1. Introduction
Breast cancer is the most frequent cancer in women worldwide [
1]. The efficacy of breast cancer therapy is associated with a number of cellular processes that in some cases lead to tumor resistance. Among other factors, inactivation of anticancer drugs by biotransformation enzymes, decreased uptake and/or increased efflux of drugs, changes in cell-cycle checkpoints, increased DNA repair or reduced cell death, and cellular compartmentalization may contribute to the development of multidrug resistance [
2].
The majority of currently used cytotoxic drugs are metabolized by biotransformation enzymes in liver and extrahepatic tissues. Biotransformation often leads to inactivation of drugs which become more polar to allow for body elimination. On the other hand, prodrugs are designed to be activated via biotransformation in the first place and then follow the same principles of metabolism as drugs. Consequently, germline genetic variability in biotransformation enzymes is considered as important factor determining individual patient sensitivity to an administered drug. These enzymes are in general divided into phase I (activation) and phase II (conjugation) enzymes [
3]. Cytochromes P450 (CYP) constitute a major group of (in)activation enzymes in phase I whereas phase II enzymes are more heterogeneous. Numerous pharmacogenomic studies in breast cancer patients concentrated mostly on analysis of selected polymorphisms in single or several genes from all biotransformation phases (reviewed in [
4]), but a comprehensive germline genetic variability screen of the majority of these enzymes in breast cancer patient cohorts is virtually missing.
Since drug efflux is mediated by membrane-bound ATP-binding cassette (ABC) transporters [
5] and drug uptake is provided by solute carrier (SLC) transporters [
6] it seems obvious that equilibrium of these exporters/importers is important for prediction of cancer drug resistance [
7,
8]. Indeed, comprehensive transcriptomic profiling studies demonstrated gene expression deregulations of a number of ABCs and SLCs between tumor and paired non-malignant tissues from patients with solid tumors, e.g., colorectal [
9], breast [
10], pancreatic [
11,
12], and ovarian [
13] suggesting their potential role in cancer progression. Moreover, these studies revealed a number of associations between gene expression levels of transporters, therapy response and survival of the patients with implications for prognosis and individualized therapy.
Data from publicly available large-scale sequencing studies have shown that genetic alterations in drug targets, cell death and major cancer driving pathways, e.g., PI3K/AKT/MTOR or RAS/MAPK, and nuclear receptors can be found across all cancer types; however, at highly variable frequencies [
14]. Very recently, highly frequent deleterious somatic mutations relevant for clinical management, including PIK3CA, RTK/RAS/MAPK and cell cycle pathway genes, were found in inflammatory breast cancer patients through next generation sequencing analysis [
15].
Pharmacogenomics represents an important tool for personalized medicine. Two major types of studies may be found in the published literature dealing with the issue of genetic susceptibility and drug response in oncology. First, studies of germline genetic variation, mainly polymorphisms, in homogeneous groups of patients treated with defined drug regimens [
16]. Second, in vitro screening of drug response in human cancer cell lines with well characterized somatic genetic profile also helped to elucidate the genetic background of chemoresistance [
17,
18]. However, recent data from analysis of sensitivity of 993 cell lines to 265 drugs show that germline genetic variability can be of the same importance as somatic one [
19]. Thus, continuing with studies in patients is imperative for further understanding and subsequent translation of these aspects into clinical setting. There are several genome-wide association studies (GWAS) in the literature demonstrating the power of pharmacogenomics in breast cancer [
20] and accelerated implementation of the next generation sequencing into clinical studies will undoubtedly bring further progress in this area. Very recently, analysis of available big data demonstrated that priority pharmacogenes for population-adjusted genetic profiling exist with highly variable distribution across populations [
21], suggesting that use of sequencing-based approaches may enable “true personalized medicine”.
Here, we explored the genetic variability of a panel of 509 genes relevant for pharmacogenomics using targeted sequencing in a testing set of patients treated with neoadjuvant or adjuvant cytotoxic therapy. Genetic variants significantly associating with therapy outcome measured as clinical response in neoadjuvant setting or disease-free survival (DFS) of the patients were evaluated in a larger validation set of patients. To our knowledge, this is the first research study providing genetic data with association to drug chemoresistance evaluated as prognosis and therapy outcome of breast cancer patients with aid of in silico prediction in the Czech population to such an extent. The validated variants may further be used for functional studies and prospective follow up trials evaluating their prognostic and predictive utility in clinical setting.
3. Discussion
There is no doubt that drug therapy tailored to individual genetic predisposition could bring substantial cost-benefit effects in terms of both enhanced drug efficacy and decreased risk of adverse drug reactions. Pharmacogenomics seem so far instrumentally more accessible than routine pharmacokinetics or pharmacodynamics in clinical day use. However, current approaches, including the state-of-the-art technological platforms such as the next generation sequencing, are still in the early evolutionary phase. Except the considerable decrease of cost per genotype in the last few years, the complexity of data management and the need for robust evaluation of results to make them clinically meaningful still hinder broader usage, especially in the pharmaceutical area. Thus, studies addressing these aspects are urgently needed.
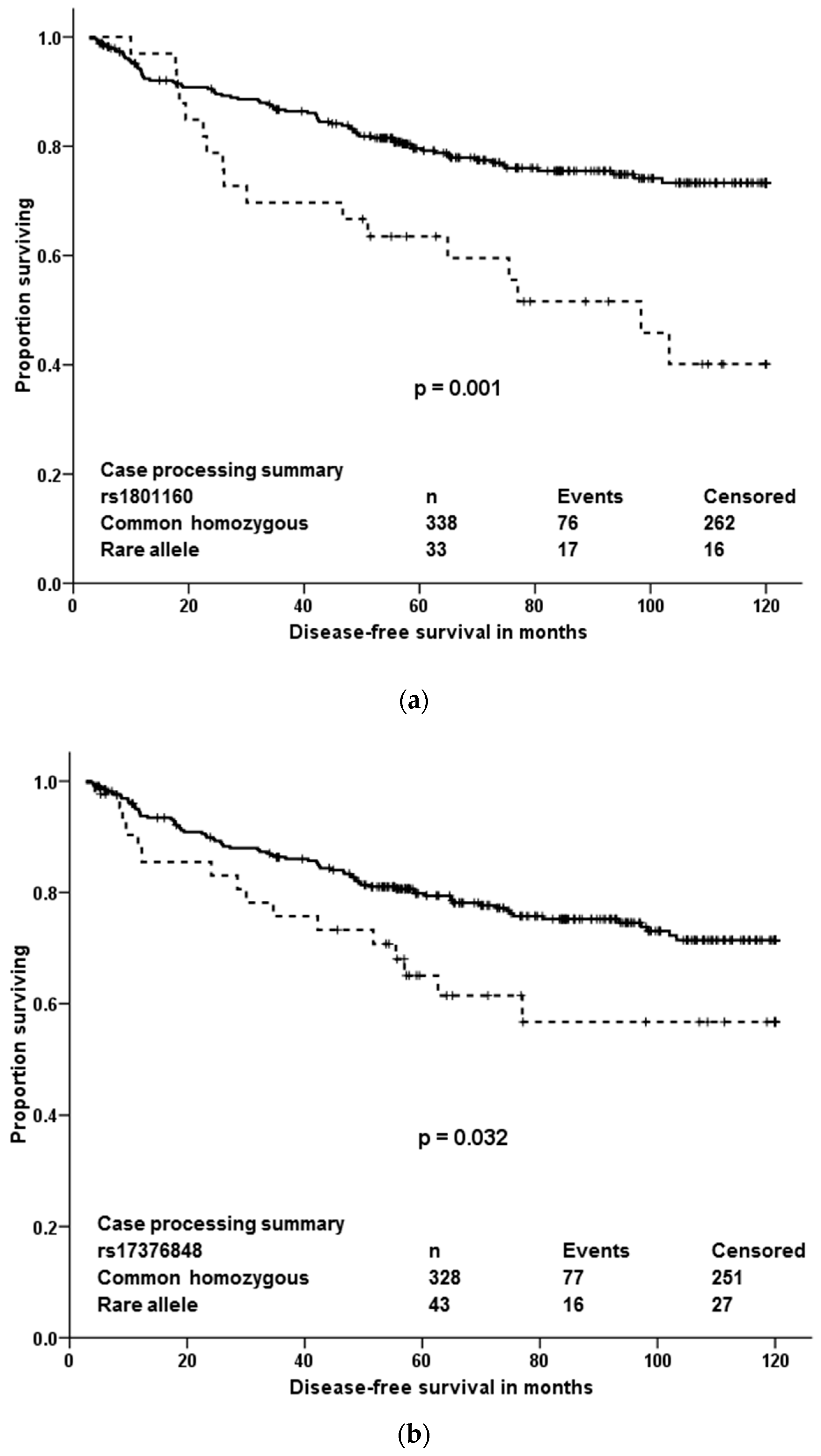
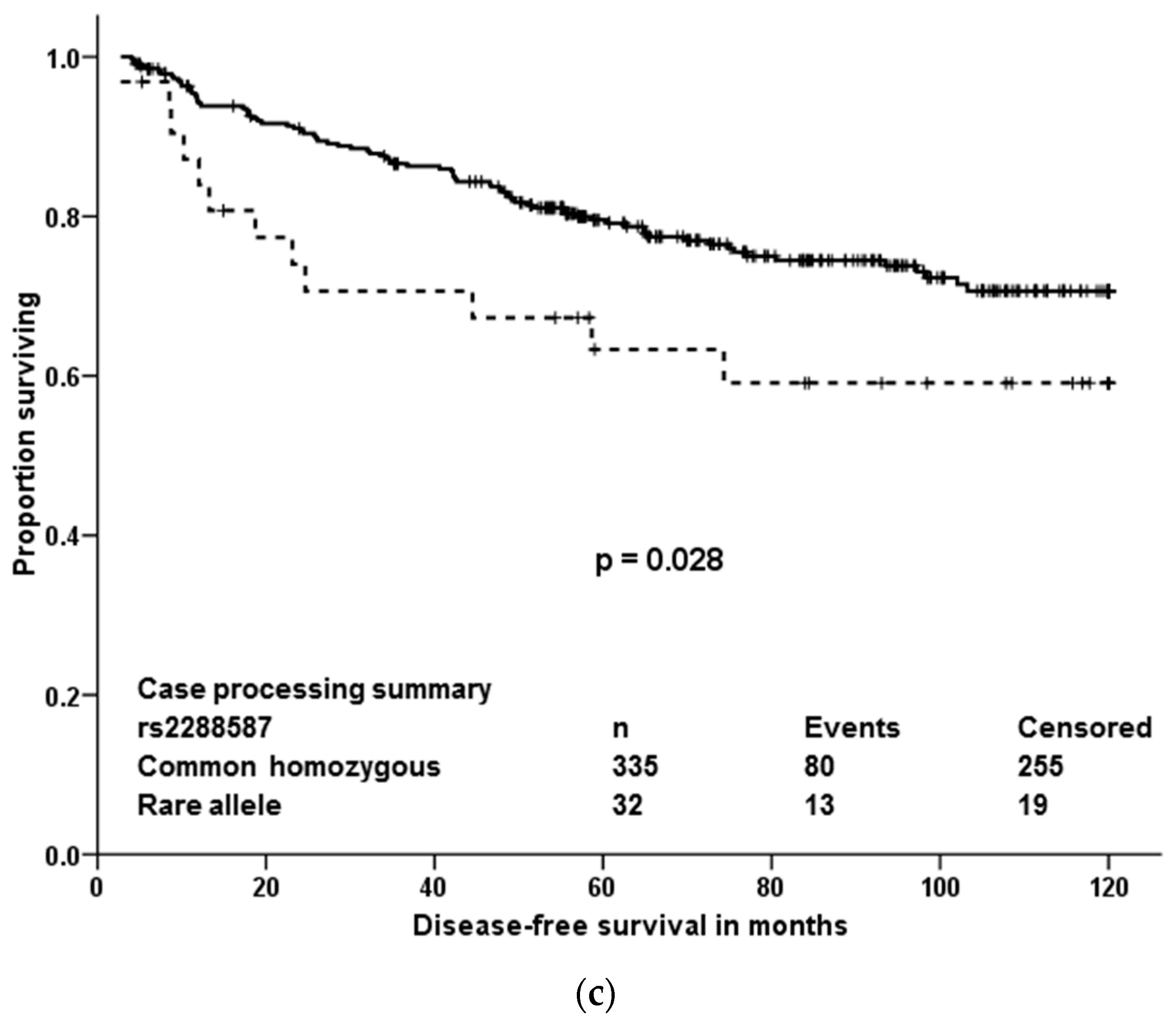
The present paper shows that out of quite large number of germline variants (18,245) detected among 509 pharmacogenes and other drug-related genes in breast cancer patients, only a few may be important from the view of individualized therapy after robust validation. Four variants associated with response of patients to neoadjuvant cytotoxic therapy and three, out of which just one was significant in multivariate analyses, were prognostic in terms of DFS after cytotoxic therapy. Responders to the neoadjuvant cytotoxic therapy carried, significantly more frequently than non-responders, the rare allele in rs10868138 of SLC28A3, the common homozygous genotype in rs2227291 (ATP7A), or rs4376673 (DFFB) or the common allele in rs2293194 (KCNAB1). However, the association of rs10868138 with response should be treated with caution while this association was non-significant after adjustment for disease stage in multivariate analysis. Rs2293194 was significantly associated with response only in patients with early stage disease 0 or I (p < 0.001). Patients with the common homozygous genotype in rs1801160 of DPYD survived longer without relapse after cytotoxic therapy than carriers of the rare allele. This effect was particularly pronounced in patients with luminal B and triple negative molecular subtypes.
Protein coding gene
SLC28A3 (Solute carrier family 28 member 3) is a sodium-dependent nucleoside transporter involved in the homeostasis of endogenous nucleosides and regulating multiple cellular processes, e.g., neurotransmission and metabolism and transport of nucleoside drugs [
22] (
https://www.genecards.org/cgi-bin/carddisp.pl?gene=SLC28A3&keywords=A-3). Genetic variability in
SLC28A3 was previously connected with pharmacokinetics of nucleoside analogs [
23] and cardiotoxicity of anthracyclines [
24] although a more recent study did not confirm the latter observation [
25]. Variation rs7867504 in
SLC28A3 was shown to be involved in gemcitabine pharmacobiology and toxicity in metastatic breast cancer patients receiving maintenance therapy [
26] or in patients with pancreatic carcinoma [
27]. The rs10868138 in
SLC28A3 is a less studied variant; however, it was recently connected with higher concentration of azathioprine in erythrocytes of patients with neuromyelitis optica [
28], suggesting that it may be functional in vivo. The other relevant gene to pharmacogenomics of nucleoside analogs is dihydropyrimidine dehydrogenase (
DPYD). The protein encoded by this gene (DPD) is a pyrimidine catabolic enzyme and the initial and rate-limiting factor in the pathway of uracil and thymidine catabolism (
https://www.genecards.org/cgi-bin/carddisp.pl?gene=DPYD). DPD is active in the catabolic pathway of 5-fluorouracil and mutations in its gene result in an increased risk of toxicity in cancer patients receiving 5-fluorouracil chemotherapy [
29].
DPYD polymorphism rs1801160, associated with survival of breast cancer patients in our study, is very frequently studied. Carriage of rs1801160 in
DPYD associated with grade 3 or 4 5-fluoropyrimidine associated adverse risk effects, e.g., neutropenia, in a recent study of colon cancer patients treated with regimens consisting of 5-fluoro-uracil or capecitabine combined with oxaliplatin [
30]. Although
DPYD genotype-guided individualized dosing for better safety of fluoropyrimidine treatment was recently suggested as a new standard of care [
31], the present study was not designed to address adverse effects and the validated association of
DPYD variant with DFS adds a new observation to the knowledge base.
Of the other validated variants associated with response to the therapy, the rs2227291 in
ATP7A raises particular attention since it is non-synonymous (V767L) and thus probably more directly functional. Notably, rs2227291 is the only association with response in the validation set that passes the false discovery rate (
p = 0.011). Copper transporter ATP7A encodes a transmembrane protein that functions in copper transport across membranes and it is frequently studied in connection with sensitivity to platinum drugs, e.g., cisplatin. Very recently, the rs2227291 was associated with cisplatin resistance in patients with epithelial ovarian cancer treated with combination of platinum and taxane [
32]. However, though the authors state that carriers of the minor allele are more sensitive to cisplatin, the functional link is missing and must be obtained using further study. GWAS studies show that regulatory non-coding variants may play a role in multiple distinct diseases such as cancer [
33] and thus the other two intronic variants associated with response (rs2293194 in
KCNAB1 and rs4376673 in
DFFB) also represent a potential target for further studies.
KCNAB1 (Potassium Voltage-Gated Channel, Shaker-Related Subfamily, Beta Member 1) gene encodes a potassium channel involved in an important dopamine pathway, chemical transmission of signal across synapses and various CYP450 pathways (
https://www.genecards.org/cgi-bin/carddisp.pl?gene=KCNAB1). In cancer genetics,
KCNAB1 variation may play a role in breast cancer pathogenesis because its overexpression was found in breast tumors in comparison to non-tumor tissues [
34]. Finally,
DFFB is a subunit Beta and active component of DNA Fragmentation Factor protein (DFF).
DFFB has been found to trigger both DNA fragmentation and chromatin condensation during the apoptosis (
https://www.genecards.org/cgi-bin/carddisp.pl?gene=DFFB). For example, enhanced expression of DFFB with doxorubicin or in combination with sulfonamides enhanced the killing of T47-D breast cancer tumor cells via apoptosis [
35,
36]. Thus, variation and deregulation of the DFFB gene in the presence of apoptosis-inducing drugs might have an impact on their efficiency in tumor cells.
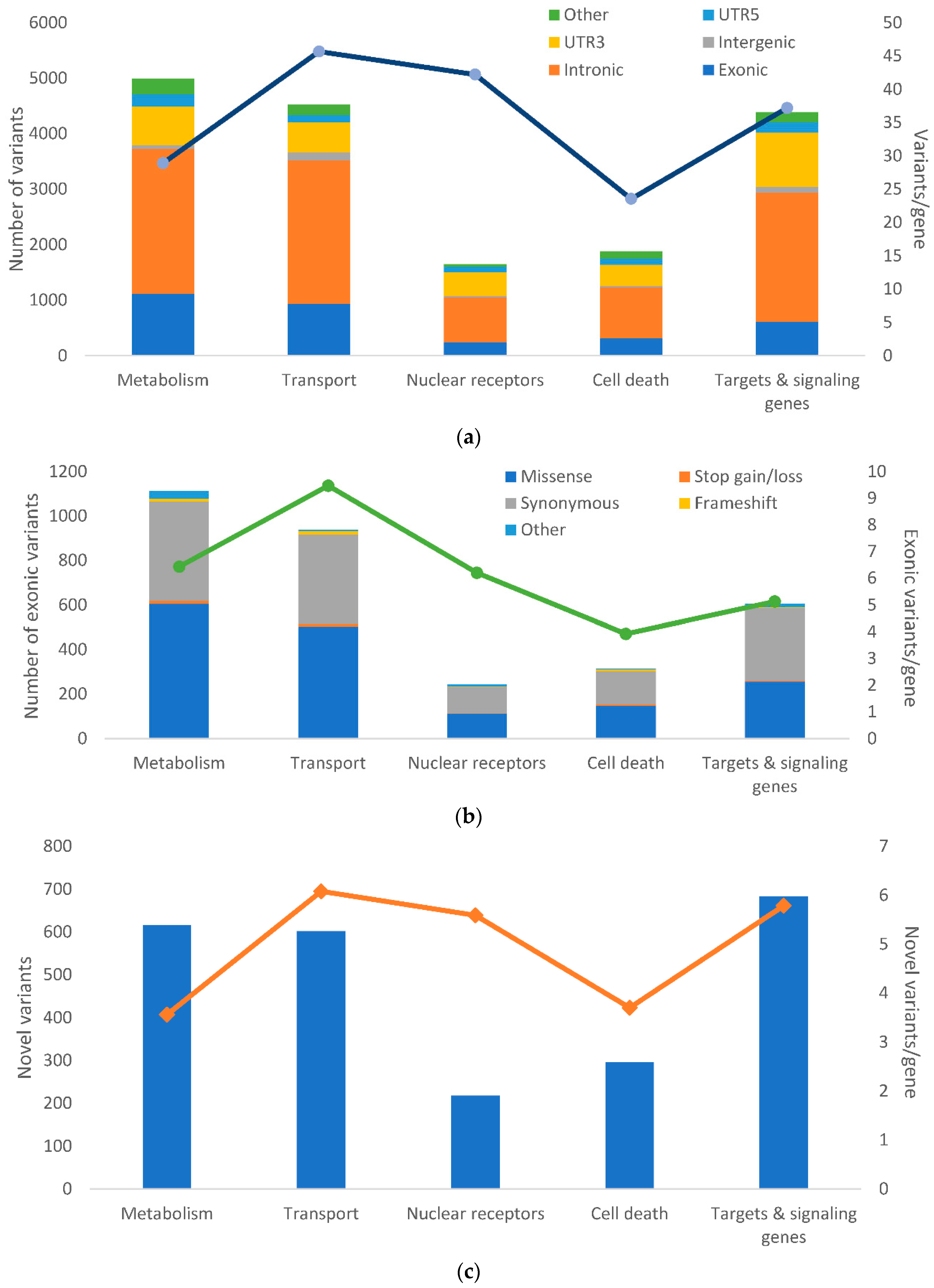
Of the 88 loss of function variants identified in our study, only seven frameshift variants had MAF above 5% ensuring the necessary statistical power to estimate the associations with DFS or response. None of the associations of frameshift indels with outcome was statistically significant. Of the genes harboring these variants, only
RRM2B, was in the first quartile of the most intolerant genes to functional variation, according to LoFtool gene score [
37]. The rest of the genes in the first quartile were
ABCA5/A6/A7/A10/A13,
ABCB4/B10,
ABCC2/C3/C5/C10/C11/C12,
DHCR7,
NR1I3,
SLC35C2 and
SLCO3A1. Of their corresponding proteins, mainly the multidrug resistance protein (MRP)2, MRP3, MRP5, and MRPs 7–9 coded by membrane transporters
ACBC2,
ABCC3,
ABCC5,
ABCC10,
ABCC11,
ABCC12 and the organic anion transporter polypeptide-related protein (OATP)3A1 coded by
SLCO3A1 are of the highest importance because of the relation of MRPs and OATPs in the chemotherapy resistance or sensitivity [
5,
6]. However, associations with response or DFS in these genes could not be assessed due to the modest size of our cohort and the low frequency of these variants in population.
Population context is currently broadly discussed, for example considerable gene-dependent variability between African and European Americans has recently been demonstrated [
21]. The present study was performed on homogeneous population of Slavic Europeans. As such, adds unique information to the existing clinically associated datasets. The only publicly available data in the Czech population on the germline whole exome level are in the National Center for Medical Genomics (NCMG) set of healthy Czech population (
n = 309 at time of writing). Of the total number of 509 genes, 503 genes (99%) contained at least one genetic alteration. No alterations were found in
ABCF1, HSPA1A, RXRB, TAP1 (ABCB2), TAP2 (ABCB3) and
VDAC1P4 genes in the present study. However, in comparison to the data in the NCMG set of healthy Czech population, these genes, except
VDAC1P4, were polymorphic. In total, 54 variants were found in
ABCF1, 13 in
HSPA1A, 16 in
RXRB, 78 in
TAP1, and 88 in
TAP2. Whether these differences are due to the different composition of both sets in terms of individual characteristics of participants (the present set contained only females while the NCMG set is composed of both genders) or due to the disease etiology (breast cancer patients
versus healthy population) remains to be elucidated. Differences between sequencing platforms, raw data management and annotation cannot be excluded either. On the other hand, the most polymorphic genes with over one hundred alterations were
NCOR2, ABCA13, RPTOR, ABCA4, CIT, BIRC6, ABCC1, ABCA1, RXRA, NCOR1, ABCA7, ABCC4 and
ABCB5 in the present study and except for
RXRA, all these genes showed more than 100 alterations in the NCMG set as well.
ABCA13 is overall the 80th most polymorphic gene in NCMG data coming from the whole exome sequencing, while the rest of the top 80 variable genes in NCMG data were not analyzed in this study. Thus, although there are some similarities in these sets, the direct comparison of data from two sets within the same population points to some differences, mainly in the low MAF variants, and thus, multiethnic cohorts must be very carefully evaluated in this regard. The recent study by Kozyra et al. [
21] reported
ABCA4, ABCA1 and
ABCC1 among genes with highest counts of variations suggesting that the most variable genes may be conserved across diverse populations.
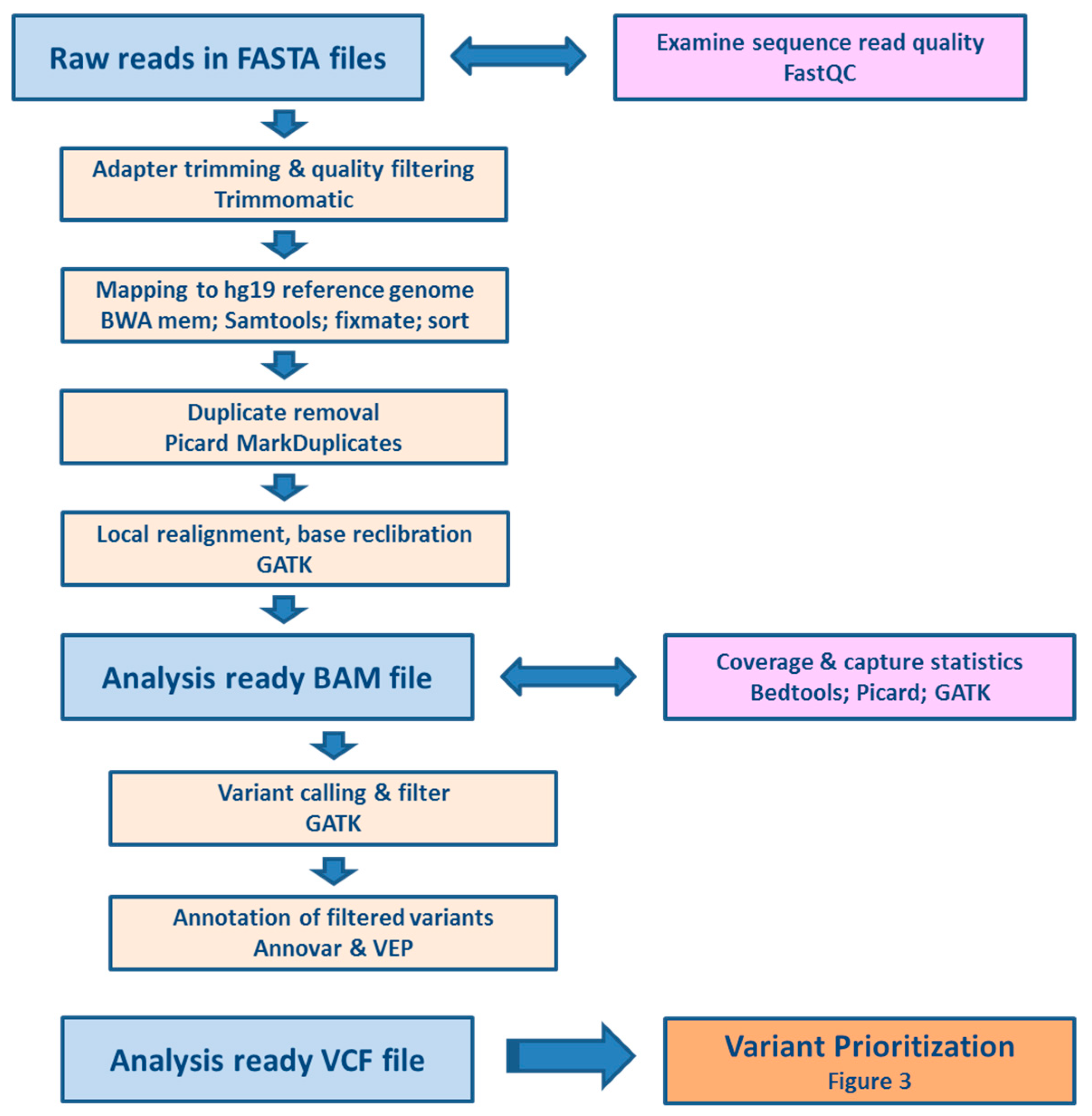
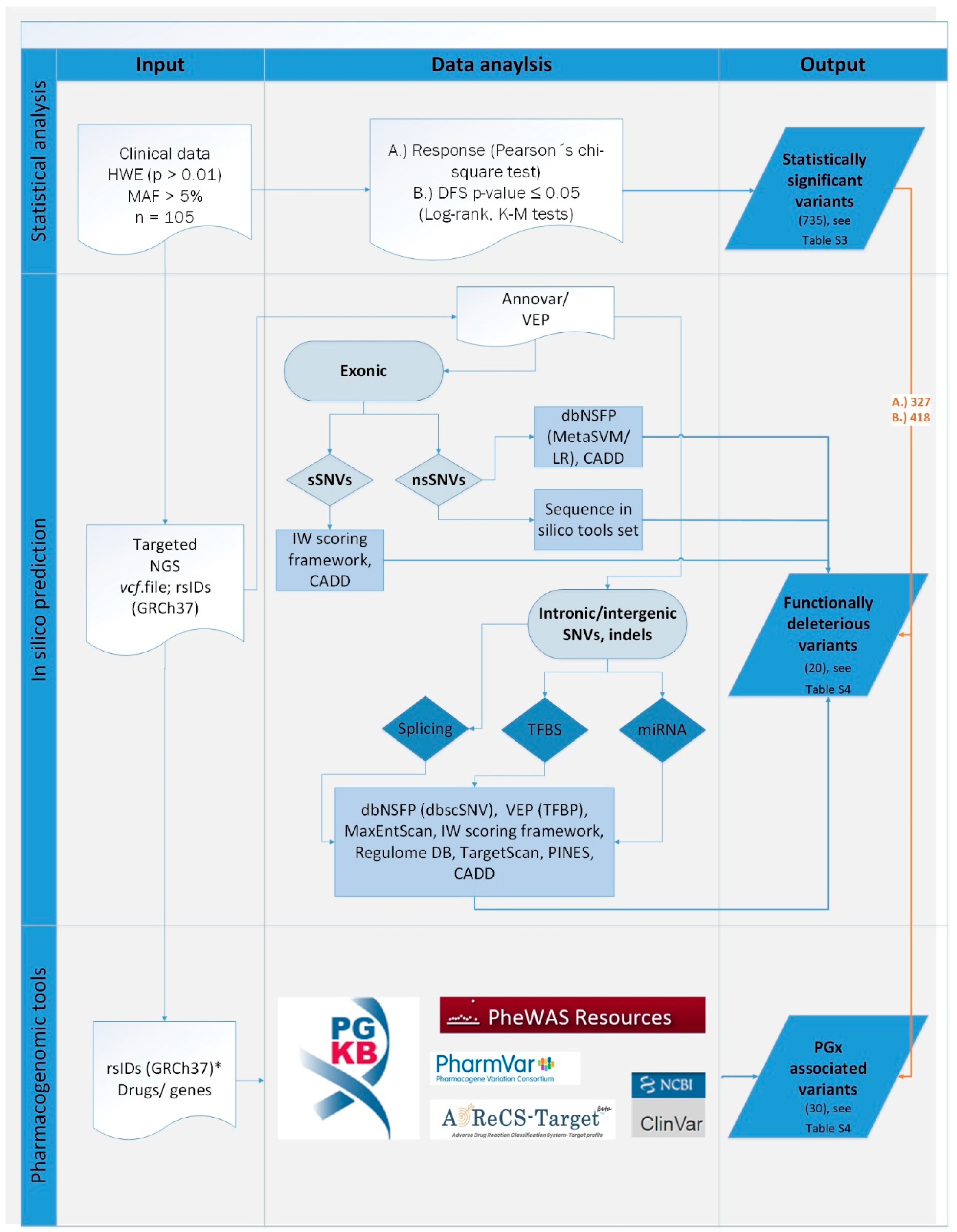
We aimed not only to contribute to the search for predictive genetic biomarkers for oncology, but also to set up a pipeline for processing of raw data generated by massively parallel gene panel sequencing, including quality controls. Last but not least, complex variant prioritization scheme including both evaluation of variants by associating them with individual patient data relevant to their pharmacological response and further filtration using in silico predictive tools and pharmacological databases is provided. Moreover, robust validation by means of comparison of results obtained by two technological platforms and two stage study evaluation using testing and validation clinical cohorts was accomplished.
Public databases such as PharmGKB are wealthy sources of germline variants which evolved from laborious curation of published studies and a strong need for systematic use of perished knowledge in personalized medicine [
38,
39]. At the time of writing of this article, 21,115 annotations in 647 drugs associated with drug response at pharmacodynamic and/or pharmacokinetic level were in PharmGKB (
https://www.pharmgkb.org/, accessed 4 November 2018). Despite the significant number of annotations available, automated prediction for drug response of sequenced variants is not available. Many in silico tools have been developed to aid with the prediction mostly for coding variants. Evolutionary characteristics of variants in pharmacogenes involved in biotransformation and transport of drugs are, however, different. This complicates accurate estimates provided by methods mostly built on Mendelian disease principles [
40]. Consequently, genomic evolutionary rate profiling or evolutionary constraint algorithms, as well as tools trained on disease pathogenic/neutral variants were not included in our in silico sequence tools set. Several attempts have been made to generate specialized tools scaled for pharmacogenes or to optimize current models for pharmacogenetic assessments [
40,
41]. Nonetheless, “gold standard” methods are still lacking in the public domain. Furthermore, even no standard recommendations on the number or types of in silico tools to be considered in analyses which may have significant impact on results are available [
42]. While this prevents to a certain extent potentially incorrect use and interpretation in clinical practice, academic research is also hindered. In our research we attempted to combine different approaches to acquire complex information for given variants. Prediction or knowledge acquired for prioritized variants was not further manually curated. The reason was to verify the ability of automated prioritization and to estimate the added value of in silico tools for our further studies.
The modest size of the testing set may be seen as a limitation of the study. Due to this fact, the importance of very rare (MAF < 0.001) and rare (<0.01) variants could not be assessed. Thus, we prioritized variants with MAF > 0.05. In the light of the recently acknowledged contribution of rare variants to inter-individual variability in drug response [
40] this limitation needs to be considered in future pharmacogenomic studies in oncology. On the other hand, ethnical homogeneity and completeness of clinical follow up is considered beneficiary. Moreover, the study may be extended by addition of more patients or compiling with similarly designed set of patients with whole exome or genome data. Another limitation of this study is the nonhomogeneity of the patient sets. The advanced disease stage and the molecular subtype can be seen as the strongest modifiers of patient prognosis. We have employed the multivariate analyses adjusted to disease stage and we have analyzed the associations of variants with molecular subtype in the testing set to circumvent these issues. We also analyzed associations with survival separately in neoadjuvantly treated patients (with predominant luminal subtype) and adjuvantly treated patients (triple negative tumors only) in the testing set and ran the full prioritization pipeline again. Despite some slight discrepancies which might be caused by chance due to small sizes of compared groups, all the major conclusions of this study remained unchanged.
Functional studies of the identified variants and genes will be the next step. Functionality of a variant may be studied using CRISPR-Cas9 gene editing in a suitable tumor cell model in vitro. Subsequently gene function, including response of the model cell line to clinically relevant drugs, e.g., taxanes, may be followed.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}