Shiga Toxin Uptake and Sequestration in Extracellular Vesicles Is Mediated by Its B-Subunit

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
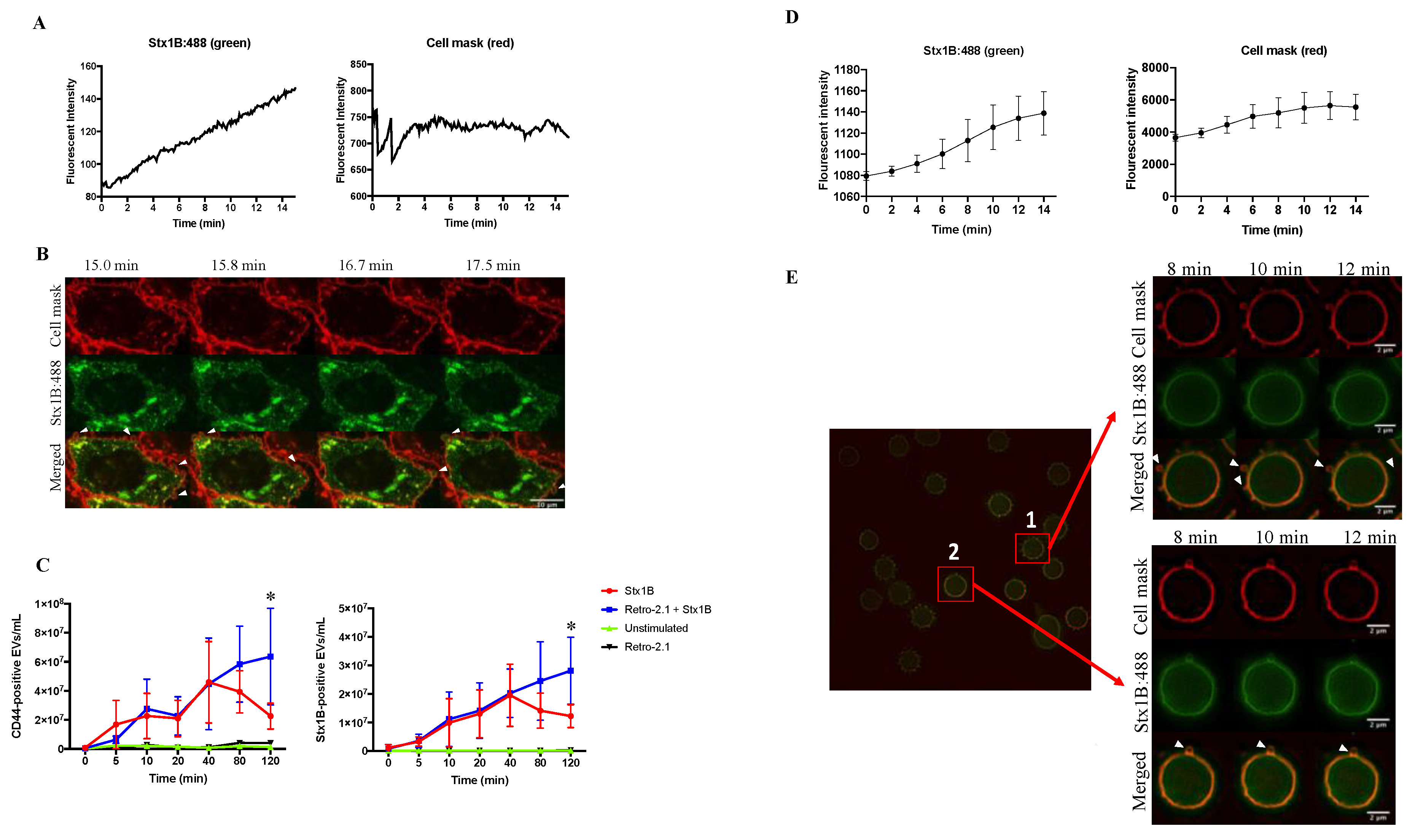
2.1. Shiga Toxin 1B that Is Rapidly Shed in Extracellular Vesicles Does Not Undergo Retrograde Transport
2.2. Shiga Toxin 1B Is Located on the Membrane and Inside Extracellular Vesicles Released by HeLa Cells
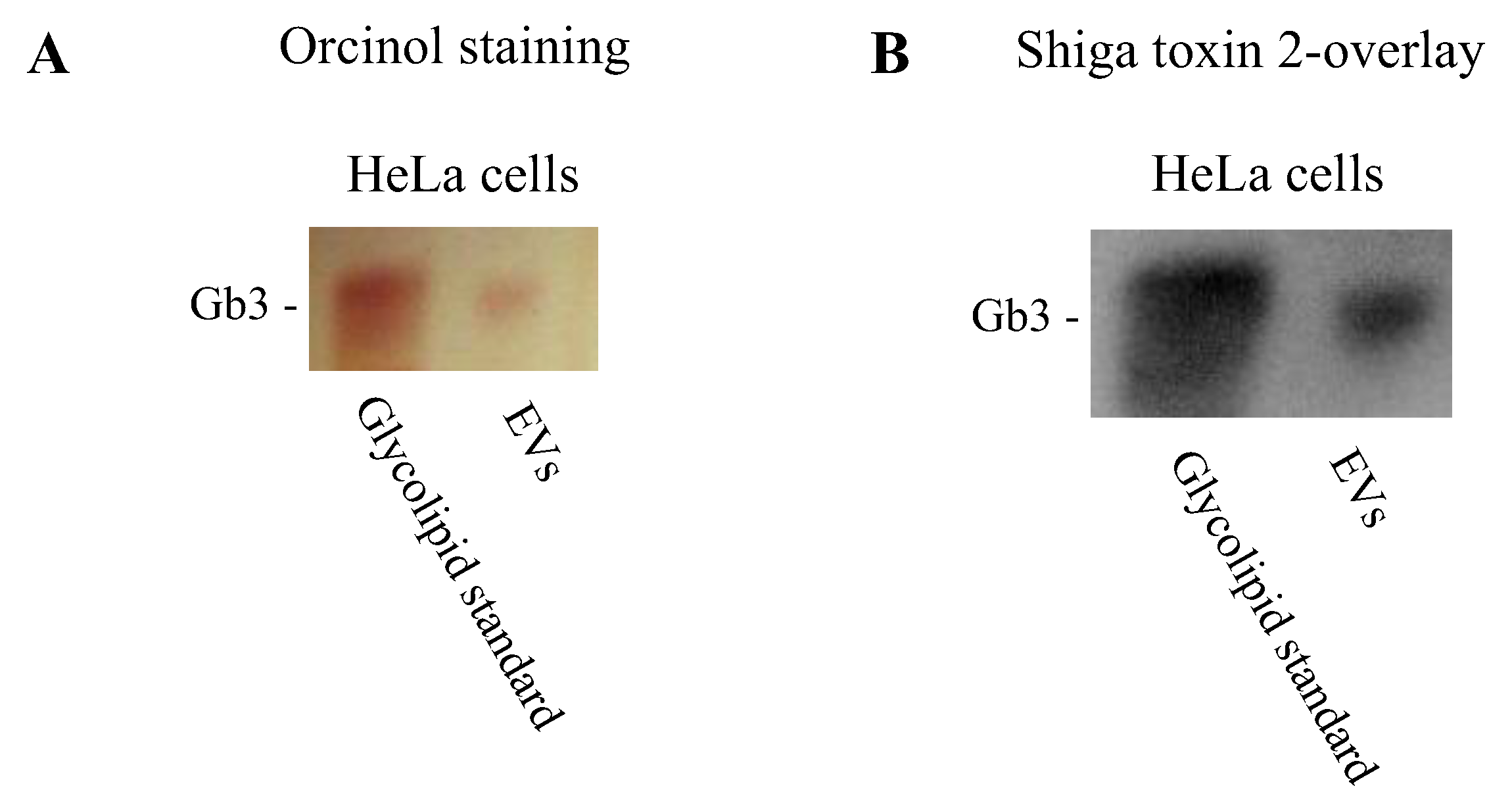
2.3. Extracellular Vesicles Possess Gb3
2.4. Shiga Toxin Binds to HeLa-Cell Derived EVs via the Gb3 Receptor
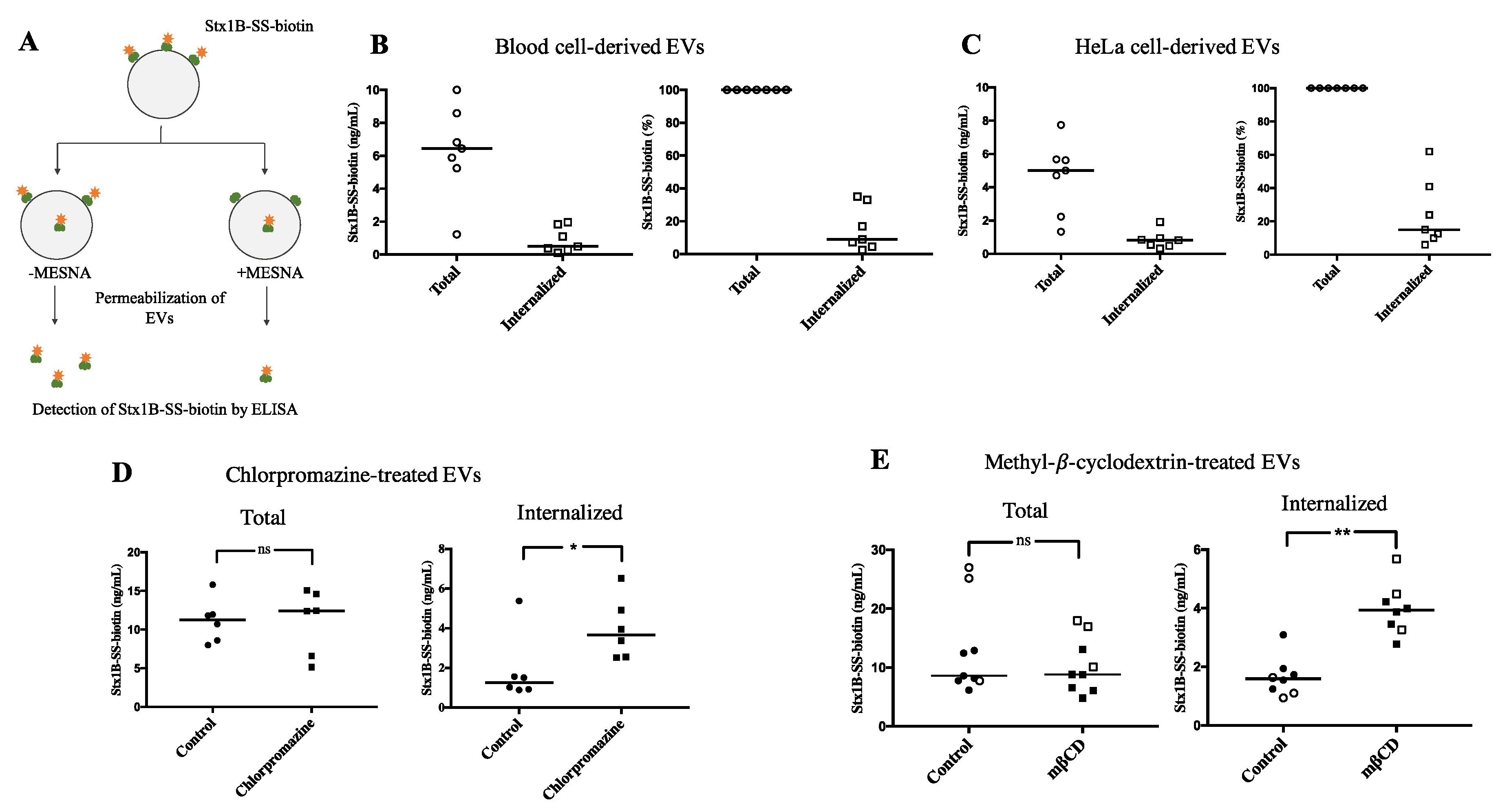
2.5. Shiga Toxin 1B Interacts with Blood Cell-Derived Extracellular Vesicles in the Absence of Cells
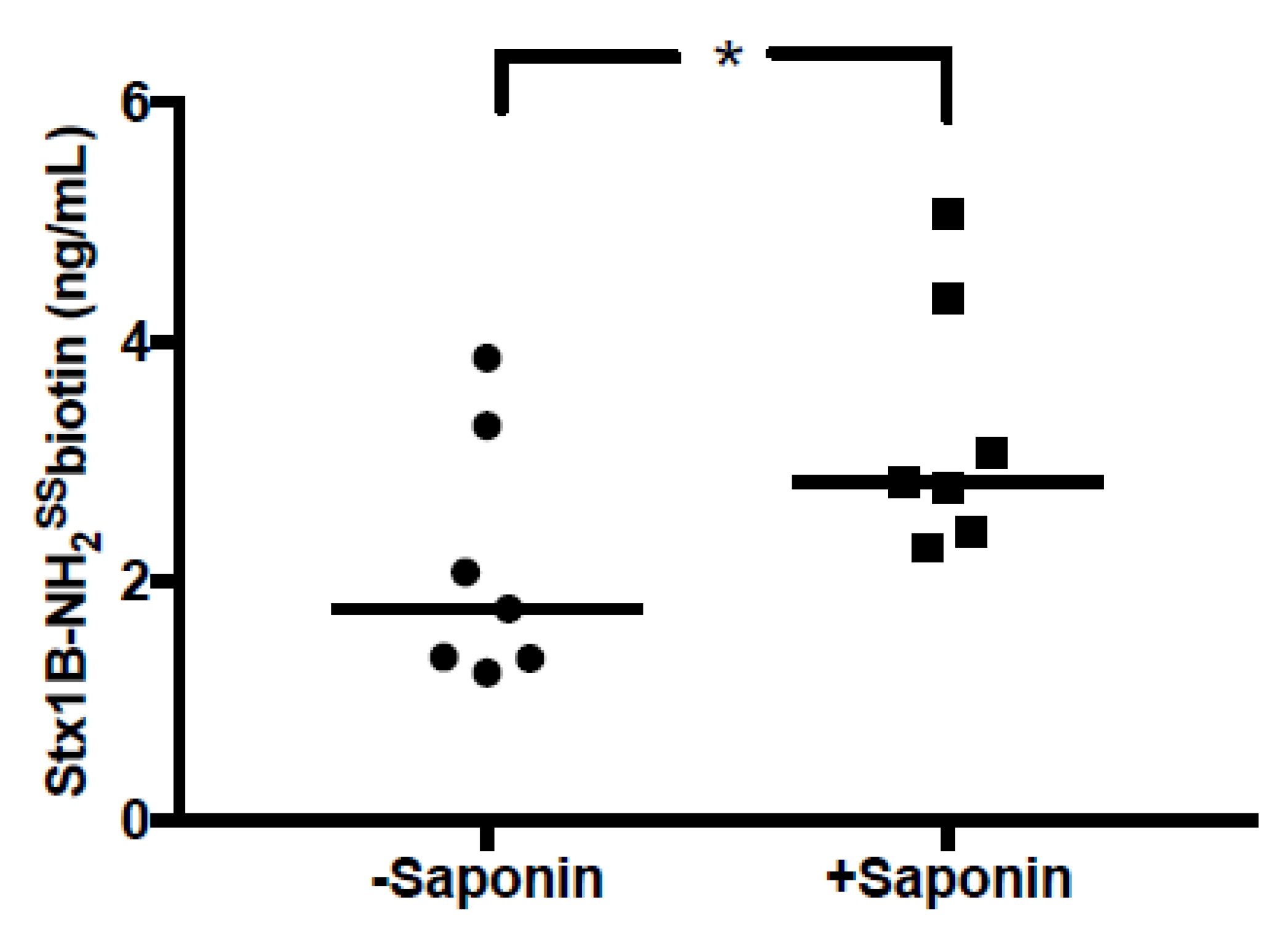
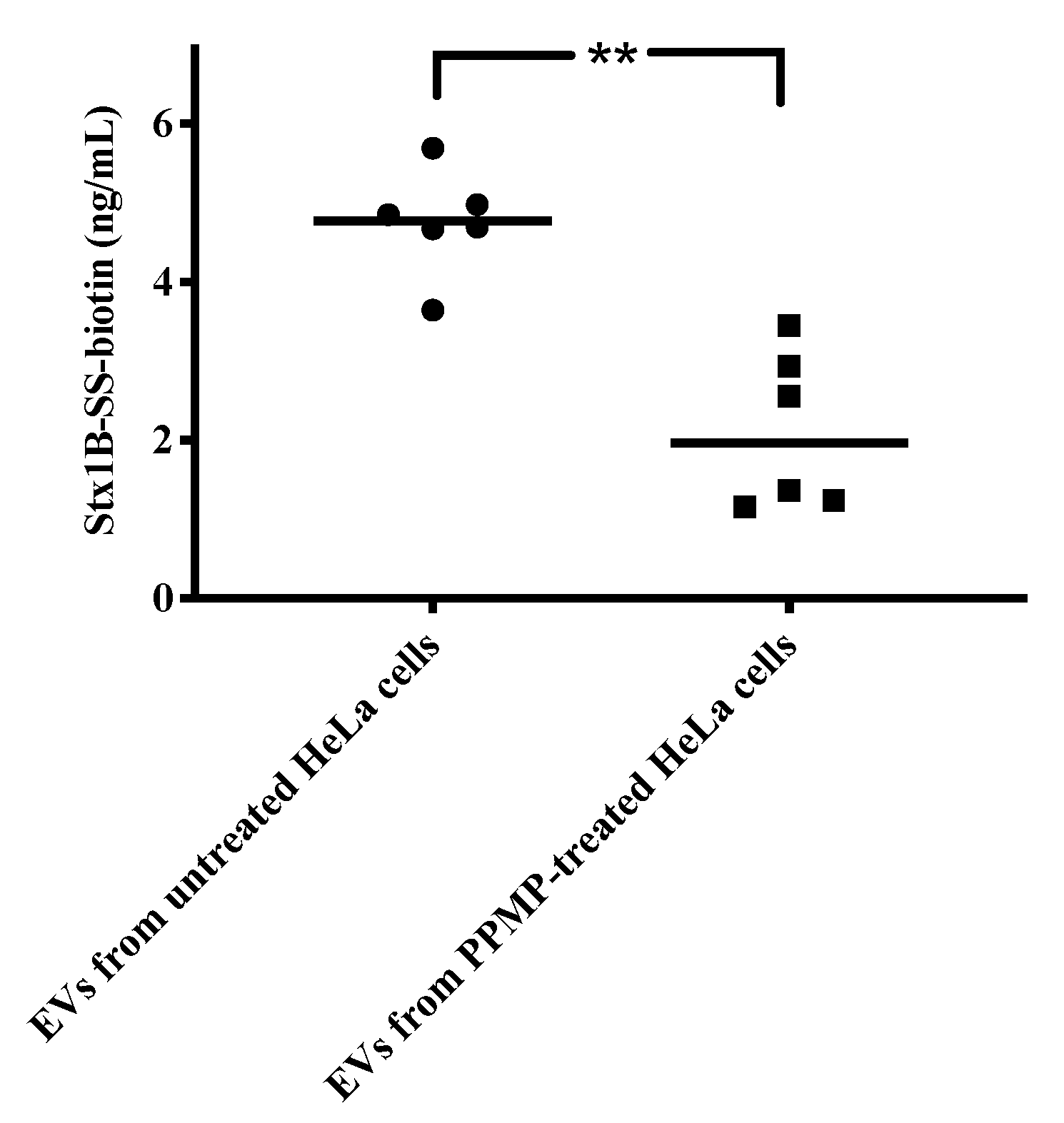
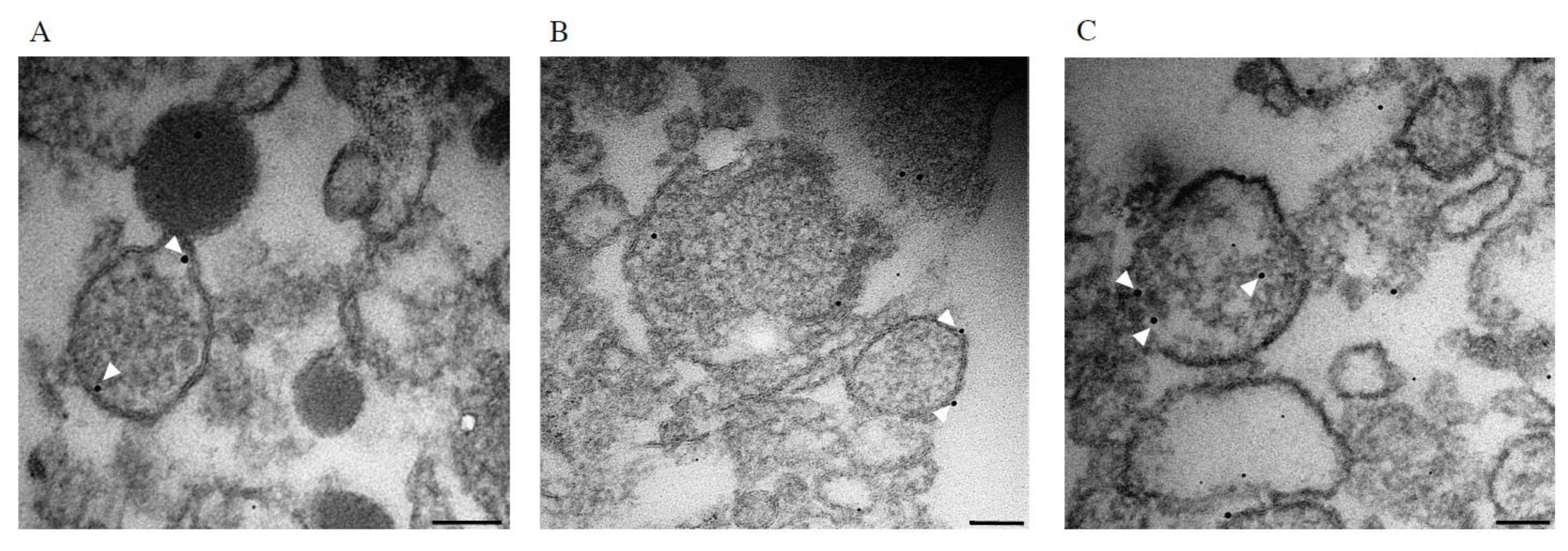
2.6. Shiga Toxin Can Be Translocated into Extracellular Vesicles
3. Discussion
4. Materials and Methods
4.1. HeLa Cell Culture and Inhibition of Gb3 Synthase
4.2. Blood Collection
4.3. Shiga Toxin 1B
4.4. Detection of Shed Vesicles from HeLa Cells by Confocal Immunofluorescence Microscopy
4.5. Isolation of Extracellular Vesicles from HeLa Cells
4.6. Treatment of Shiga Toxin 1B-Stimulated HeLa Cells with Retro-2.1 and Analysis of Extracellular Vesicles by Flow Cytometry
4.7. Detection of Shed Vesicles on Red Blood Cells by Super Illumination Microscopy
4.8. Detection of Shiga Toxin 1B by ELISA
4.9. Thin Layer Chromatography for Detection of Gb3 on Extracellular Vesicles
4.10. Stimulation of Whole Blood and Isolation of Blood Cell-Derived Extracellular Vesicles
4.11. Transmission Electron Microscopy
4.12. Membrane Translocation of Shiga Toxin
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olsnes, S.; Reisbig, R.; Eiklid, K. Subunit structure of Shigella cytotoxin. J. Biol. Chem. 1981, 256, 8732–8738. [Google Scholar] [PubMed]
- Lindberg, A.A.; Brown, J.E.; Strömberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar] [PubMed]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The a-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Renard, H.F.; Simunovic, M.; Lemiere, J.; Boucrot, E.; Garcia-Castillo, M.D.; Arumugam, S.; Chambon, V.; Lamaze, C.; Wunder, C.; Kenworthy, A.K.; et al. Endophilin-a2 functions in membrane scission in clathrin-independent endocytosis. Nature 2015, 517, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (vt2) from Escherichia coli o157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA n-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef]
- Sandvig, K. Shiga toxins. Toxicon 2001, 39, 1629–1635. [Google Scholar] [CrossRef]
- Gupta, N.; Pons, V.; Noel, R.; Buisson, D.A.; Michau, A.; Johannes, L.; Gillet, D.; Barbier, J.; Cintrat, J.C. (s)-n-methyldihydroquinazolinones are the active enantiomers of retro-2 derived compounds against toxins. ACS Med. Chem. Lett. 2014, 5, 94–97. [Google Scholar] [CrossRef] [Green Version]
- Forrester, A.; Rathjen, S.J.; Daniela Garcia-Castillo, M.; Bachert, C.; Couhert, A.; Tepshi, L.; Pichard, S.; Martinez, J.; Munier, M.; Sierocki, R.; et al. Functional dissection of the retrograde Shiga toxin trafficking inhibitor retro-2. Nat. Chem. Biol. 2020, 16, 327–336. [Google Scholar] [CrossRef]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef]
- Karpman, D.; Ståhl, A.L. Enterohemorrhagic Escherichia coli pathogenesis and the host response. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, A.L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.C.; Bekassy, Z.D.; Morgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.; Hertel, B.; Emden, S.H.; Beneke, J.; Menne, J.; Haller, H.; von Vietinghoff, S. Microparticle generation and leucocyte death in Shiga toxin-mediated hus. Nephrol. Dial. Transplant. 2012, 27, 2768–2775. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, I.; Ståhl, A.L.; Hedström, M.M.; Kristoffersson, A.C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Invest. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Little, K.M.; Smalley, D.M.; Harthun, N.L.; Ley, K. The plasma microparticle proteome. Semin. Thromb. Hemost. 2010, 36, 845–856. [Google Scholar] [CrossRef]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Karpman, D.; Ståhl, A.L.; Arvidsson, I. Extracellular vesicles in renal disease. Nat. Rev. Nephrol. 2017, 13, 545–562. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga toxin (stx) classification, structure, and function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- McKee, M.L.; O’Brien, A.D. Investigation of enterohemorrhagic Escherichia coli o157:H7 adherence characteristics and invasion potential reveals a new attachment pattern shared by intestinal E. coli. Infect. Immun. 1995, 63, 2070–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr Infect Dis J. 2011, 30, 486–490. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Quinones, B.; Loo, M.T.; Loos, S.; Scavia, G.; Brigotti, M.; Levtchenko, E.; Monnens, L. Serum Shiga toxin 2 values in patients during acute phase of diarrhoea-associated haemolytic uraemic syndrome. Acta Paediatr. 2015, 104, e564–e568. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, A.L.; Sartz, L.; Nelsson, A.; Bekassy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS ONE 2009, 4, e6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, K.; Willysson, A.; Kristoffersson, A.C.; Tontanahal, A.; Gillet, D.; Ståhl, A.L.; Karpman, D. Shiga toxin-bearing microvesicles exert a cytotoxic effect on recipient cells only when the cells express the toxin receptor. Front Cell Infect. Microbiol. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Puri, A.; Hug, P.; Jernigan, K.; Rose, P.; Blumenthal, R. Role of glycosphingolipids in HIV-1 entry: Requirement of globotriosylceramide (Gb3) in CD4/CXCR4-dependent fusion. Biosci. Rep. 1999, 19, 317–325. [Google Scholar] [CrossRef]
- Mallard, F.; Antony, C.; Tenza, D.; Salamero, J.; Goud, B.; Johannes, L. Direct pathway from early/recycling endosomes to the golgi apparatus revealed through the study of Shiga toxin b-fragment transport. J. Cell Biol. 1998, 143, 973–990. [Google Scholar] [CrossRef]
- MacKenzie, A.; Wilson, H.L.; Kiss-Toth, E.; Dower, S.K.; North, R.A.; Surprenant, A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 2001, 15, 825–835. [Google Scholar] [CrossRef]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J. Cell Biol. 1989, 108, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Buzas, E.I.; Toth, E.A.; Sodar, B.W.; Szabo-Taylor, K.E. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.H.; Rothberg, K.G.; Anderson, R.G. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 1993, 123, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, N.; Murata, T.; Omata, N.; Takashima, Y.; Tanii, H.; Yonekura, Y.; Fujibayashi, Y.; Wada, Y. Effects of chlorpromazine on plasma membrane permeability and fluidity in the rat brain: A dynamic positron autoradiography and fluorescence polarization study. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2007, 31, 178–186. [Google Scholar] [CrossRef]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Castillo, M.D.; Tran, T.; Bobard, A.; Renard, H.F.; Rathjen, S.J.; Dransart, E.; Stechmann, B.; Lamaze, C.; Lord, M.; Cintrat, J.C.; et al. Retrograde transport is not required for cytosolic translocation of the b-subunit of Shiga toxin. J. Cell Sci. 2015, 128, 2373–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falguieres, T.; Romer, W.; Amessou, M.; Afonso, C.; Wolf, C.; Tabet, J.C.; Lamaze, C.; Johannes, L. Functionally different pools of Shiga toxin receptor, globotriaosyl ceramide, in HeLa cells. FEBS J. 2006, 273, 5205–5218. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Ruter, C.; Bauwens, A.; Greune, L.; Jarosch, K.A.; Steil, D.; Zhang, W.; He, X.; Lloubes, R.; Fruth, A.; et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli o157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017, 13, e1006159. [Google Scholar] [CrossRef]
- Ståhl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef] [Green Version]
- Thuresson, B.; Westman, J.S.; Olsson, M.L. Identification of a novel A4GALT exon reveals the genetic basis of the p1/p2 histo-blood groups. Blood 2011, 117, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Falguieres, T.; Mallard, F.; Baron, C.; Hanau, D.; Lingwood, C.; Goud, B.; Salamero, J.; Johannes, L. Targeting of Shiga toxin b-subunit to retrograde transport route in association with detergent-resistant membranes. Mol. Biol. Cell. 2001, 12, 2453–2468. [Google Scholar] [CrossRef] [Green Version]
- Nutikka, A.; Binnington-Boyd, B.; Lingwood, C.A. Methods for the identification of host receptors for Shiga toxin. Methods Mol. Med. 2003, 73, 197–208. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Shiga Toxin | Conjugate | Concentration | Media | Cells | Experiment | Detection Method |
|---|---|---|---|---|---|---|
| Stx1B | Alexa-488 | 1 μg/mL | DMEM 1 | HeLa | Incubation 0–30 min with cells | Live cell imaging |
| Stx1B | Alexa-488 | 10 ng/mL | DMEM 1 | RBC 2 | Incubation 0–15 min with RBC in suspension | Live cell imaging |
| Stx1B | Alexa-488 | 10 ng/mL | DMEM 3 | HeLa | Incubation 0–120 min with cells 4 | Flow cytometry |
| Stx1B | SS-biotin | 200 ng/mL | OptiMEM | HeLa | Incubation with cells for 40 min 4 | Stx1B-ELISA |
| Stx2 | - | 200 ng/mL | HeLa | Gb3 5 overlay on extracted membrane lipids | Thin layer chromatography | |
| Stx1B | Nanogold | 200 ng/mL | DMEM | Blood | Extracellular vesicles incubated for 1 h 4 | Electron microscopy |
| Stx1B 6 | SS-biotin | 1 µg/mL | OptiMEM or DMEM 7 | HeLa and blood cells | Extracellular vesicles incubated for 1 h 4 | ELISA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willysson, A.; Ståhl, A.-l.; Gillet, D.; Barbier, J.; Cintrat, J.-C.; Chambon, V.; Billet, A.; Johannes, L.; Karpman, D. Shiga Toxin Uptake and Sequestration in Extracellular Vesicles Is Mediated by Its B-Subunit. Toxins 2020, 12, 449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070449
Willysson A, Ståhl A-l, Gillet D, Barbier J, Cintrat J-C, Chambon V, Billet A, Johannes L, Karpman D. Shiga Toxin Uptake and Sequestration in Extracellular Vesicles Is Mediated by Its B-Subunit. Toxins. 2020; 12(7):449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070449
Chicago/Turabian StyleWillysson, Annie, Anne-lie Ståhl, Daniel Gillet, Julien Barbier, Jean-Christophe Cintrat, Valérie Chambon, Anne Billet, Ludger Johannes, and Diana Karpman. 2020. "Shiga Toxin Uptake and Sequestration in Extracellular Vesicles Is Mediated by Its B-Subunit" Toxins 12, no. 7: 449. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12070449