Scalable Reporter Assays to Analyze the Regulation of stx2 Expression in Shiga Toxin-Producing Enteropathogens

 , ,
, ,
Abstract
:1. Introduction
2. Results
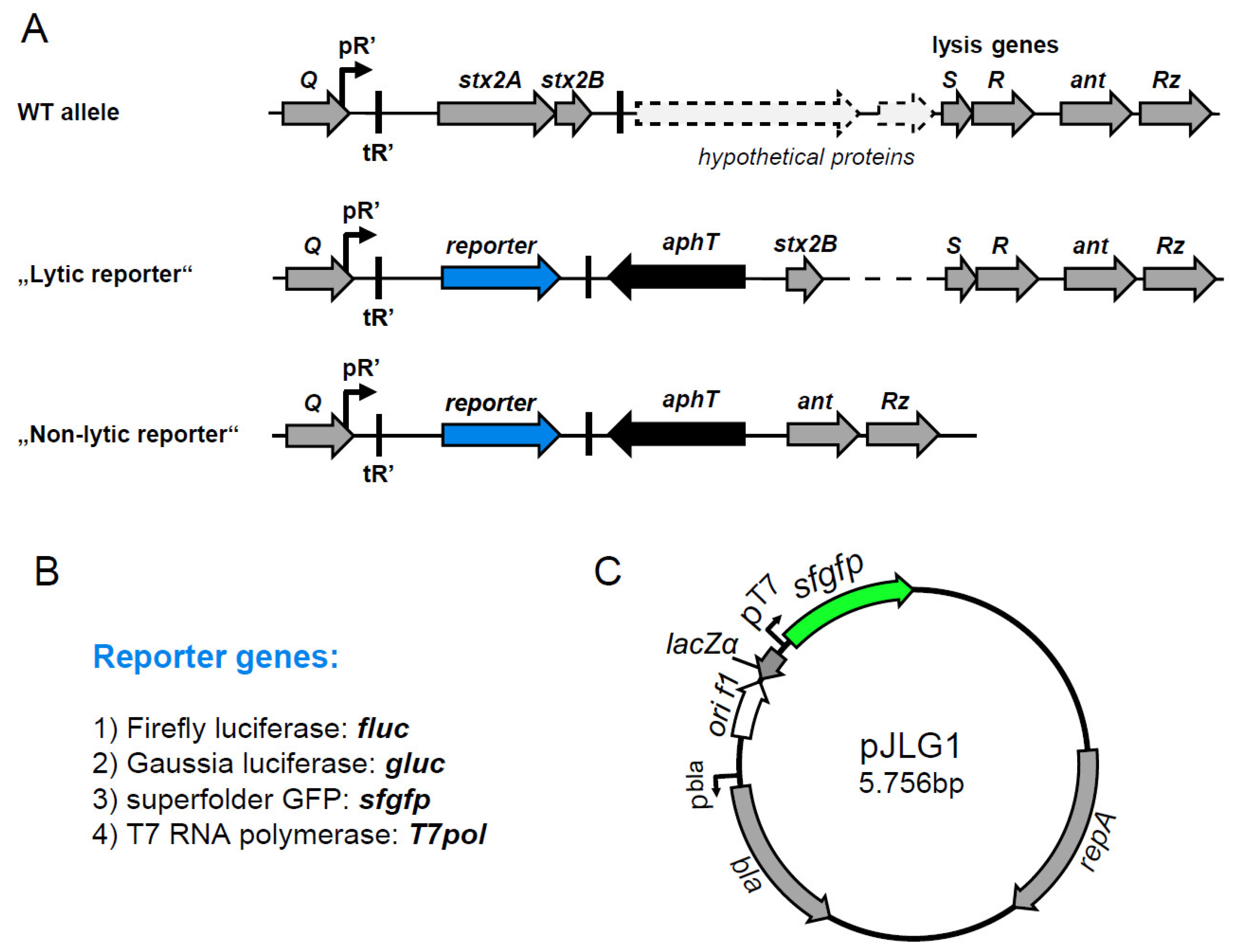
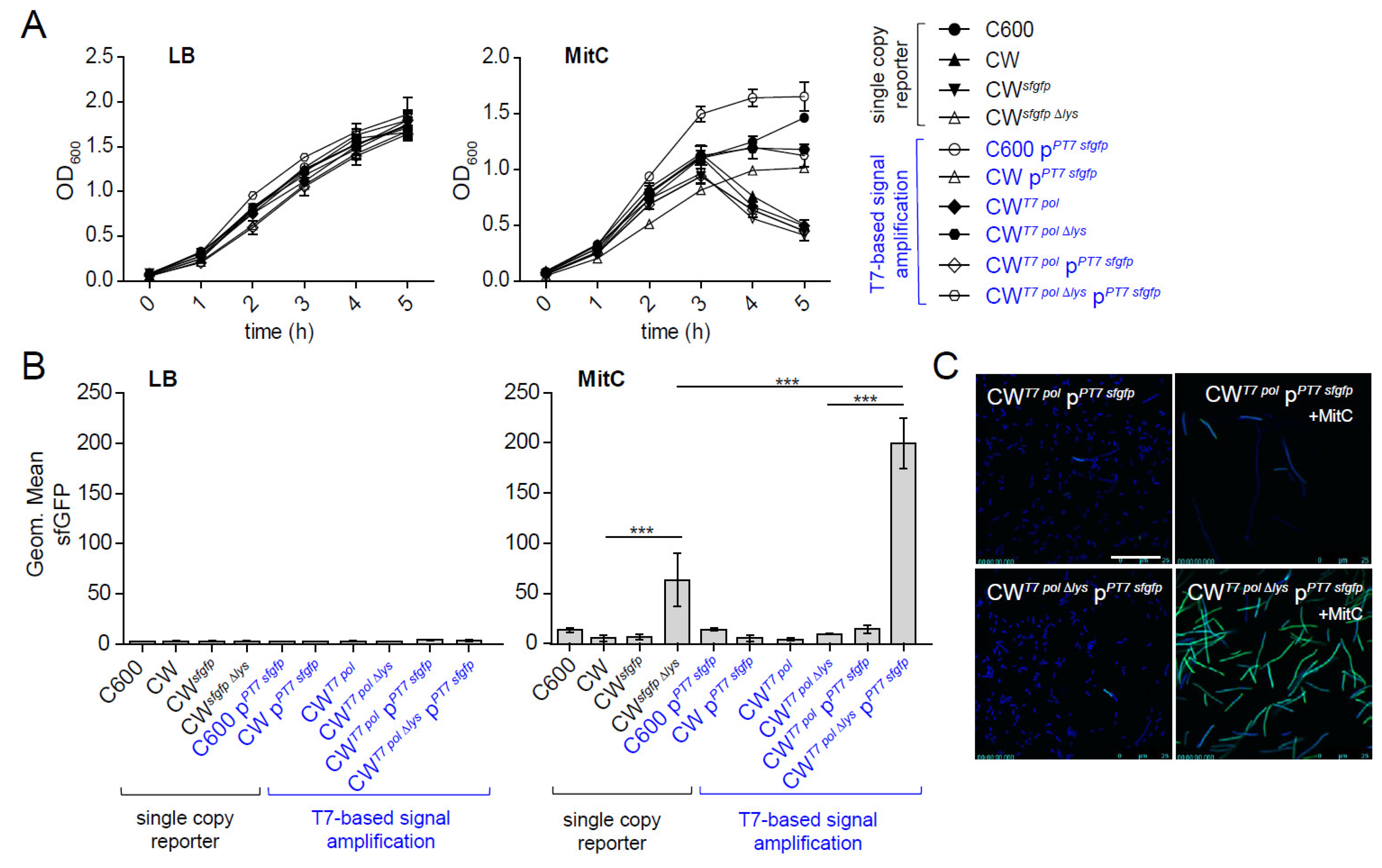
2.1. Generation of a Signal-Amplified sfgfp Reporter to Monitor stx2AB Expression at the Single-Cell Level
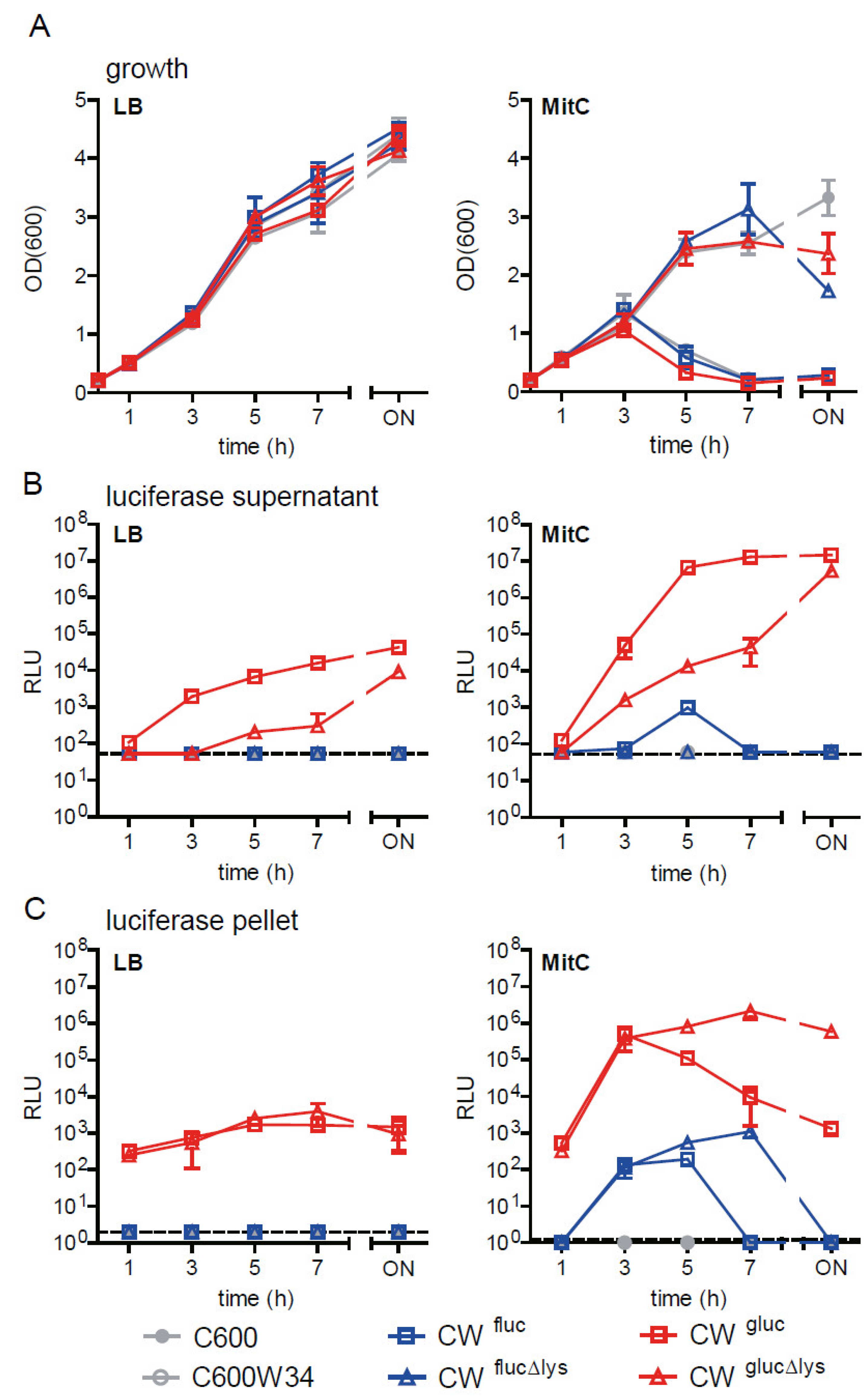
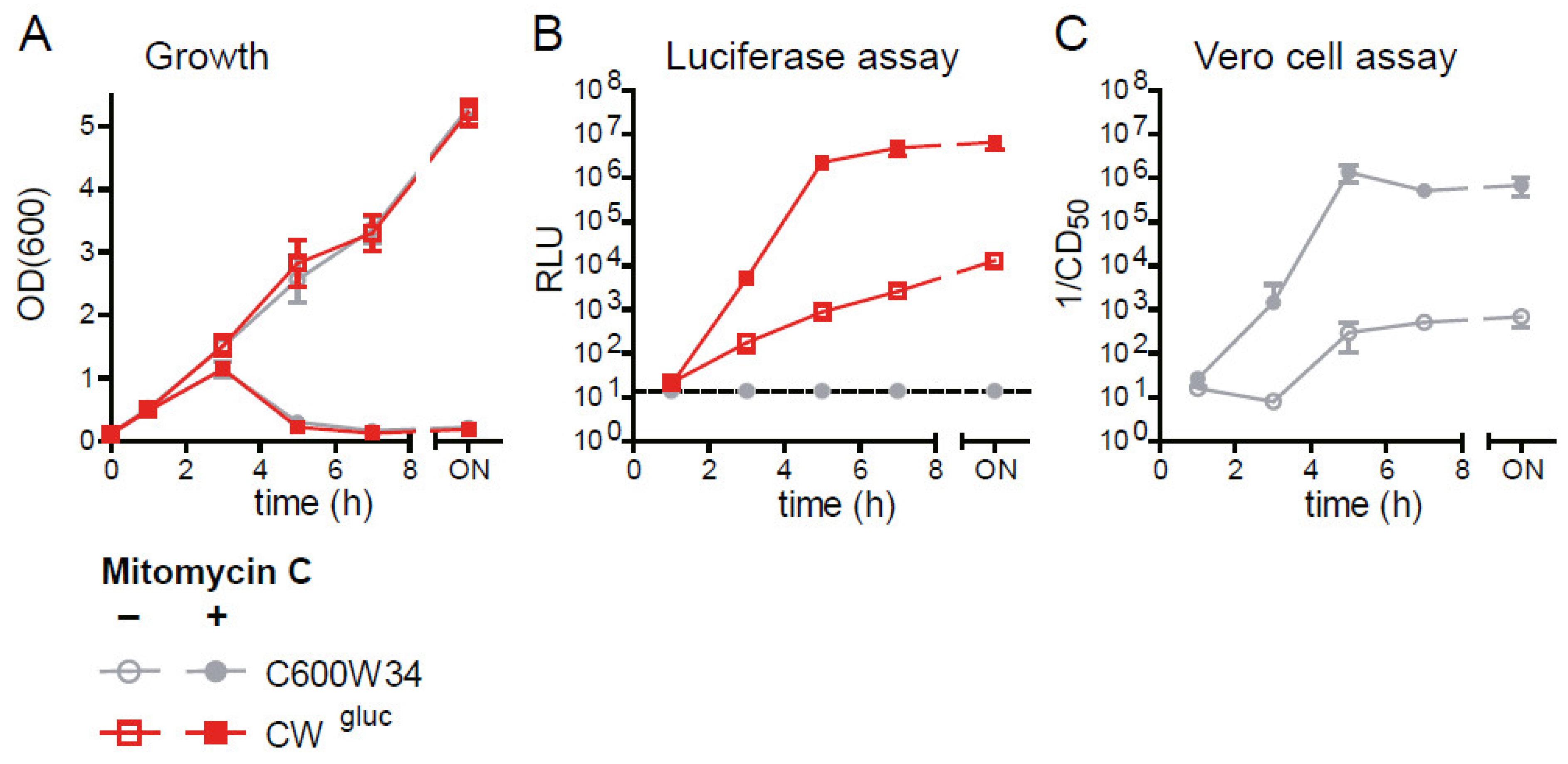
2.2. Comparison of Fluc and Gluc luciferase Reporter Strains to Monitor stx2AB Induction and Φstx-Induced Lysis as a Proxy for Stx2 Release
2.3. The Lytic Gluc Reporter Mirrors Kinetics of Stx Release from E. coli
2.4. The Gluc Reporter Assay for Φstx-Induced Lysis Can Be Scaled Up to a 96-Well Format
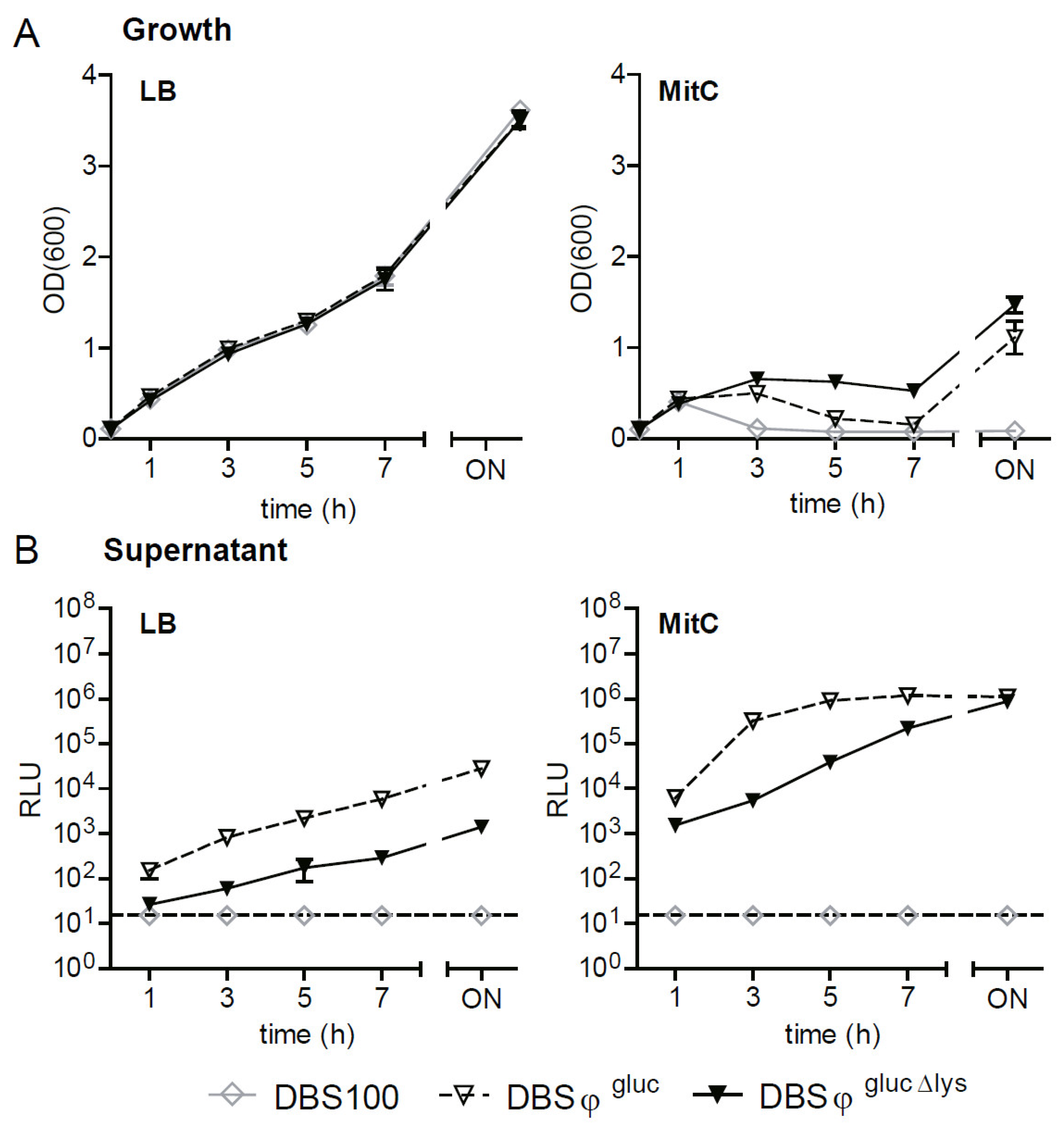
2.5. Generation of BSL-2 Gluc Reporter Strains in a BSL-3** Pathogen
3. Discussion
4. Materials and Methods
4.1. Generation of Bacterial Mutant Strains and Plasmids
4.2. Bacterial Growth Conditions
4.3. Luciferase Assays
4.4. Preparation of Samples for Confocal Fluorescence Microscopy
4.5. Flow Cytometry
4.6. Vero Cell Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Michino, H.; Araki, K.; Minami, S.; Takaya, S.; Sakai, N.; Miyazaki, M.; Ono, A.; Yanagawa, H. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am. J. Epidemiol. 1999, 150, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.S.; Mooney, J.C.; Brandt, J.R.; Staples, A.O.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Tarr, P.I. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: A multivariable analysis. Clin. Infect. Dis. 2012, 55, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Tserenpuntsag, B.; Chang, H.G.; Smith, P.F.; Morse, D.L. Hemolytic uremic syndrome risk and Escherichia coli O157:H7. Emerg. Infect. Dis. 2005, 11, 1955–1957. [Google Scholar] [CrossRef]
- Wong, C.S.; Jelacic, S.; Habeeb, R.L.; Watkins, S.L.; Tarr, P.I. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 2000, 342, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Shiga toxin-induced haemolytic uraemic syndrome and the role of antibiotics: A global overview. J. Infect. 2019, 79, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Kock, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Kitov, P.I.; Sadowska, J.M.; Bundle, D.R. Discovery of inhibitors of Shiga toxin type 2 by on-plate generation and screening of a focused compound library. Angew. Chem. Int. Ed. Engl. 2014, 53, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R.; O’Brien, A.D. New Therapeutic Developments against Shiga Toxin-Producing Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Tu, W.; Liu, Y.; Zhou, P.; Cai, K.; Li, Z.; Liu, X.; Ning, N.; Huang, J.; Wang, S.; et al. A potential therapeutic peptide-based neutralizer that potently inhibits Shiga toxin 2 in vitro and in vivo. Sci. Rep. 2016, 6, 21837. [Google Scholar] [CrossRef] [Green Version]
- Luz, D.; Amaral, M.M.; Sacerdoti, F.; Bernal, A.M.; Quintilio, W.; Moro, A.M.; Palermo, M.S.; Ibarra, C.; Piazza, R.M.F. Human Recombinant Fab Fragment Neutralizes Shiga Toxin Type 2 Cytotoxic Effects in vitro and in vivo. Toxins (Basel) 2018, 10, 508. [Google Scholar] [CrossRef] [Green Version]
- Kavaliauskiene, S.; Dyve Lingelem, A.B.; Skotland, T.; Sandvig, K. Protection against Shiga Toxins. Toxins (Basel) 2017, 9, 44. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef]
- Bergan, J.; Dyve Lingelem, A.B.; Simm, R.; Skotland, T.; Sandvig, K. Shiga toxins. Toxicon 2012, 60, 1085–1107. [Google Scholar] [CrossRef]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisen, N.; Melton-Celsa, A.R.; Scheutz, F.; O’Brien, A.D.; Nataro, J.P. Shiga toxin 2a and Enteroaggregative Escherichia coli—A deadly combination. Gut Microbes 2015, 6, 272–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, B.K.; Wasteson, Y.; Granum, P.E.; Brynestad, S. Mosaic structure of Shiga-toxin-2-encoding phages isolated from Escherichia coli O157:H7 indicates frequent gene exchange between lambdoid phage genomes. Microbiology 2001, 147, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Neely, M.N.; Friedman, D.I. Functional and genetic analysis of regulatory regions of coliphage H-19B: Location of shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol. Microbiol. 1998, 28, 1255–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, J.M.; Los, M.; Wegrzyn, A.; Wegrzyn, G. Hydrogen peroxide-mediated induction of the Shiga toxin-converting lambdoid prophage ST2-8624 in Escherichia coli O157:H7. FEMS Immunol. Med. Microbiol. 2010, 58, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; McDaniel, A.D.; Wolf, L.E.; Keusch, G.T.; Waldor, M.K.; Acheson, D.W. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 2000, 181, 664–670. [Google Scholar] [CrossRef]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielaszewska, M.; Idelevich, E.A.; Zhang, W.; Bauwens, A.; Schaumburg, F.; Mellmann, A.; Peters, G.; Karch, H. Effects of antibiotics on Shiga toxin 2 production and bacteriophage induction by epidemic Escherichia coli O104:H4 strain. Antimicrob. Agents Chemother. 2012, 56, 3277–3282. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, A.R.; Sperandio, V. Shiga toxin in enterohemorrhagic E.coli: Regulation and novel anti-virulence strategies. Front. Cell Infect. Microbiol. 2012, 2, 81. [Google Scholar] [CrossRef] [Green Version]
- Rasko, D.A.; Moreira, C.G.; Li de, R.; Reading, N.C.; Ritchie, J.M.; Waldor, M.K.; Williams, N.; Taussig, R.; Wei, S.; Roth, M.; et al. Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321, 1078–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicki, D.; Rodzik, O.; Herman-Antosiewicz, A.; Szalewska-Palasz, A. Isothiocyanates as effective agents against enterohemorrhagic Escherichia coli: Insight to the mode of action. Sci. Rep. 2016, 6, 22263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pifer, R.; Sperandio, V. The Interplay between the Microbiota and Enterohemorrhagic Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Licznerska, K.; Dydecka, A.; Bloch, S.; Topka, G.; Nejman-Falenczyk, B.; Wegrzyn, A.; Wegrzyn, G. The Role of the Exo-Xis Region in Oxidative Stress-Mediated Induction of Shiga Toxin-Converting Prophages. Oxid. Med. Cell. Longev. 2016, 2016, 8453135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Ohta, Y.; Tsutsuki, H.; Noda, M. Construction of a novel bioluminescent reporter system for investigating Shiga toxin expression of enterohemorrhagic Escherichia coli. Gene 2011, 478, 1–10. [Google Scholar] [CrossRef]
- Livny, J.; Friedman, D.I. Characterizing spontaneous induction of Stx encoding phages using a selectable reporter system. Mol. Microbiol. 2004, 51, 1691–1704. [Google Scholar] [CrossRef] [Green Version]
- Tyler, J.S.; Beeri, K.; Reynolds, J.L.; Alteri, C.J.; Skinner, K.G.; Friedman, J.H.; Eaton, K.A.; Friedman, D.I. Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog. 2013, 9, e1003236. [Google Scholar] [CrossRef] [Green Version]
- Pedelacq, J.D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Tannous, B.A.; Kim, D.E.; Fernandez, J.L.; Weissleder, R.; Breakefield, X.O. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 2005, 11, 435–443. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; Newland, J.W.; Miller, S.F.; Holmes, R.K.; Smith, H.W.; Formal, S.B. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef]
- Spriewald, S.; Stadler, E.; Hense, B.A.; Munch, P.C.; McHardy, A.C.; Weiss, A.S.; Obeng, N.; Muller, J.; Stecher, B. Evolutionary Stabilization of Cooperative Toxin Production through a Bacterium-Plasmid-Phage Interplay. mBio 2020, 11. [Google Scholar] [CrossRef]
- Konowalchuk, J.; Speirs, J.I.; Stavric, S. Vero response to a cytotoxin of Escherichia coli. Infect. Immun. 1977, 18, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Mallick, E.M.; McBee, M.E.; Vanguri, V.K.; Melton-Celsa, A.R.; Schlieper, K.; Karalius, B.J.; O’Brien, A.D.; Butterton, J.R.; Leong, J.M.; Schauer, D.B. A novel murine infection model for Shiga toxin-producing Escherichia coli. J. Clin. Investig. 2012, 122, 4012–4024. [Google Scholar] [CrossRef] [PubMed]
- Lenz, A.; Tomkins, J.; Fabich, A.J. Draft Genome Sequence of Citrobacter rodentium DBS100 (ATCC 51459), a Primary Model of Enterohemorrhagic Escherichia coli Virulence. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Muhlen, S.; Ramming, I.; Pils, M.C.; Koeppel, M.; Glaser, J.; Leong, J.; Flieger, A.; Stecher, B.; Dersch, P. Identification of Antibiotics That Diminish Disease in a Murine Model of Enterohemorrhagic Escherichia coli Infection. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Magaziner, S.J.; Zeng, Z.; Chen, B.; Salmond, G.P.C. The Prophages of Citrobacter rodentium Represent a Conserved Family of Horizontally Acquired Mobile Genetic Elements Associated with Enteric Evolution towards Pathogenicity. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, R.H.; Falkow, S. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 1997, 277, 2007–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunstan, S.J.; Simmons, C.P.; Strugnell, R.A. Use of in vivo-regulated promoters to deliver antigens from attenuated Salmonella enterica var. Typhimurium. Infect. Immun. 1999, 67, 5133–5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviv, G.; Gal-Mor, O. lacZ Reporter System as a Tool to Study Virulence Gene Regulation in Bacterial Pathogens. Methods Mol. Biol. 2018, 1734, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Tyler, J.S.; Mills, M.J.; Friedman, D.I. The operator and early promoter region of the Shiga toxin type 2-encoding bacteriophage 933W and control of toxin expression. J. Bacteriol. 2004, 186, 7670–7679. [Google Scholar] [CrossRef] [Green Version]
- Suardana, I.W.; Pinatih, K.J.P.; Widiasih, D.A.; Artama, W.T.; Asmara, W.; Daryono, B.S. Regulatory elements of stx2 gene and the expression level of Shiga-like toxin 2 in Escherichia coli O157:H7. J. Microbiol. Immunol. Infect. 2018, 51, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, D.I.; Court, D.L. Transcription antitermination: The lambda paradigm updated. Mol. Microbiol. 1995, 18, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Spriewald, S.; Glaser, J.; Beutler, M.; Koeppel, M.B.; Stecher, B. Reporters for Single-Cell Analysis of Colicin Ib Expression in Salmonella enterica Serovar Typhimurium. PLoS ONE 2015, 10, e0144647. [Google Scholar] [CrossRef]
- Aertsen, A.; Van Houdt, R.; Michiels, C.W. Construction and use of an stx1 transcriptional fusion to gfp. FEMS Microbiol. Lett. 2005, 245, 73–77. [Google Scholar] [CrossRef]
- Huerta-Uribe, A.; Marjenberg, Z.R.; Yamaguchi, N.; Fitzgerald, S.; Connolly, J.P.; Carpena, N.; Uvell, H.; Douce, G.; Elofsson, M.; Byron, O.; et al. Identification and Characterization of Novel Compounds Blocking Shiga Toxin Expression in Escherichia coli O157:H7. Front. Microbiol. 2016, 7, 1930. [Google Scholar] [CrossRef] [Green Version]
- Wagner, P.L.; Livny, J.; Neely, M.N.; Acheson, D.W.; Friedman, D.I.; Waldor, M.K. Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol. Microbiol. 2002, 44, 957–970. [Google Scholar] [CrossRef]
- Calderwood, S.B.; Mekalanos, J.J. Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. J. Bacteriol. 1987, 169, 4759–4764. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.; Aijaz, I.; Berger, P.; Dobrindt, U.; Koudelka, G. Transcriptional and Translational Inhibitors Block SOS Response and Shiga Toxin Expression in Enterohemorrhagic Escherichia coli. Sci. Rep. 2019, 9, 18777. [Google Scholar] [CrossRef]
- McGannon, C.M.; Fuller, C.A.; Weiss, A.A. Different classes of antibiotics differentially influence shiga toxin production. Antimicrob. Agents Chemother. 2010, 54, 3790–3798. [Google Scholar] [CrossRef] [Green Version]
- Wiles, S.; Ferguson, K.; Stefanidou, M.; Young, D.B.; Robertson, B.D. Alternative luciferase for monitoring bacterial cells under adverse conditions. Appl. Environ. Microbiol. 2005, 71, 3427–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, Y.; Mondal, S.I.; Islam, M.R.; Mako, T.; Arisawa, K.; Katsura, K.; Ooka, T.; Gotoh, Y.; Murase, K.; Ohnishi, M.; et al. The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 2015, 5, 16663. [Google Scholar] [CrossRef] [Green Version]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, A.K.; Leatham, M.P.; Conway, T.; Nuijten, P.J.; de Haan, L.A.; Krogfelt, K.A.; Cohen, P.S. An Escherichia coli MG1655 lipopolysaccharide deep-rough core mutant grows and survives in mouse cecal mucus but fails to colonize the mouse large intestine. Infect. Immun. 2003, 71, 2142–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleyard, R.K. Segregation of New Lysogenic Types during Growth of a Doubly Lysogenic Strain Derived from Escherichia coli K12. Genetics 1954, 39, 440–452. [Google Scholar] [CrossRef]
- Schauer, D.B.; Zabel, B.A.; Pedraza, I.F.; O’Hara, C.M.; Steigerwalt, A.G.; Brenner, D.J. Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J. Clin. Microbiol. 1995, 33, 2064–2068. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, R.G.; Holzer, S.U.; Jackel, D.; Hensel, M. Rapid engineering of bacterial reporter gene fusions by using Red recombination. Appl. Environ. Microbiol. 2007, 73, 4234–4242. [Google Scholar] [CrossRef] [Green Version]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [Green Version]
- Mirold, S.; Rabsch, W.; Rohde, M.; Stender, S.; Tschape, H.; Russmann, H.; Igwe, E.; Hardt, W.D. Isolation of a temperate bacteriophage encoding the type III effector protein SopE from an epidemic Salmonella typhimurium strain. Proc. Natl. Acad. Sci. USA 1999, 96, 9845–9850. [Google Scholar] [CrossRef] [Green Version]
- Wille, T.; Blank, K.; Schmidt, C.; Vogt, V.; Gerlach, R.G. Gaussia princeps luciferase as a reporter for transcriptional activity, protein secretion, and protein-protein interactions in Salmonella enterica serovar typhimurium. Appl. Environ. Microbiol. 2012, 78, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.F.; Kushner, S.R. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 1991, 100, 195–199. [Google Scholar] [CrossRef]
- Gentry, M.K.; Dalrymple, J.M. Quantitative microtiter cytotoxicity assay for Shigella toxin. J. Clin. Microbiol. 1980, 12, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.L.; Billoud, L.; Lewis, S.B.; Phillips, A.D.; Schuller, S. Shiga toxin production and translocation during microaerobic human colonic infection with Shiga toxin-producing E. coli O157:H7 and O104:H4. Cell. Microbiol. 2014, 16, 1255–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli Strains | Designation | Description/Genotype | Reference |
|---|---|---|---|
| DH5α | EcDH5α | F− Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17(rk−, mk+) phoA supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| MG1655 | EcMG1655 | E. coli K-12 strain MG1655, F− λ− ilvG− rfb-50 rph-1; RifR, SmR | [55,56] |
| C600 | C600 | [57] | |
| C600W34 | C600W34 | E. coli C600 transduced with phage 933W from EHEC strain EDL933 | [33] |
| MBK1 | CW sfgfp | C600W34 stx2A::sfgfp aphT | This study |
| MBK4 | CW sfgfpΔlys | C600W34 stx2AB SR::sfgfp aphT, phage lysis-deficient strain | This study |
| MBK6 | CW fluc | C600W34 stx2A::fluc aphT | This study |
| MKB7 | CW flucΔlys | C600W34 stx2AB SR::fluc aphT, phage lysis-deficient strain | This study |
| JLG5 | CW gluc | C600W34 stx2A::gluc aphT | [38] |
| JLG6 | CW glucΔlys | C600W34 stx2AB SR::gluc aphT, phage lysis-deficient strain | This study |
| JLG11 | CW T7pol | C600W34 stx2A::T7pol aphT | This study |
| JLG12 | CW T7polΔlys | C600W34 stx2AB SR::T7pol aphT, phage lysis-deficient strain | This study |
| C. rodentium Strains | |||
| DBS100 | DBS100 | Citrobacter rodentium wild-type strain | [58] |
| DBS770 | DBSφ | C. rodentium transduced with phage stx2φ17220 | [36] |
| MBK22 | DBSφ glucΔlys | DBS770 stx2AB SR::gluc aphT, phage lysis-deficient strain | This study |
| MBK23 | DBSφ gluc | DBS770 stx2A::gluc aphT | [38] |
| Plasmids | |||
| p3121 | High-copy vector, colE1-replicon; carries firefly luc aphT flanked by FRT sequences; ampicillin resistance | [59] | |
| pKD46 | Temperature-sensitive replication (repA101ts); encodes λ-Red genes (exo, bet, gam); native terminator (tL3) after exo gene; arabinose-inducible promoter for expression (ParaB); encodes araC for repression of ParaB promoter; ampicillin resistance | [60] | |
| pMBK3 | pSB377stx2AB SR Cm::gluc,aphT | This study | |
| pMBK4 | pSB377stx2aA::gluc,aphT | [38] | |
| pSB377 | tetAB oriR6K | [61] | |
| pJLG1 | pM955, PT7::sfgfp Amp100 | [34] | |
| pJLG2 | p2795, T7 gene 1 aphT Amp100, Kan30 | [34] | |
| pACYC184 | New England Biolabs | ||
| pWRG7 | High-copy vector, colE1-replicon; carries sfgfp aphT flanked by FRT sequences; ampicillin resistance | [46] | |
| pWRG215 | High-copy vector, colE1-replicon; carries Gaussia luciferase (flash kinetics) GlucM43 aphT flanked by FRT sequences; ampicillin resistance | [62] | |
| pWRG701 | High-copy vector, colE1-replicon; carries Gaussia luciferase (glow kinetics) GlucM43LM110L aphT flanked by FRT sequences; ampicillin resistance | [38] | |
| pWKS30 | Low-copy vector; pSC101-based replicon; ampicillin-resistance marker | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koeppel, M.B.; Glaser, J.; Baumgartner, T.; Spriewald, S.; Gerlach, R.G.; von Armansperg, B.; Leong, J.M.; Stecher, B. Scalable Reporter Assays to Analyze the Regulation of stx2 Expression in Shiga Toxin-Producing Enteropathogens. Toxins 2021, 13, 534. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080534
Koeppel MB, Glaser J, Baumgartner T, Spriewald S, Gerlach RG, von Armansperg B, Leong JM, Stecher B. Scalable Reporter Assays to Analyze the Regulation of stx2 Expression in Shiga Toxin-Producing Enteropathogens. Toxins. 2021; 13(8):534. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080534
Chicago/Turabian StyleKoeppel, Martin B., Jana Glaser, Tobias Baumgartner, Stefanie Spriewald, Roman G. Gerlach, Benedikt von Armansperg, John M. Leong, and Bärbel Stecher. 2021. "Scalable Reporter Assays to Analyze the Regulation of stx2 Expression in Shiga Toxin-Producing Enteropathogens" Toxins 13, no. 8: 534. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080534