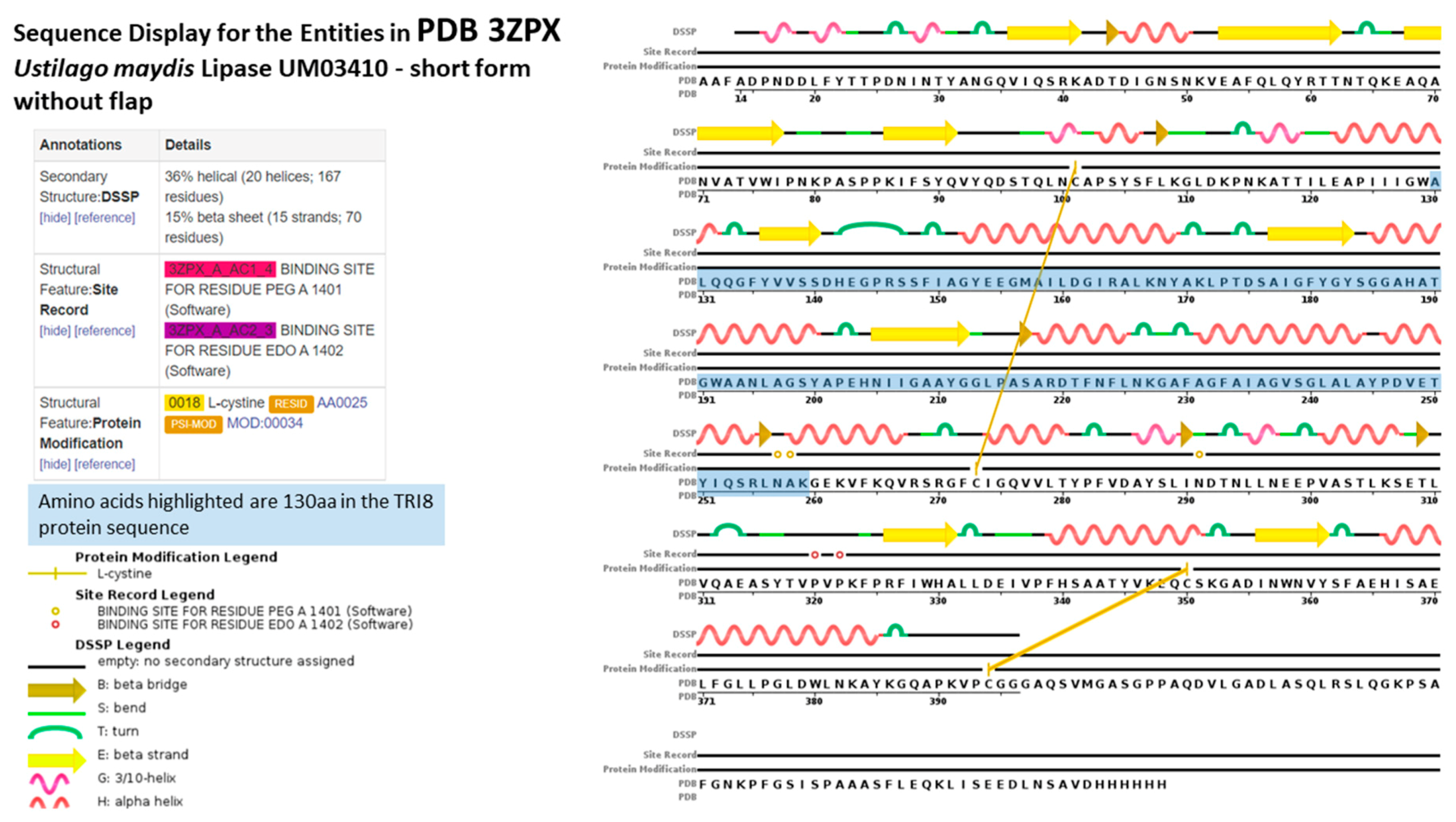
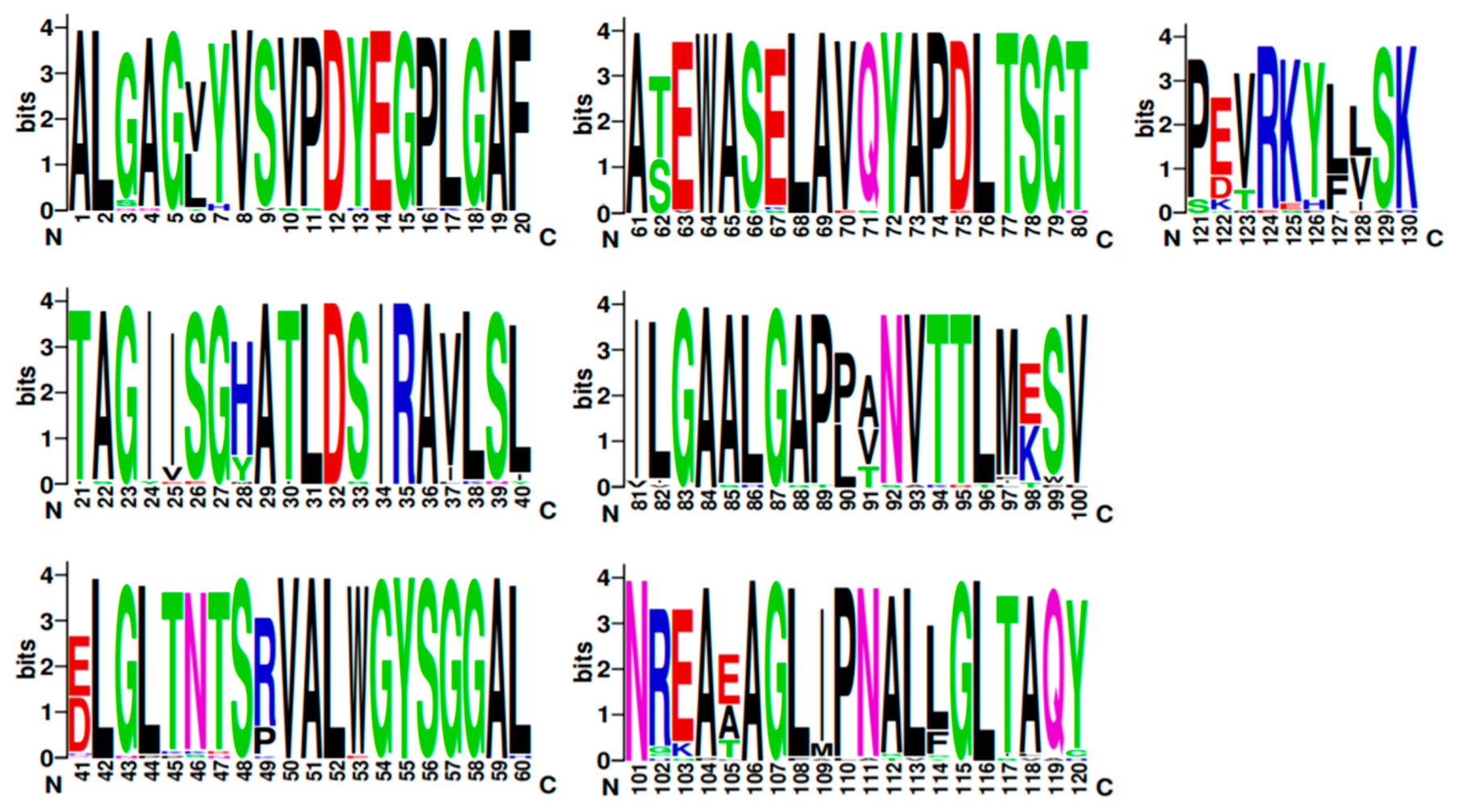
2.5. TRI8p and PDB Mapping
3. Discussion
3.1. Singletons
3.2. Protein Order and Disorder
3.3. Use of Protein Trees over Nucleotide Trees
3.4. TRI5—Trichodiene Synthase
3.5. TRI8 Trichothecene C-3 Esterase
3.6. TRI11—Trichothecene C-15 Hydroxylase
3.7. Use of TRI Protein Sequence Data
4. Materials and Methods
4.1. Collection and Identification of FIESC Isolates
4.2. DNA Extraction, PCR Amplification and Sequencing
4.3. TRI Gene Sequence Analysis
4.4. TRI Protein Sequence Analysis
4.5. Trichodiene Synthase and Trichothecene C-3 Esterase Protein Structure Conservation Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calvo, A.M.; Wilson, R.A.; Bok, J.W.; Keller, N.P. Relationship between secondary metabolism and fungal development. Microbiol. Mol. Biol. Rev. 2002, 66, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism-from biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.E.; Hohn, T.M.; McCormick, S.P. Effect of gene disruption of trichodiene synthase on the virulence of Gibberella pulicaris. Mol. Plant Microbe Interact. 1992, 5, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, A.E.; Proctor, R.H.; Bai, G.; McCormick, S.P.; Shaner, G.; Buechley, G.; Hohn, T.M. Reduced virulence of trichothecene-nonproducing mutants of Gibberella zeae in wheat field tests. Mol. Plant Microbe Interact. 1996, 9, 775–781. [Google Scholar] [CrossRef]
- Proctor, R.H.; Hohn, T.M.; McCormick, S.P. Reduced virulence of Gibberella zeae caused by disruption of a trichothecene toxin biosynthetic gene. Mol. Plant Microbe Interact. 1995, 8, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desjardins, A.E. Fusarium Mycotoxins: Chemistry, Genetics, and Biology; American Phytopathological Society: St. Paul, MN, USA, 2006. [Google Scholar] [CrossRef]
- Cuzick, A.; Urban, M.; Hammond-Kosack, K. Fusarium graminearum gene deletion mutants map1 and TRI5 reveal similarities and differences in the pathogenicity requirements to cause disease on Arabidopsis and wheat floral tissue. New Phytol. 2008, 177, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Z.; Yang, X.; Yang, J.; Gong, A.; Zhang, J.; Chen, L.; Zhou, C. Fusarium graminearum Species Complex and Trichothecene Genotype. In Mycotoxins and Food Safety; IntechOpen: London, UK, 2019; pp. 1–19. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.P.; Rezanoor, H.N.; Holden, D.; Desjardins, A.E.; Plattner, R.D.; Nicholson, P. Variation in pathogenicity associated with the genetic diversity of Fusarium graminearum. Eur. J. Plant Pathol. 2002, 108, 573–583. [Google Scholar] [CrossRef]
- Zhang, H.; Van der Lee, T.; Waalwijk, C.; Chen, W.; Xu, J.; Xu, J.; Zhang, Y.; Feng, J. Population analysis of the Fusarium graminearum species complex from wheat in China show a shift to more aggressive isolates. PLoS ONE 2012, 7, e31722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cundliffe, E.; Cannon, M.; Davies, J. Mechanism of inhibition of eukaryotic protein synthesis by trichothecene fungal toxins. Proc. Natl. Acad. Sci. USA 1974, 71, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, O.; Ansari, K.; Doohan, F.M. Effects of trichothecene mycotoxins on eukaryotic cells: A review. Food Addit. Contam. 2005, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.H.; McCormick, S.P.; Alexander, N.J.; Desjardins, A.E. Evidence that a secondary metabolic biosynthetic gene cluster has grown by gene relocation during evolution of the filamentous fungus Fusarium. Mol. Microbiol. 2009, 74, 1128–1142. [Google Scholar] [CrossRef] [PubMed]
- Alexander, N.J.; Proctor, R.H.; McCormick, S.P. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev. 2009, 28, 198–215. [Google Scholar] [CrossRef]
- Kimura, M.; Tokai, T.; Takahashi-Ando, N.; Ohsato, S.; Fujimura, M. Molecular and genetic studies of Fusarium trichothecene biosynthesis: Pathways, genes, and evolution. Biosci. Biotechnol. Biochem. 2007, 71, 2105–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wawrzyn, G.T.; Bloch, S.E.; Schmidt-Dannert, C. Chapter Five: Discovery and Characterization of Terpenoid Biosynthetic Pathways of Fungi. In Methods in Enzymology; Hopwood, D.A., Ed.; Academic Press: San Diego, CA, USA, 2012; pp. 83–105. [Google Scholar] [CrossRef]
- Proctor, R.H.; McCormick, S.P.; Kim, H.S.; Cardoza, R.E.; Stanley, A.M.; Lindo, L.; Kelly, A.; Brown, D.W.; Lee, T.; Vaughan, M.M.; et al. Evolution of structural diversity of trichothecenes, a family of toxins produced by plant pathogenic and entomopathogenic fungi. PLoS Pathog. 2018, 14, e1006946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoza, R.E.; Malmierca, M.G.; Hermosa, M.R.; Alexander, N.J.; McCormick, S.P.; Proctor, R.H.; Tijerino, A.M.; Rumbero, A.; Monte, E.; Gutiérrez, S. Identification of loci and functional characterization of trichothecene biosynthesis genes in filamentous fungi of the genus Trichoderma. Appl. Environ. Microbiol. 2011, 77, 4867–4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.; Holtom, A.M.; Hanson, J.R. Biosynthesis of 2-cis-farnesol. J. Chem. Soc. Chem. Commun. 1973, 14, 465a. [Google Scholar] [CrossRef]
- McCormick, S.P.; Stanley, A.M.; Stover, N.A.; Alexander, N.J. Trichothecenes: From simple to complex mycotoxins. Toxins 2011, 3, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Degenkolb, T.; Dieckmann, R.; Nielsen, K.F.; Gräfenhan, T.; Theis, C.; Zafari, D.; Chaverri, P.; Ismaiel, A.; Brückner, H.; von Döhren, H.; et al. The Trichoderma brevicompactum clade: A separate lineage with new species, new peptaibiotics, and mycotoxins. Mycol. Prog. 2008, 7, 177–219. [Google Scholar] [CrossRef] [Green Version]
- McCormick, S.P.; Alexander, N.J. Fusarium TRI8 encodes a trichothecene C-3 esterase. Appl. Environ. Microbiol. 2002, 68, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, R.; Levander, D.; Adams, W.; Miller, J.D.; Blackwell, B.A.; McAlees, A.J.; Taylor, A. Production and characterization of deoxynivalenol and other secondary metabolites of Fusarium culmorum (CMI 14764, HLX 1503). J. Agric. Food Chem. 1986, 34, 98–102. [Google Scholar] [CrossRef]
- Alexander, N.J.; Hohn, T.M.; McCormick, S.P. The TRI11 gene of Fusarium sporotrichioides encodes a cytochrome P-450 monooxygenase required for C-15 hydroxylation in trichothecene biosynthesis. Appl. Environ. Microbiol. 1998, 64, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tag, A.G.; Garifullina, G.F.; Peplow, A.W.; Ake, C., Jr.; Phillips, T.D.; Hohn, T.M.; Beremand, M.N. A novel regulatory gene, TRI10, controls trichothecene toxin production and gene expression. Appl. Environ. Microbiol. 2001, 67, 5294–5302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdial, H.; Rampersad, S.N. Characterization of Colletotrichum spp. causing anthracnose of bell pepper (Capsicum annuum L.) in Trinidad. Phytoparasitica 2015, 43, 37–49. [Google Scholar] [CrossRef]
- O’Donnell, K.; Sutton, D.A.; Rinaldi, M.G.; Sarver, B.A.J.; Balajee, S.A.; Schroers, H.J.; Summerbell, R.C.; Robert, V.A.R.; Crous, P.W.; Zhang, N.; et al. Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. J. Clin. Microbiol. 2010, 48, 3708–3718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Diepeningen, A.D.; Brankovics, B.; Iltes, J.; van der Lee, T.A.J.; Waalwijk, C. Diagnosis of Fusarium infections, approaches to identification by the clinical mycology laboratory. Curr. Fungal Infect. Rep. 2015, 9, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, K.; Sutton, D.A.; Rinaldi, M.G.; Gueidan, C.; Crous, P.W.; Geiser, D.M. A novel multi-locus sequence typing scheme reveals high genetic diversity of human pathogenic members of the Fusarium incarnatum-F. equiseti and F. chlamydosporum species complexes within the U.S. J. Clin. Microbiol. 2009, 47, 3851–3861. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Rooney, A.P.; Proctor, R.H.; Brown, D.W.; McCormick, S.P.; Ward, T.J.; Frandsen, R.J.; Lysøe, E.; Rehner, S.A.; Aoki, T.; et al. Phylogenetic analyses of RPB1 and RPB2 support a middle cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet. Biol. 2013, 52, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.; Moretti, A.; De Saeger, S.; Han, Z.; Di Mavungu, J.D.; Soares, C.M.; Proctor, R.H.; Venâncio, A.; Lima, N.; Stea, G.; et al. A polyphasic approach for characterization of a collection of cereal isolates of the Fusarium incarnatum-equiseti species complex. Int. J. Food Microbiol. 2016, 234, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villafana, R.T.; Rampersad, S.N. Three-locus sequence identification and differential tebuconazole sensitivity suggest novel Fusarium equiseti haplotype from Trinidad. Pathogens 2020, 9, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickie, I.A. Insidious effects of sequencing errors on perceived diversity in molecular surveys. New Phytol. 2010, 188, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.H.; Chao, A. Estimating and comparing microbial diversity in the presence of sequencing errors. PeerJ 2016, 4, e1634. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, P.C.; Françoso, E.; Arias, M.C. Fidelity of DNA polymerases in the detection of intra-individual variation of mitochondrial DNA. Mitochondrial DNA B Resour. 2020, 5, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Willis, A. Species Richness Estimation with High Diversity but Spurious Singletons. Available online: https://arxiv.org/abs/1604.02598 (accessed on 13 March 2020).
- Brown, S.P.; Veach, A.M.; Rigdon-Huss, A.R.; Grond, K.; Lickteig, S.K.; Lothamer, K.; Oliver, A.K.; Jumpponen, A. Scraping the bottom of the barrel: Are rare high throughput sequences artifacts? Fungal Ecol. 2015, 13, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, G.J.; Porter, C.T.; Borkakoti, N.; Thornton, J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002, 324, 105–121. [Google Scholar] [CrossRef]
- Yang, Z.R.; Thomson, R.; Mcneil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Romero, P.; Obradovic, Z.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genom. Inform. 2000, 11, 161–171. [Google Scholar] [CrossRef]
- Opperdoes, F.R. Phylogenetic analysis using protein sequences. In The Phylogenetics Handbook: A Practical Approach to DNA and Protein Phylogeny, 1st ed.; Salemi, M., Vandamme, A.-M., Eds.; Cambridge University Press: Cambridge/London, UK, 2003; pp. 207–235. [Google Scholar]
- Lemey, P.; Salemi, M.; Vandamme, A.M. The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing; Cambridge University Press: Cambridge/London, UK, 2009. [Google Scholar]
- Rokas, A. Phylogenetic analysis of protein sequence data using the randomized axelerated maximum likelihood (RAXML) program. Curr. Protoc. Mol. Biol. 2011, 96, 19-11. [Google Scholar] [CrossRef] [PubMed]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, I.M.; Chaney, J.L.; Clark, P.L. Expanding anfinsen’s principle: Contributions of synonymous codon selection to rational protein design. J. Am. Chem. Soc. 2014, 36, 858–861. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Ciryam, P.; Vendruscolo, M.; Dobson, C.M. Understanding the influence of codon translation rates on cotranslational protein folding. Acc. Chem. Res. 2014, 47, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Bali, V.; Bebok, Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int. J. Biochem. Cell Biol. 2015, 64, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athey, J.; Alexaki, A.; Osipova, E.; Rostovtsev, A.; Santana-Quintero, L.V.; Katneni, U.; Simonyan, V.; Kimchi-Sarfaty, C. A new and updated resource for codon usage tables. BMC Bioinform. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cane, D.E.; Xue, Q.; Fitzsimons, B.C. Trichodiene synthase. Probing the role of the highly conserved aspartate-rich region by site-directed mutagenesis. Biochemistry 1996, 35, 12369–12376. [Google Scholar] [CrossRef] [PubMed]
- Vedula, L.S.; Rynkiewicz, M.J.; Pyun, H.J.; Coates, R.M.; Cane, D.E.; Christianson, D.W. Molecular recognition of the substrate diphosphate group governs product diversity in trichodiene synthase mutants. Biochemistry 2005, 44, 6153–6163. [Google Scholar] [CrossRef] [PubMed]
- Vedula, L.S.; Zhao, Y.; Coates, R.M.; Koyama, T.; Cane, D.E.; Christianson, D.W. Exploring biosynthetic diversity with trichodiene synthase. Arch. Biochem. Biophys. 2007, 466, 260–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rynkiewicz, M.J.; Cane, D.E.; Christianson, D.W. X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry 2002, 41, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Cane, D.E.; Shim, J.H.; Xue, Q.; Fitzsimons, B.C.; Hohn, T.M. Trichodiene synthase. Identification of active site residues by site-directed mutagenesis. Biochemistry 1995, 34, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Vedula, L.S.; Jiang, J.; Zakharian, T.; Cane, D.E.; Christianson, D.W. Structural and mechanistic analysis of trichodiene synthase using site-directed mutagenesis: Probing the catalytic function of tyrosine-295 and the asparagine-225/serine-229/glutamate-233–Mg2+ B motif. Arch. Biochem. Biophys. 2008, 469, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.W.; McCormick, S.P.; Alexander, N.J.; Proctor, R.H.; Desjardins, A.E. A genetic and biochemical approach to study trichothecene diversity in Fusarium sporotrichioides and Fusarium graminearum. Fungal Genet. Biol. 2001, 32, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peplow, A.W.; Tag, A.G.; Garifullina, G.F.; Beremand, M.N. Identification of new genes positively regulated by TRI10 and a regulatory network for trichothecene mycotoxin production. Appl. Environ. Microbiol. 2003, 69, 2731–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasrayan, A.; Bocola, M.; Sandström, A.G.; Lavén, G.; Bäckvall, J.E. Prediction of the Candida antarctica lipase a protein structure by comparative modeling and site-directed mutagenesis. ChemBioChem 2007, 8, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P.; Hohn, T.M. Accumulation of trichothecenes in liquid cultures of a Fusarium sporotrichioides mutant lacking a functional trichothecene C-15 hydroxylase. Appl. Environ. Microbiol. 1997, 63, 1685–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M. Fuzziness in protein interactions- a historical perspective. J. Mol. Biol. 2018, 430, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic plasticity, bet-hedging, and androgen independence in prostate cancer: Role of non-genetic heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Shanthirabalan, S.; Chomilier, J.; Carpentier, M. Structural effects of point mutations in proteins. Proteins 2018, 86, 853–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Jang, H. Protein ensembles link genotype to phenotype. PLoS Comput. Biol. 2019, 15, e1006648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J. DNA Sequence polymorphism analysis using DnaSP. In Bioinformatics for DNA Sequence Analysis; Methods in Molecular Biology Series; Posada, D., Ed.; Humana Press: Totowa, NJ, USA, 2009; Volume 537, pp. 337–350. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapp, S.C.; Hohn, T.M.; McCormick, S.P.; Jarvis, B.B. Characterization of the gene cluster for biosynthesis of macrocyclic trichothecenes in Myrothecium roridum. Mol. Gen. Genet. 1998, 257, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Ismail, Y.; McCormick, S.; Hijri, M. A fungal symbiont of plant-roots modulates mycotoxin gene expression in the pathogen Fusarium sambucinum. PLoS ONE 2011, 6, e17990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. 2016. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y. A Fast Computation of Pairwise Sequence Alignment Scores between a Protein and a Set of Single-Locus Variants of another Protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB ′12), Orlando, FL, USA, October 2012; Association for Computing Machinery: New York, NY, USA, 2012; pp. 414–417. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Vedula, L.S.; Cane, D.E.; Christianson, D.W. Role of arginine-304 in the diphosphate-triggered active site closure mechanism of trichodiene synthase. Biochemistry 2005, 44, 12719–12727. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.M.; Nymalm-Rejstrom, Y.; Airenne, T.; Salminen, T. The Crystal Structure of Lipase A from Candida antarctica. In Acta Crystallographica A—Foundation and Advances; Int Union Crystallography: Chester, UK, 2008; Volume 64, p. C245. [Google Scholar] [CrossRef] [Green Version]
- Ben Chorin, A.; Masrati, G.; Kessel, A.; Narunsky, A.; Sprinzak, J.; Lahav, S.; Ashkenazy, H.; Ben-Tal, N. ConSurf-DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020, 29, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, O.; Erez, E.; Nimrod, G.; Ben-Tal, N. The ConSurf-DB: Pre-calculated evolutionary conservation profiles of protein structures. Nucleic Acids Res. 2009, 37, D323–D327. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Alignment Statistic | TRI5nt | TRI8nt | TRI11nt | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Trinidad | Reference Sequences | Total | Trinidad | Reference Sequences | Total | Trinidad | Reference Sequences | Total | |
| N | 32 | 8 | 40 | 32 | 10 | 42 | 32 | 12 | 44 |
| No. of sites analyzed | 394 | 394 | 394 | 470 | 470 | 470 | 705 | 705 | 705 |
| No. of singleton variable sites | 0 | 16 | 13 | 0 | 62 | 33 | 0 | 93 | 60 |
| Total number of singleton mutations, Eta(s) | 0 | 18 | 14 | 0 | 65 | 36 | 0 | 118 | 86 |
| Total number of mutations, Eta | 55 | 56 | 86 | 108 | 102 | 176 | 78 | 237 | 264 |
| Number of polymorphic sites, S | 52 | 53 | 81 | 100 | 97 | 158 | 77 | 203 | 217 |
| Average no. nucleotide differences, k | 17.091 | 24.214 | 20.023 | 29.734 | 28.089 | 40.11 | 26.938 | 64.061 | 49.969 |
| Nucleotide diversity, Pi | 0.04498 | 0.06372 | 0.05269 | 0.06326 | 0.05976 | 0.08534 | 0.03881 | 0.09244 | 0.07211 |
| Number of haplotypes, h | 7 | 6 | 13 | 7 | 8 | 15 | 4 | 12 | 16 |
| Haplotype (gene) diversity, Hd | 0.792 | 0.893 | 0.864 | 0.738 | 0.956 | 0.847 | 0.651 | 1 | 0.817 |
| Fu and Li’s D test statistic, FLD; p < 0.02 | 1.72888 | 0.4508 | 0.57684 | 1.87228 | −1.1548 | 0.29559 | 1.90862 | −0.63889 | −0.82709 |
| Fu and Li’s F test statistic, FLF; p < 0.02 | 1.72973 | 0.5375 | 0.41238 | 1.63219 | −1.28824 | 0.19375 | 2.08398 | −0.79709 | −0.91004 |
| Recombination | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TRI5p | Mutation Data |
|---|---|
| N, total no. of mutations | 32 |
| No. of neutral mutations | 28 |
| No. of non-neutral mutations | 4 |
| Non-neutral mutations | Isolates (strains) |
| A54L | LN995579 (15511); LN995580 (11348) |
| Y56E | ACZ56393 (13381),ACZ56398 (31160),ACZ56401 (FRC R-06979), AXP09554 (64414) |
| I57T | TRI5_5,TRI5_6,TRI5_7,TRI5_8,TRI5_9,TRI5_11,TRI5_13 (Trinidad strains) |
| I57Q | ACZ56393 (13381),ACZ56398 (31160),ACZ56401 (FRC R-06979), AXP09554 (64414) |
| TRI8p | Mutation Data |
|---|---|
| N, total no. of mutations | 73 |
| No. of neutral mutations | 62 |
| No. of non-neutral mutations | 11 |
| Non-neutral mutations | Isolates (strains) |
| Y20H | TRI8_5, TRI8_9 (Trinidad strains) |
| H41Y | TRI8_5,TRI8_6,TRI8_7,TRI8_8,TRI8_9,TRI8_11,TRI8_13 (Trinidad strains) |
| R62P | TRI8_26, TRI8_29, TRI8_30, TRI8_31, TRI8_32, TRI8_33, TRI8_34 (Trinidad strains) |
| S112W | TRI8_ 5, TRI8_9 (Trinidad strains) |
| R115H | LN995594 (11363) |
| A125T | LN995587 (15511) |
| Y133C | TRI8_15, TRI8_19 (Trinidad strains) |
| P134S | TRI8_6, TRI8_7, TRI8_8, TRI8_13 (Trinidad strains) |
| E135D | LN995588 (11348) |
| L140F | TRI8_ 1, TRI8_12, TRI8_16, TRI8_17, TRI8_18, TRI8_23, TRI8_28 (Trinidad strains) |
| N187D | LN995587 (15511), LN995588 (11348), LN995591 (10395) |
| TRI11p | Mutation Data |
|---|---|
| N, total no. of mutations | 28 |
| No. of neutral mutations | 22 |
| No. of non-neutral mutations | 6 |
| Non-neutral mutations | Isolates (strains) |
| E4V | GQ915566 (FRC R-06979) |
| G47D | LN995598 (11401), LN995599 (11407) |
| P50N | LN995603 (11363) |
| P50V | TRI11_5,TRI11_6,TRI11_7,TRI11_8,TRI11_9,TRI11_11,TRI11_13 (Trinidad strains) |
| G75S | LN995596 (15511), LN995597 (11348), LN995598 (11401), LN995599 (11407), LN995600 (10395), LN995601 (11294), LN995602 (11345), LN995603 (11363) |
| P117S | LN995596 (15511) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villafana, R.T.; Rampersad, S.N. Signatures of TRI5, TRI8 and TRI11 Protein Sequences of Fusarium incarnatum-equiseti Species Complex (FIESC) Indicate Differential Trichothecene Analogue Production. Toxins 2020, 12, 386. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060386
Villafana RT, Rampersad SN. Signatures of TRI5, TRI8 and TRI11 Protein Sequences of Fusarium incarnatum-equiseti Species Complex (FIESC) Indicate Differential Trichothecene Analogue Production. Toxins. 2020; 12(6):386. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060386
Chicago/Turabian StyleVillafana, Ria T., and Sephra N. Rampersad. 2020. "Signatures of TRI5, TRI8 and TRI11 Protein Sequences of Fusarium incarnatum-equiseti Species Complex (FIESC) Indicate Differential Trichothecene Analogue Production" Toxins 12, no. 6: 386. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12060386