Okadaic Acid Is at Least as Toxic as Dinophysistoxin-1 after Repeated Administration to Mice by Gavage



Abstract
:1. Introduction
2. Results and Discussion
2.1. Acute Oral Toxicity Study of OA and DTX-1
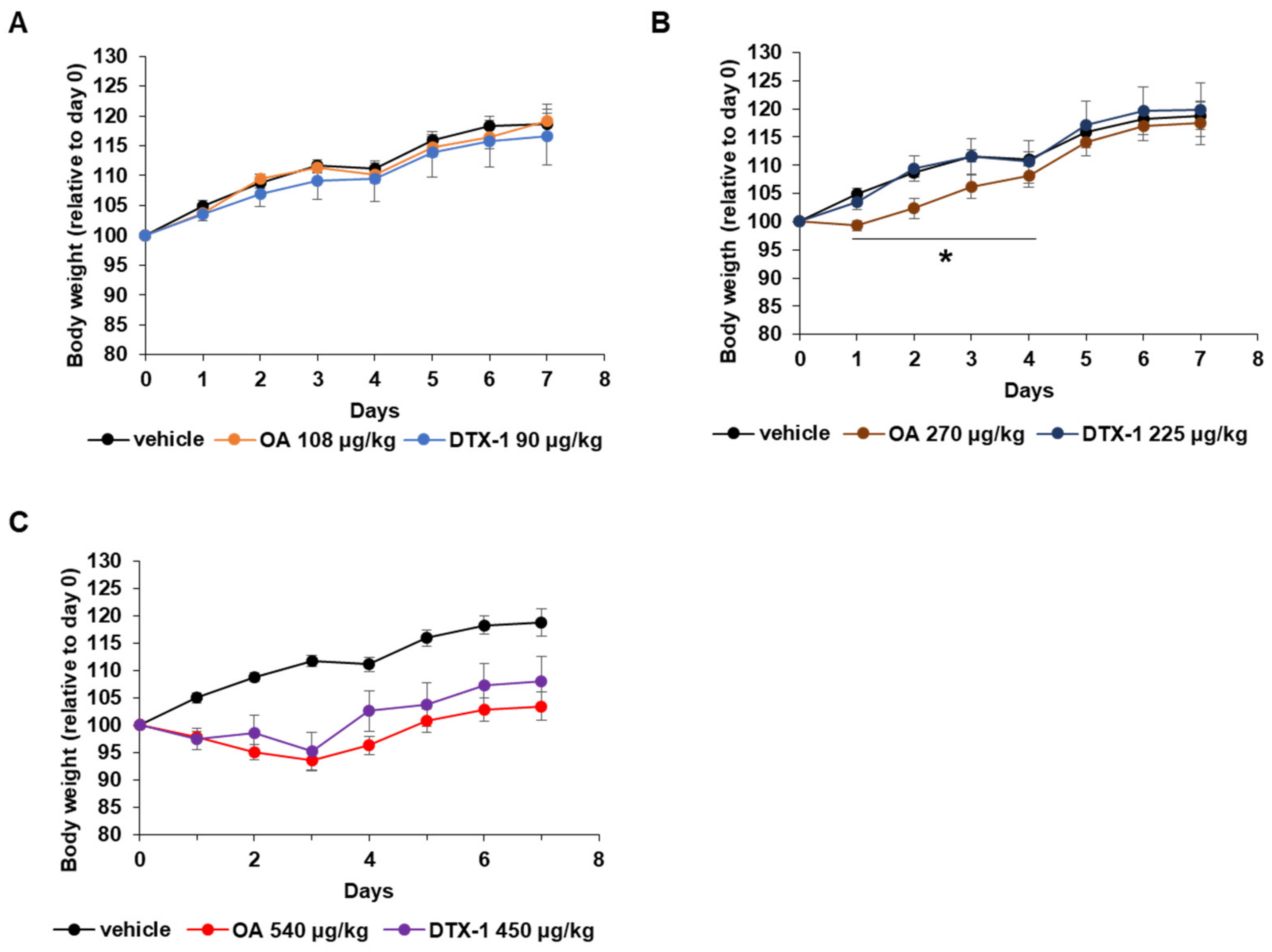
2.2. Physiological Changes during the Repeated Oral Dose Study
2.3. Pathological Changes in the Small Intestine after Repeated Oral Doses of OA and DTX-1
2.4. Other Toxic Effects of Repeated Oral OA and DTX-1 Administrations
3. Conclusions
4. Materials and Methods
4.1. Diarrhetic Shellfish Poisoning Toxins
4.2. Animals
4.3. Acute Oral Dose Toxicity Study
4.4. Repeated Oral Dose Toxicity Study
4.5. Histological Evaluation
4.6. In Vitro Experiment
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nielsen, L.T.; Hansen, P.J.; Krock, B.; Vismann, B. Accumulation, transformation and breakdown of DSP toxins from the toxic dinoflagellate Dinophysis acuta in blue mussels, Mytilus edulis. Toxicon 2016, 117, 84–93. [Google Scholar] [CrossRef]
- Suarez-Gomez, B.; Souto, M.L.; Norte, M.; Fernandez, J.J. Isolation and structural determination of DTX-6, a new okadaic acid derivative. J. Nat. Prod. 2001, 64, 1363–1364. [Google Scholar] [CrossRef]
- Morris, J.G. 286—Human Illness Associated with Harmful Algal Blooms. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 3192–3195. [Google Scholar]
- Takai, A.; Murata, M.; Torigoe, K.; Isobe, M.; Mieskes, G.; Yasumoto, T. Inhibitory effect of okadaic acid derivatives on protein phosphatases. A study on structure-affinity relationship. Biochem. J. 1992, 284 Pt 2, 539–544. [Google Scholar] [CrossRef]
- Yadav, L.; Tamene, F.; Goos, H.; van Drogen, A.; Katainen, R.; Aebersold, R.; Gstaiger, M.; Varjosalo, M. Systematic Analysis of Human Protein Phosphatase Interactions and Dynamics. Cell Syst. 2017, 4, 430–444.e435. [Google Scholar] [CrossRef]
- Reynhout, S.; Janssens, V. Physiologic functions of PP2A: Lessons from genetically modified mice. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 31–50. [Google Scholar] [CrossRef]
- Dzulko, M.; Pons, M.; Henke, A.; Schneider, G.; Kramer, O.H. The PP2A subunit PR130 is a key regulator of cell development and oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188453. [Google Scholar] [CrossRef]
- Verbinnen, I.; Vaneynde, P.; Reynhout, S.; Lenaerts, L.; Derua, R.; Houge, G.; Janssens, V. Protein Phosphatase 2A (PP2A) mutations in brain function, development, and neurologic disease. Biochem. Soc. Trans. 2021, 49, 1567–1588. [Google Scholar] [CrossRef]
- Messner, D.J.; Romeo, C.; Boynton, A.; Rossie, S. Inhibition of PP2A, but not PP5, mediates p53 activation by low levels of okadaic acid in rat liver epithelial cells. J. Cell. Biochem. 2006, 99, 241–255. [Google Scholar] [CrossRef]
- Fujiki, H.; Sueoka, E.; Watanabe, T.; Komori, A.; Suganuma, M. Cancer progression by the okadaic acid class of tumor promoters and endogenous protein inhibitors of PP2A, SET and CIP2A. J. Cancer Res. Clin. Oncol. 2023, 149, 9425–9433. [Google Scholar] [CrossRef]
- Ikehara, T.; Imamura, S.; Yoshino, A.; Yasumoto, T. PP2A inhibition assay using recombinant enzyme for rapid detection of okadaic acid and its analogs in shellfish. Toxins 2010, 2, 195–204. [Google Scholar] [CrossRef]
- Abal, P.; Louzao, M.C.; Suzuki, T.; Watanabe, R.; Vilarino, N.; Carrera, C.; Botana, A.M.; Vieytes, M.R.; Botana, L.M. Toxic Action Reevaluation of Okadaic Acid, Dinophysistoxin-1 and Dinophysistoxin-2: Toxicity Equivalency Factors Based on the Oral Toxicity Study. Cell. Physiol. Biochem. 2018, 49, 743–757. [Google Scholar] [CrossRef]
- Munday, R. Is protein phosphatase inhibition responsible for the toxic effects of okadaic Acid in animals? Toxins 2013, 5, 267–285. [Google Scholar] [CrossRef]
- Seth, A.; Sheth, P.; Elias, B.C.; Rao, R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J. Biol. Chem. 2007, 282, 11487–11498. [Google Scholar] [CrossRef]
- Dunagan, M.; Chaudhry, K.; Samak, G.; Rao, R.K. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1356–G1364. [Google Scholar] [CrossRef]
- Louzao, M.C.; Fernandez, D.A.; Abal, P.; Fraga, M.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Diarrhetic effect of okadaic acid could be related with its neuronal action: Changes in neuropeptide Y. Toxicol. Lett. 2015, 237, 151–160. [Google Scholar] [CrossRef]
- Louzao, M.C.; Costas, C.; Abal, P.; Suzuki, T.; Watanabe, R.; Vilarino, N.; Carrera, C.; Boente-Juncal, A.; Vale, C.; Vieytes, M.R.; et al. Serotonin involvement in okadaic acid-induced diarrhoea in vivo. Arch. Toxicol. 2021, 95, 2797–2813. [Google Scholar] [CrossRef]
- Botana, L.M.; Hess, P.; Munday, R.; Nathalie, A.; DeGrasse, S.L.; Feeley, M.; Suzuki, T.; van den Berg, M.; Fattori, V.; Garrido Gamarro, E.; et al. Derivation of toxicity equivalency factors for marine biotoxins associated with Bivalve Molluscs. Trends Food Sci. Technol. 2017, 59, 15–24. [Google Scholar] [CrossRef]
- EFSA. Scientific Opinion of the Panel on Biological Hazards on a request from DG SANCO on the assessment of the possible effect of the four antimicrobial treatment substances on the emergence of antimicrobial resistance. EFSA J. 2008, 6, 659. [Google Scholar] [CrossRef]
- World Health Organization. Toxicity Equivalence Factors for Marine Biotoxins Associated with Bivalve Molluscs; World Health Organization: Geneva, Switzerland, 2016.
- Garcia, C.; Truan, D.; Lagos, M.; Santelices, J.P.; Diaz, J.C.; Lagos, N. Metabolic transformation of dinophysistoxin-3 into dinophysistoxin-1 causes human intoxication by consumption of O-acyl-derivatives dinophysistoxins contaminated shellfish. J. Toxicol. Sci. 2005, 30, 287–296. [Google Scholar] [CrossRef]
- Abal, P.; Louzao, M.C.; Cifuentes, J.M.; Vilarino, N.; Rodriguez, I.; Alfonso, A.; Vieytes, M.R.; Botana, L.M. Characterization of the dinophysistoxin-2 acute oral toxicity in mice to define the Toxicity Equivalency Factor. Food Chem. Toxicol. 2017, 102, 166–175. [Google Scholar] [CrossRef]
- Louzao, M.C.; Abal, P.; Costas, C.; Suzuki, T.; Watanabe, R.; Vilarino, N.; Botana, A.M.; Vieytes, M.R.; Botana, L.M. DSP Toxin Distribution across Organs in Mice after Acute Oral Administration. Mar. Drugs 2021, 19, 23. [Google Scholar] [CrossRef]
- Ito, E.; Yasumoto, T.; Takai, A.; Imanishi, S.; Harada, K. Investigation of the distribution and excretion of okadaic acid in mice using immunostaining method. Toxicon 2002, 40, 159–165. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Carbonatto, M.; Altinier, G.; Vita, F.; Melato, M.; Satake, M.; Yasumoto, T. Oral and intraperitoneal acute toxicity studies of yessotoxin and homoyessotoxins in mice. Toxicon 2003, 41, 783–792. [Google Scholar] [CrossRef]
- Ogino, H.; Kumagai, M.; Yasumoto, T. Toxicologic evaluation of yessotoxin. Nat. Toxins 1997, 5, 255–259. [Google Scholar] [CrossRef]
- Ito, E.; Terao, K. Injury and recovery process of intestine caused by okadaic acid and related compounds. Nat. Toxins 1994, 2, 371–377. [Google Scholar]
- Le Hégarat, L.; Jacquin, A.G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar] [CrossRef]
- Dewi, S.; Aune, T.; Bunaes, J.A.; Smith, A.J.; Larsen, S. The development of response surface pathway design to reduce animal numbers in toxicity studies. BMC Pharmacol. Toxicol. 2014, 15, 18. [Google Scholar] [CrossRef]
- Ferron, P.J.; Hogeveen, K.; Fessard, V.; Le Hegarat, L. Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar. Drugs 2014, 12, 4616–4634. [Google Scholar] [CrossRef]
- Solino, L.; Sureda, F.X.; Diogene, J. Evaluation of okadaic acid, dinophysistoxin-1 and dinophysistoxin-2 toxicity on Neuro-2a, NG108-15 and MCF-7 cell lines. Toxicol. In Vitro 2015, 29, 59–62. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.W.; Peng, X.C.; Li, H.Y.; Huang, L.; Li, D.W.; Liu, J.S.; Yang, W.D. Changes in colonic microbiotas in rat after long-term exposure to low dose of okadaic acid. Chemosphere 2020, 254, 126874. [Google Scholar] [CrossRef]
- O’Mahony, M. EU Regulatory Risk Management of Marine Biotoxins in the Marine Bivalve Mollusc Food-Chain. Toxins 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, H.; Suganuma, M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: The okadaic acid class of compounds. Adv. Cancer Res. 1993, 61, 143–194. [Google Scholar] [CrossRef] [PubMed]
- Terao, K.; Ito, E.; Yanagi, T.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning. I. Ultrastructural changes in the small intestine and liver of suckling mice induced by dinophysistoxin-1 and pectenotoxin-1. Toxicon 1986, 24, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Gong, J.; Hu, Y.; Tan, Q.L.; Liu, B.; Yu, X.W.; Hao, X.L.; Guo, Q.N. Long-term exposure to low levels of okadaic acid accelerates cell cycle progression in colonic epithelial cells via p53 and Jak/Stat3 signaling pathways. Heliyon 2022, 8, e10444. [Google Scholar] [CrossRef] [PubMed]
- del Campo, M.; Toledo, H.; Lagos, N. Okadaic acid toxin at sublethal dose produced cell proliferation in gastric and colon epithelial cell lines. Mar. Drugs 2013, 11, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Gordon, F.D. Ascites. Clin. Liver Dis. 2012, 16, 285–299. [Google Scholar] [CrossRef]
- Tubaro, A.; Sosa, S.; Altinier, G.; Soranzo, M.R.; Satake, M.; Della Loggia, R.; Yasumoto, T. Short-term oral toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar] [CrossRef]
- Sosa, S.; Ardizzone, M.; Beltramo, D.; Vita, F.; Dell’Ovo, V.; Barreras, A.; Yasumoto, T.; Tubaro, A. Repeated oral co-exposure to yessotoxin and okadaic acid: A short term toxicity study in mice. Toxicon 2013, 76, 94–102. [Google Scholar] [CrossRef]
- Sosa, S.; Pelin, M.; Ponti, C.; Carlin, M.; Tubaro, A. Acute Toxicity by Oral Co-Exposure to Palytoxin and Okadaic Acid in Mice. Mar. Drugs 2022, 20, 735. [Google Scholar] [CrossRef]
- Aune, T.; Espenes, A.; Aasen, J.A.; Quilliam, M.A.; Hess, P.; Larsen, S. Study of possible combined toxic effects of azaspiracid-1 and okadaic acid in mice via the oral route. Toxicon 2012, 60, 895–906. [Google Scholar] [CrossRef]
- Carrier, P.; Jacques, J.; Debette-Gratien, M.; Legros, R.; Sarabi, M.; Vidal, E.; Sautereau, D.; Bezanahary, H.; Ly, K.H.; Loustaud-Ratti, V. Non-cirrhotic ascites: Pathophysiology, diagnosis and etiology. Rev. Med. Interne 2014, 35, 365–371. [Google Scholar] [CrossRef]
- Hernaez, R.; Hamilton, J.P. Unexplained ascites. Clin. Liver Dis. 2016, 7, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, S.F.; Vilarino, N.; Carrera, C.; Louzao, M.C.; Cantalapiedra, A.G.; Santamarina, G.; Cifuentes, J.M.; Vieira, A.C.; Botana, L.M. Subacute Cardiovascular Toxicity of the Marine Phycotoxin Azaspiracid-1 in Rats. Toxicol. Sci. 2016, 151, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Zhao, X.Y.; Ji, L.D.; Xu, J. Okadaic acid (OA): Toxicity, detection and detoxification. Toxicon 2019, 160, 1–7. [Google Scholar] [CrossRef]
- Gaginella, T.S.; Bass, P. Laxatives: An update on mechanism of action. Life Sci. 1978, 23, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Ewe, K. Intestinal transport in constipation and diarrhoea. Pharmacology 1988, 36 (Suppl. 1), 73–84. [Google Scholar] [CrossRef]
- Costas, C.; Louzao, M.C.; Raposo-Garcia, S.; Vale, C.; Vieytes, M.R.; Botana, L.M. Intestinal secretory mechanisms in Okadaic acid induced diarrhoea. Food Chem. Toxicol. 2022, 169, 113449. [Google Scholar] [CrossRef]
- Edebo, L.; Lange, S.; Li, X.P.; Allenmark, S. Toxic mussels and okadaic acid induce rapid hypersecretion in the rat small intestine. APMIS 1988, 96, 1029–1035. [Google Scholar] [CrossRef]
- Hamano, Y.; Kinoshita, Y.; Yasumoto, T. Enteropathogenicity of Diarrhetic Shellfish Toxins in Intestinal Models Studies on Diarrhetic Shellfish Toxins. I. Food Hyg. Saf. Sci. (Shokuhin Eiseigaku Zasshi) 1986, 27, 375–379_1. [Google Scholar] [CrossRef]
- Tripuraneni, J.; Koutsouris, A.; Pestic, L.; De Lanerolle, P.; Hecht, G. The toxin of diarrheic shellfish poisoning, okadaic acid, increases intestinal epithelial paracellular permeability. Gastroenterology 1997, 112, 100–108. [Google Scholar] [CrossRef]
- OECD. Test No. 425: Acute oral toxicity: Up-and-down procedure. In OECD Guidelines for the Testing of Chemicals, Section 4; OECD Publishing: Berlin, Germany, 2008. [Google Scholar] [CrossRef]
- Dobson, A.J.; Barnett, A.G. An Introduction to Generalized Linear Models; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Barron, L.; Sun, R.C.; Aladegbami, B.; Erwin, C.R.; Warner, B.W.; Guo, J. Intestinal Epithelial-Specific mTORC1 Activation Enhances Intestinal Adaptation after Small Bowel Resection. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 231–244. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | Okadaic Acid | Dinophysistoxin-1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Level | B.W. (g) | No. Mice | Dose (μg/kg) | Lethality (%) | B.W. (g) | No. Mice | Dose (μg/kg) | Lethality (%) |
| 1 | 20.4 ± 0.2 | 3 | 1200 | 66.7 | 19.6 ± 0.1 | 3 | 650 | 0.0 |
| 2 | 20.3 ± 0.2 | 5 | 950 | 20.0 | 20.4 ± 0.3 | 5 | 902 | 60.0 |
| 3 | 20.3 ± 0.4 | 7 | 1064 | 57.1 | 20.4 ± 0.3 | 7 | 887 | 28.6 |
| LD50 (μg/kg) | 1069 | 897 | ||||||
| Group | Percentage of LD50 (%) | Okadaic Acid (μg/kg) | Dinophysistoxin-1 (μg/kg) |
|---|---|---|---|
| L; Low dose | 10 | 108 | 90 |
| M; Moderate dose | 25 | 270 | 225 |
| H; High dose | 50 | 540 | 450 |
| Vehicle | Okadaic Acid (μg/kg) | Dinophysistoxin-1 (μg/kg) | |||||
|---|---|---|---|---|---|---|---|
| 108 | 270 | 540 | 90 | 225 | 450 | ||
| Incidence of ascites (%) | 0 | 0 | 80 | 100 | 0 | 60 | 100 |
| Ascites score | 0 | 0 | 0.8 ± 0.2 ** | 2.2 ± 0.2 ***,# | 0 | 0.6 ± 0.2 * | 1.4 ± 0.2 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Kang, J.-H.; Jung, H.J.; Hwang, J.H.; Chun, H.S.; Yoon, Y.S.; Oh, S.H. Okadaic Acid Is at Least as Toxic as Dinophysistoxin-1 after Repeated Administration to Mice by Gavage. Toxins 2023, 15, 587. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins15100587
Park SY, Kang J-H, Jung HJ, Hwang JH, Chun HS, Yoon YS, Oh SH. Okadaic Acid Is at Least as Toxic as Dinophysistoxin-1 after Repeated Administration to Mice by Gavage. Toxins. 2023; 15(10):587. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins15100587
Chicago/Turabian StylePark, Se Yong, Ju-Hee Kang, Hyun Jin Jung, Jung Ho Hwang, Hyang Sook Chun, Yeo Sung Yoon, and Seung Hyun Oh. 2023. "Okadaic Acid Is at Least as Toxic as Dinophysistoxin-1 after Repeated Administration to Mice by Gavage" Toxins 15, no. 10: 587. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins15100587