CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer

 ,
,
Abstract
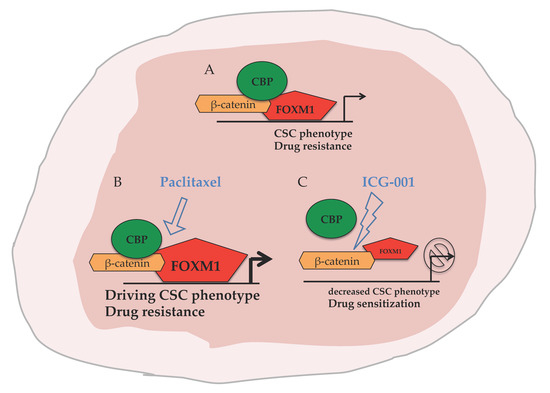
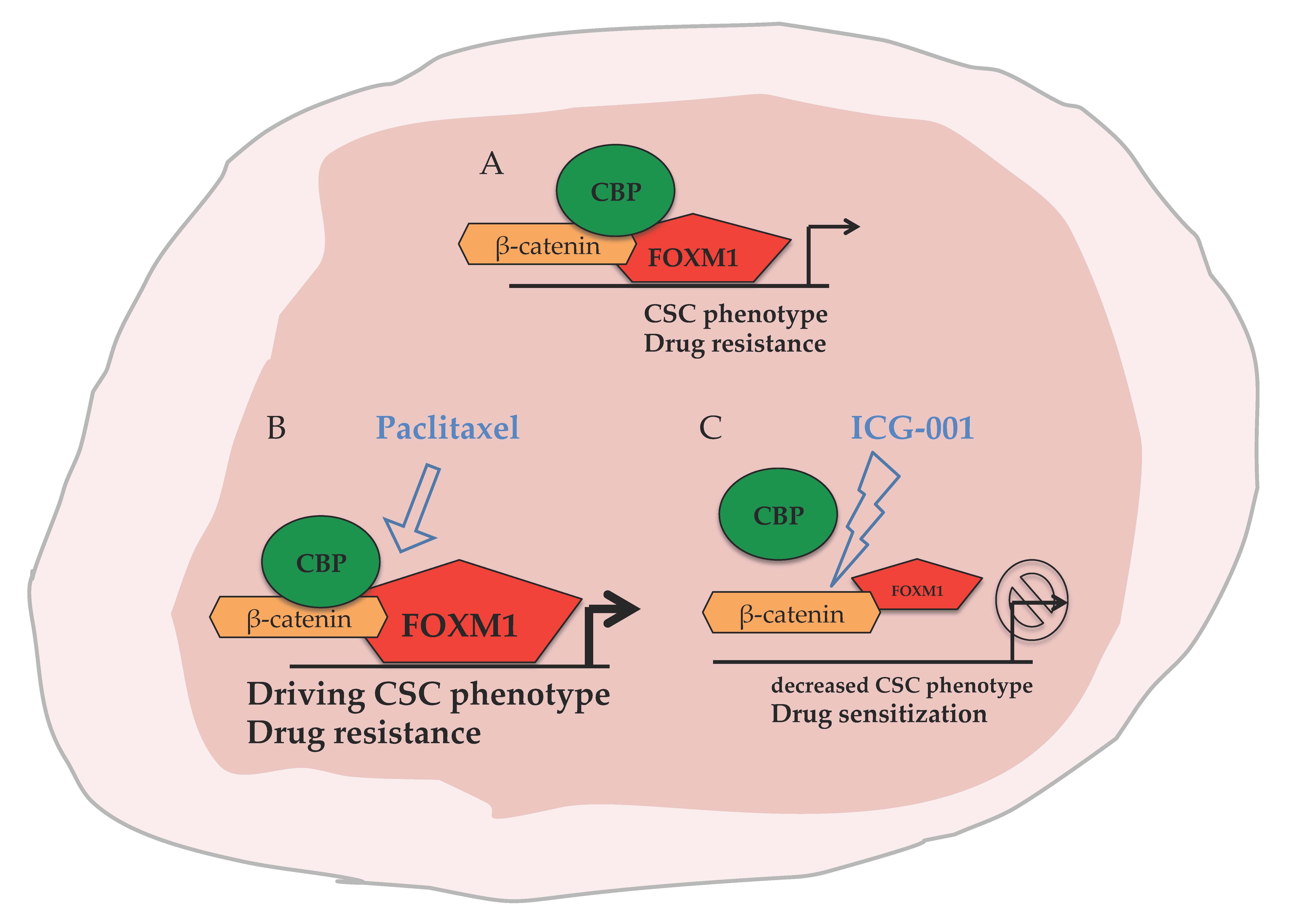
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
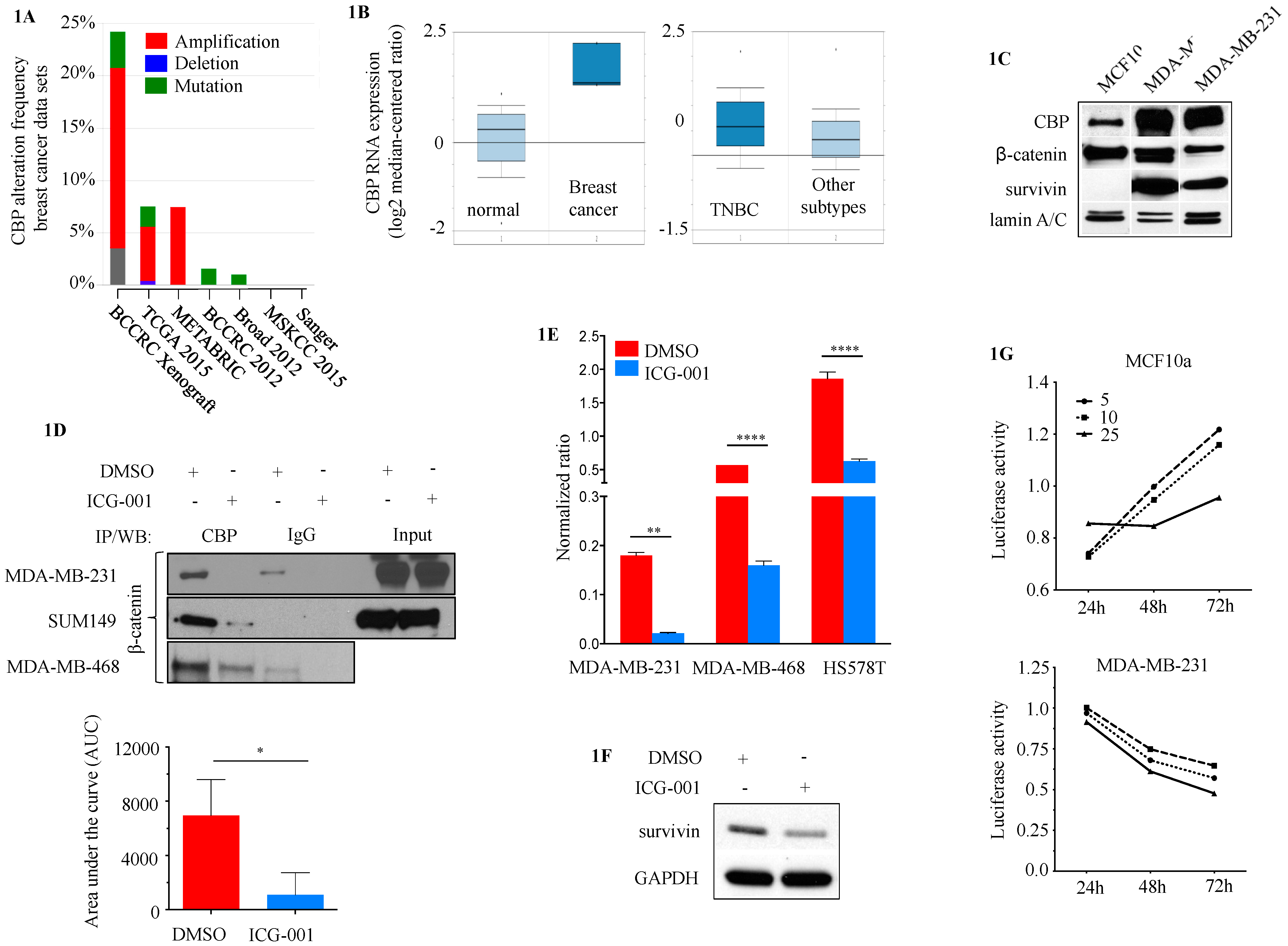
2.1. CBP as a Potential Target in TNBC
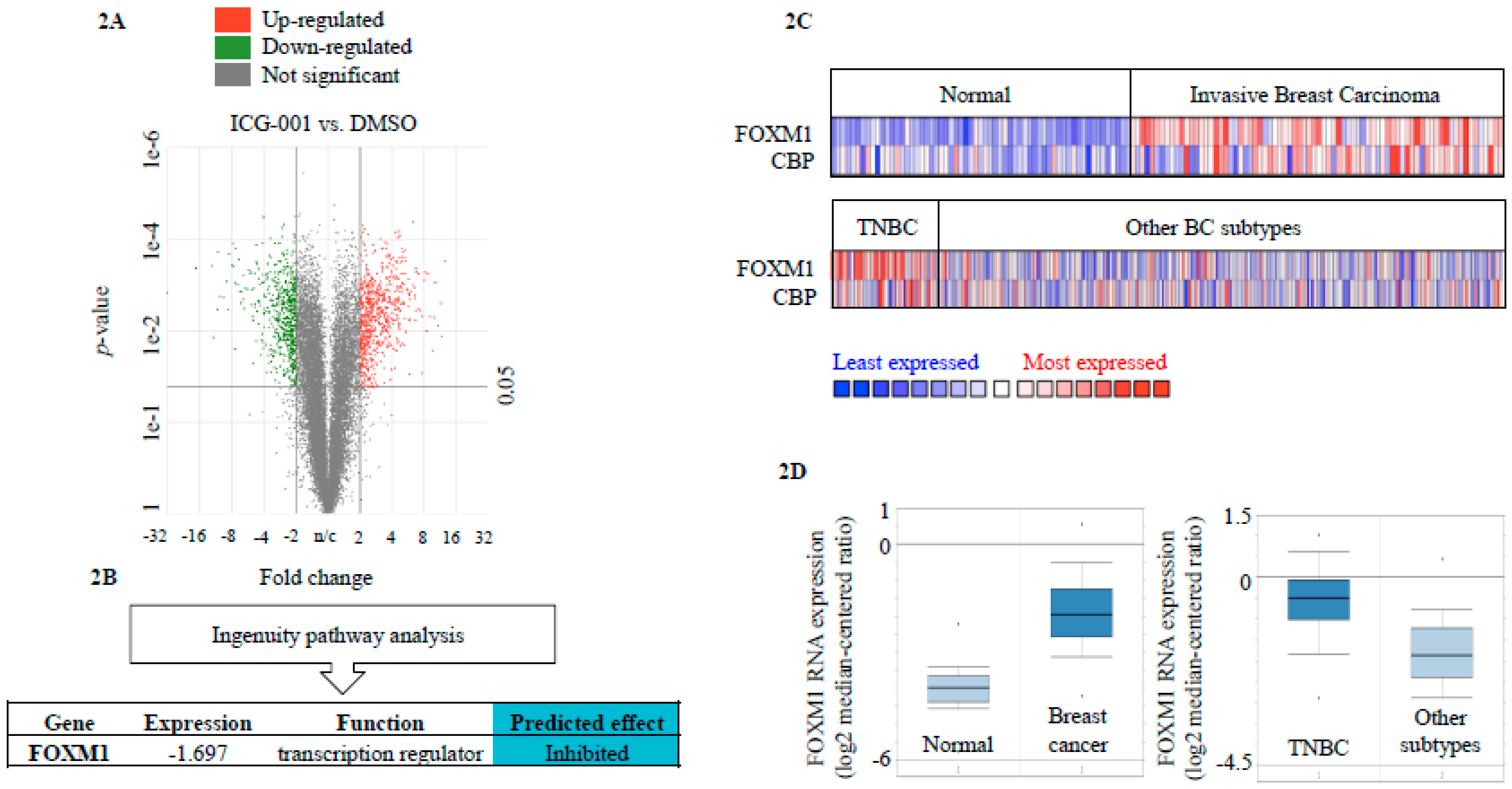
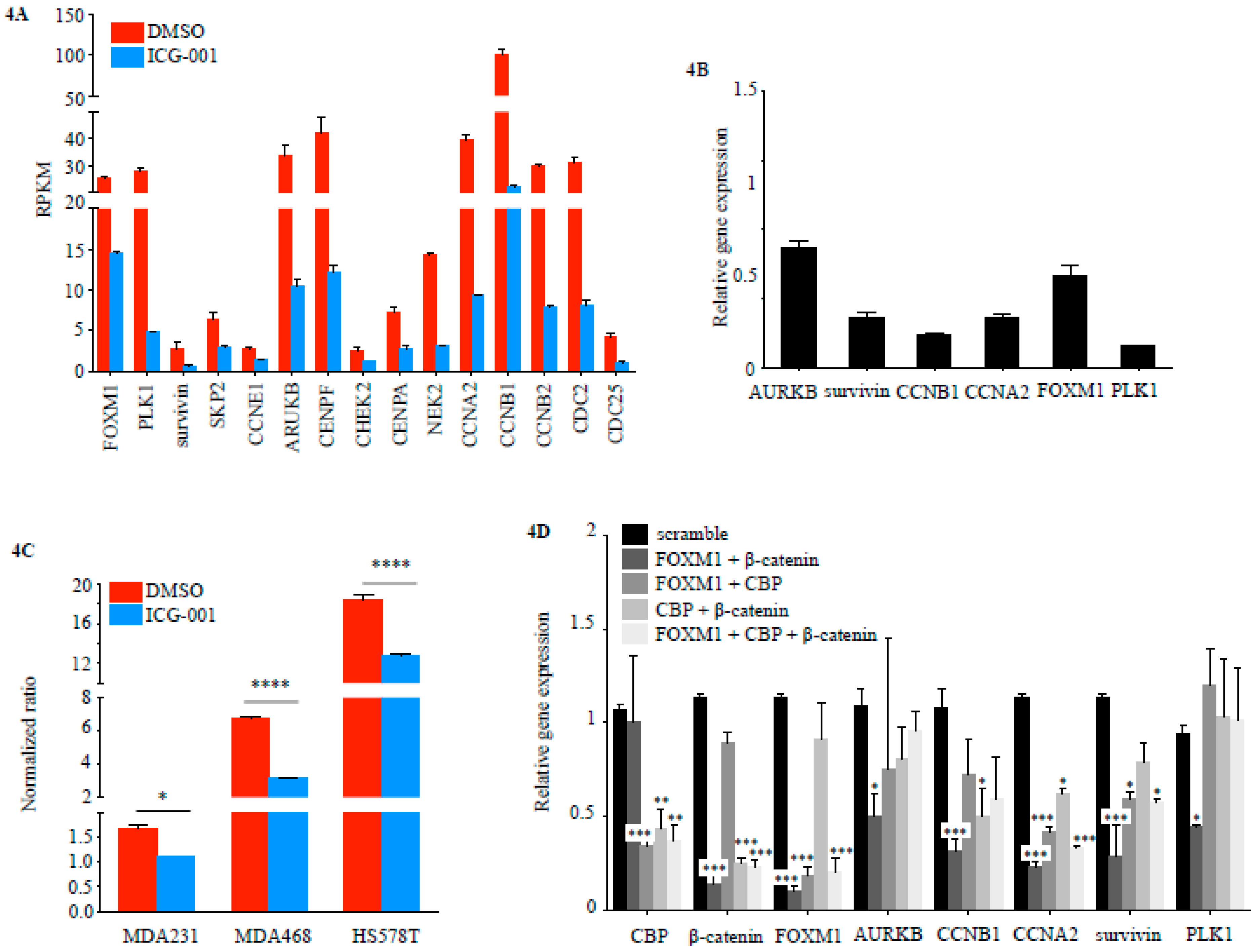
2.2. FOXM1 is a Downstream Effector of CBP-Signaling in TNBC
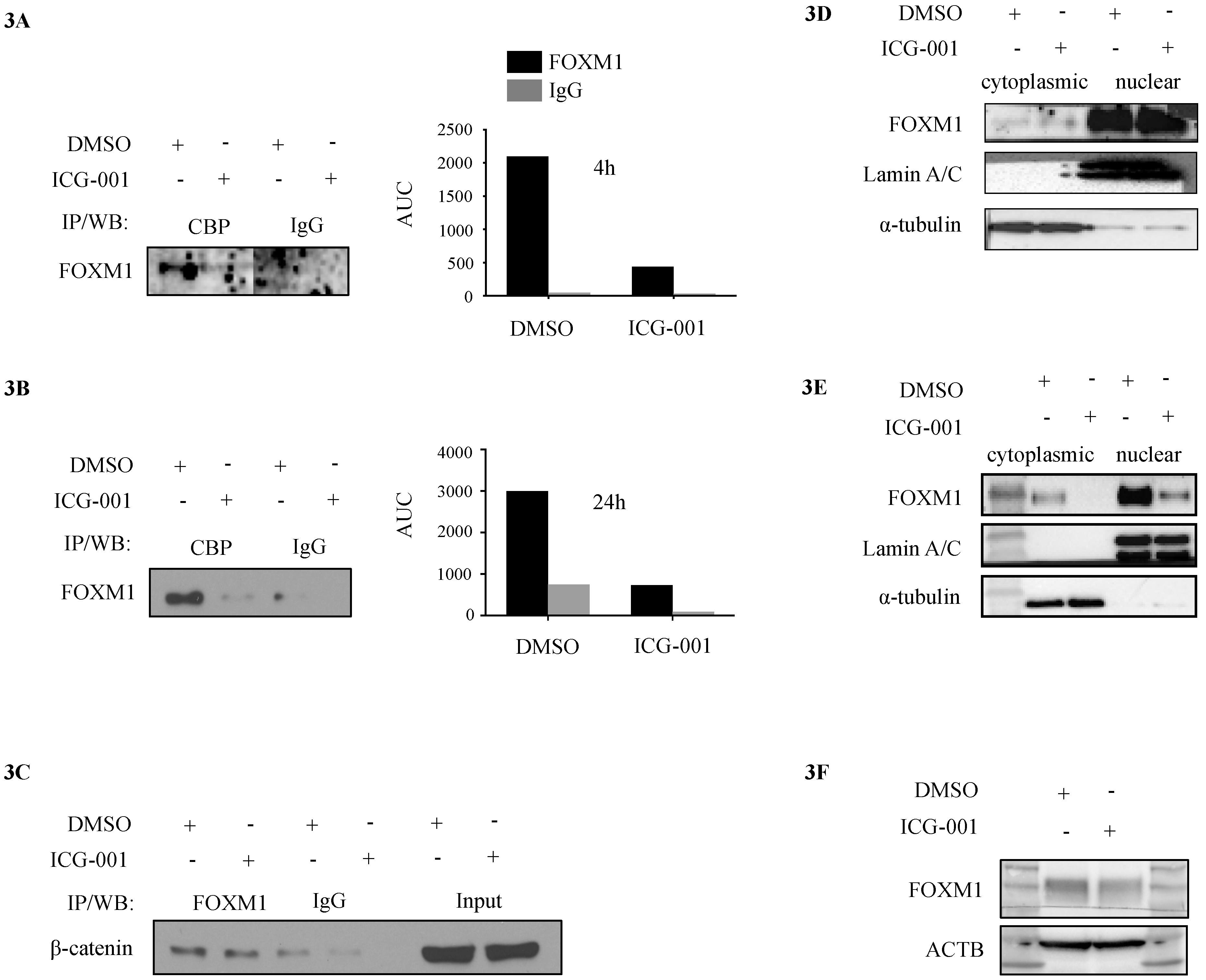
2.3. CBP, FOXM1 and β-Catenin form a Transcriptionally Active Complex That Can be Targeted with ICG-001
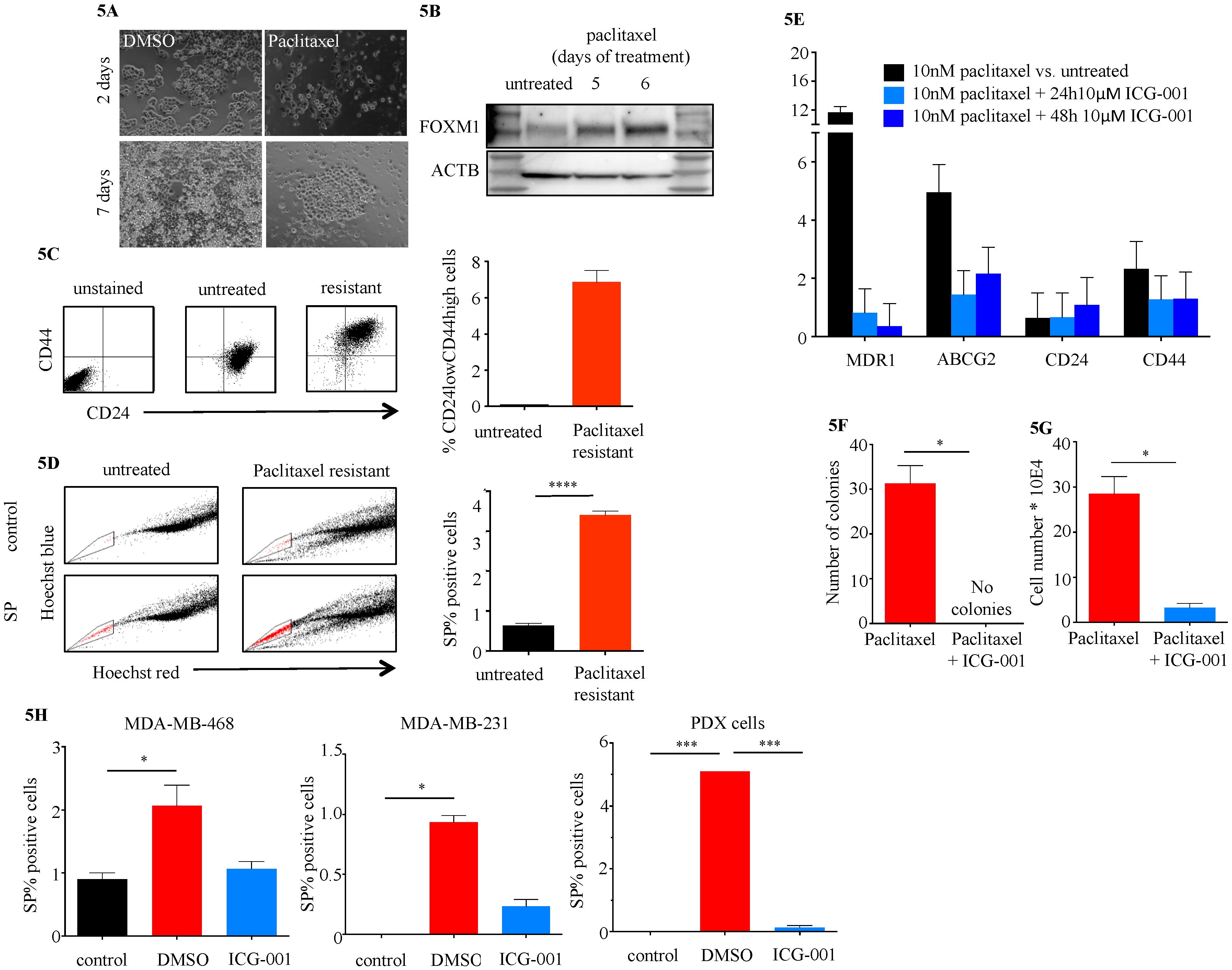
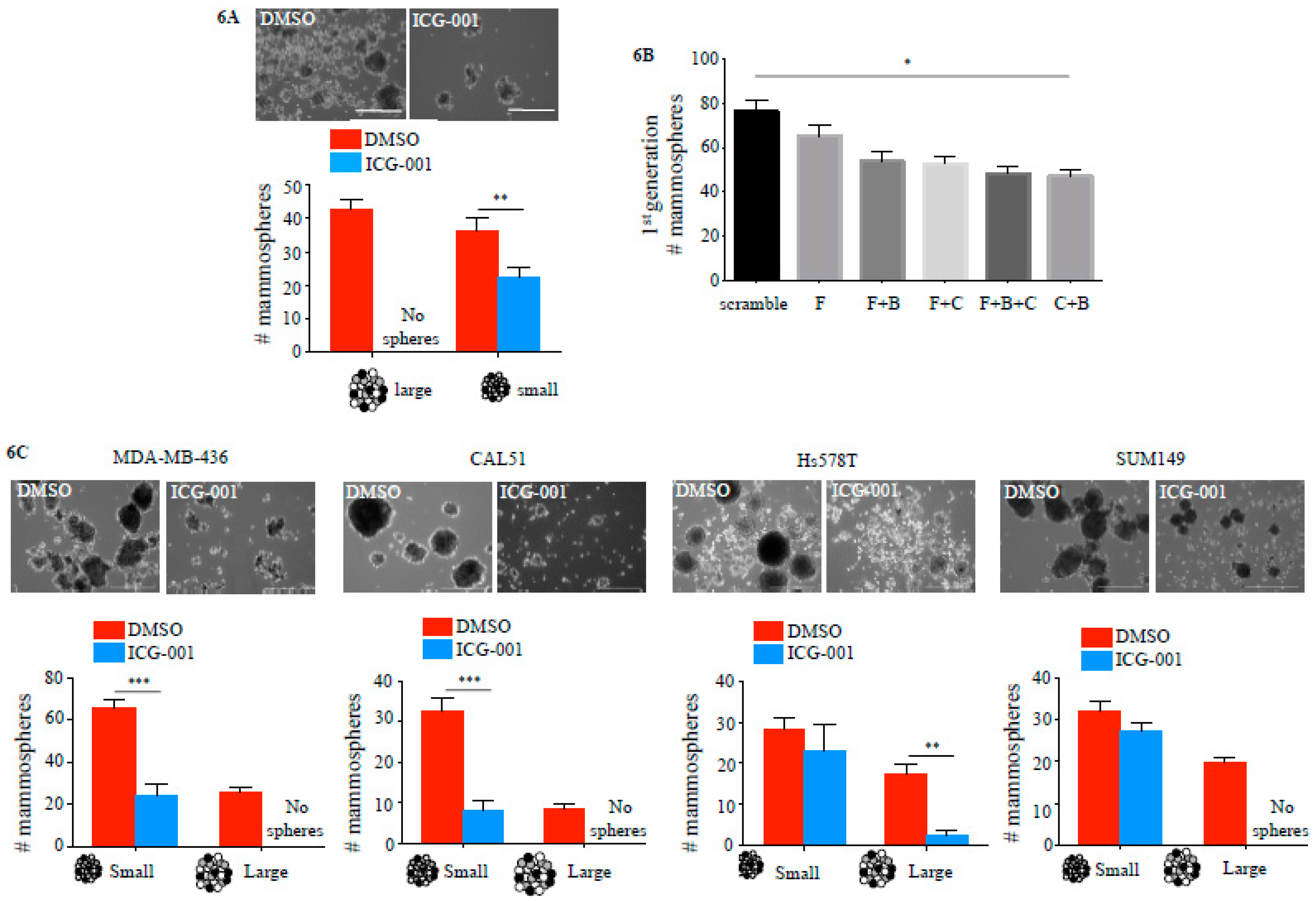
2.4. ICG-001 Ameliorates Paclitaxel-Induced Increases in FOXM1 Expression and CSC Phenotype as Well as Tumor Initiation Capacity In Vitro
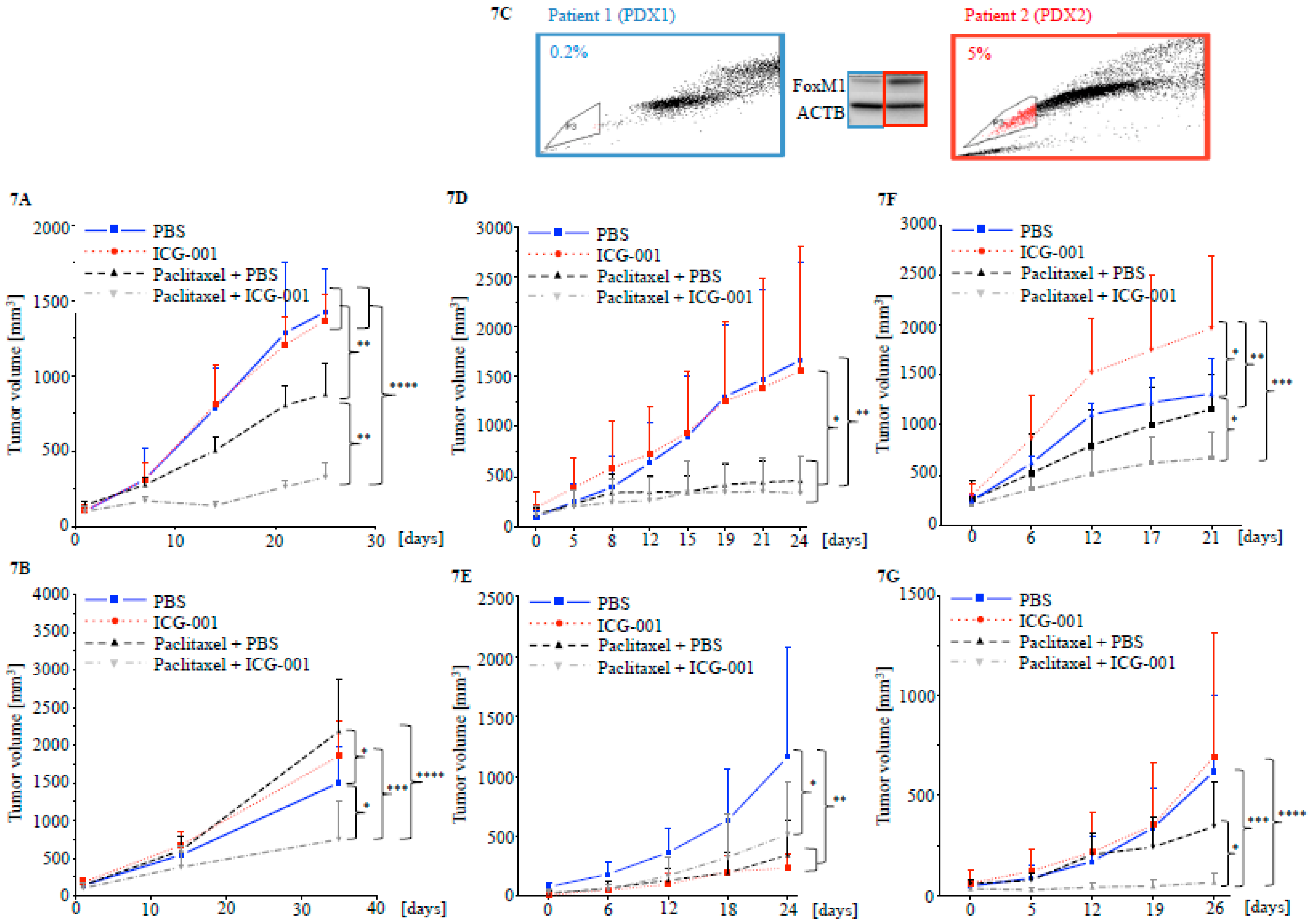
2.5. Targeting CBP Enhances the Response to Paclitaxel In Vivo Dependent on FOXM1 Expression
2.6. Tissue Micro Arrays (TMA) Stained for FOXM1 Demonstrated Correlation with TNBC Subtype and Tumor Grade
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transient Transfection and siRNA-Mediated Gene Knockdown (KD)
4.3. Xenografts of TNBC
4.4. Drug Application and Treatment Conditions
4.5. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.6. RNA Seq Library Preparation and Data Analysis
4.7. Public Breast Cancer Gene Expression Data Repositories
4.8. Fluorescence Activated Cell Sorting (FACS) and Side Population Assay
4.9. Protein Quantification
4.10. Co-Immunoprecipitation (CoIP)
4.11. Tissue Microarrays (TMAs)
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, L.N.; Ismaila, N.; McShane, L.M.; Andre, F.; Collyar, D.E.; Gonzalez-Angulo, A.M.; Hammond, E.H.; Kuderer, N.M.; Liu, M.C.; Mennel, R.G.; et al. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women With Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 1134–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Martin, M.; Nielsen, T.O.; Prat, A.; Voduc, D.; Rodriguez-Lescure, A.; Ruiz, A.; Chia, S.; Shepherd, L.; Ruiz-Borrego, M.; et al. Defining breast cancer intrinsic subtypes by quantitative receptor expression. Oncologist 2015, 20, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef]
- DiMeo, T.A.; Anderson, K.; Phadke, P.; Fan, C.; Perou, C.M.; Naber, S.; Kuperwasser, C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009, 69, 5364–5373. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; van Roy, F.; Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar]
- Ma, H.; Nguyen, C.; Lee, K.S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Masiello, D.; McMillian, M.; Nguyen, C.; Wu, Y.; Melendez, E.; Smbatyan, G.; Kida, A.; He, Y.; Teo, J.L.; et al. CBP/catenin antagonist safely eliminates drug-resistant leukemia-initiating cells. Oncogene 2016, 35, 3705–3717. [Google Scholar] [CrossRef] [PubMed]
- Major, M.L.; Lepe, R.; Costa, R.H. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol. Cell. Biol. 2004, 24, 2649–2661. [Google Scholar] [CrossRef] [PubMed]
- Pilarsky, C.; Wenzig, M.; Specht, T.; Saeger, H.D.; Grutzmann, R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 2004, 6, 744–750. [Google Scholar] [CrossRef]
- Korver, W.; Roose, J.; Clevers, H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fernandez, M.; Medema, R.H. Novel functions of FoxM1: From molecular mechanisms to cancer therapy. Front. Oncol. 2013, 3, 30. [Google Scholar] [CrossRef]
- Yang, N.; Wang, C.; Wang, Z.; Zona, S.; Lin, S.X.; Wang, X.; Yan, M.; Zheng, F.M.; Li, S.S.; Xu, B.; et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 2017. [Google Scholar] [CrossRef]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.L.; Kim, H.Y.; Moon, S.H.; et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Chan, L.S.; Ip, J.C.; Lo, C.; Yip, T.T.; Ngan, R.K.; Wong, R.N.; Lo, K.W.; Ng, W.T.; Lee, A.W.; et al. Therapeutic targeting of CBP/beta-catenin signaling reduces cancer stem-like population and synergistically suppresses growth of EBV-positive nasopharyngeal carcinoma cells with cisplatin. Sci. Rep. 2015, 5, 9979. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef]
- Cioce, M.; Gherardi, S.; Viglietto, G.; Strano, S.; Blandino, G.; Muti, P.; Ciliberto, G. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like- and early progenitor-targeting drugs. Cell Cycle 2010, 9, 2878–2887. [Google Scholar] [CrossRef]
- King, T.D.; Suto, M.J.; Li, Y. The Wnt/beta-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef]
- Lenz, H.J.; Kahn, M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Hallett, R.M.; Kondratyev, M.K.; Giacomelli, A.O.; Nixon, A.M.; Girgis-Gabardo, A.; Ilieva, D.; Hassell, J.A. Small molecule antagonists of the Wnt/beta-catenin signaling pathway target breast tumor-initiating cells in a Her2/Neu mouse model of breast cancer. PLoS ONE 2012, 7, e33976. [Google Scholar] [CrossRef]
- Koehler, A.N. A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol. 2010, 14, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Yang, C.; Yu, L.; Xie, L.; Hu, J.; Zeng, L.; Tan, Y. Adenovirus-mediated RNA interference targeting FOXM1 transcription factor suppresses cell proliferation and tumor growth of nasopharyngeal carcinoma. J. Gene Med. 2012, 14, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gartel, A.L. The suppression of FOXM1 and its targets in breast cancer xenograft tumors by siRNA. Oncotarget 2011, 2, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.M.; Myatt, S.S.; Marson, C.M.; Coombes, R.C.; Constantinidou, D.; Lam, E.W. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008, 7, 2022–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ging, N.C.; Komoda, T.; Hanada, T.; Suzuki, T.; Watanabe, K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett. 2005, 579, 6423–6427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapp, A.K.; Pricer, R.; Sturlis, S. Targeting transcription is no longer a quixotic quest. Nat. Chem. Biol. 2015, 11, 891–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halasi, M.; Gartel, A.L. Targeting FOXM1 in cancer. Biochem. Pharmacol. 2013, 85, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Gang, E.J.; Hsieh, Y.T.; Pham, J.; Zhao, Y.; Nguyen, C.; Huantes, S.; Park, E.; Naing, K.; Klemm, L.; Swaminathan, S.; et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17. [Google Scholar] [CrossRef]
- Saba, R.; Alsayed, A.; Zacny, J.P.; Dudek, A.Z. The Role of Forkhead Box Protein M1 in Breast Cancer Progression and Resistance to Therapy. Int. J. Breast Cancer 2016, 2016, 9768183. [Google Scholar] [CrossRef] [PubMed]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.S.; Myatt, S.S.; Kwok, J.M.; Sivanandan, K.; Coombes, R.C.; Medema, R.H.; Hartman, J.; et al. FOXM1 is a transcriptional target of ERalpha and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. (N. Y.) 2013, 42, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Alman, B.A. Side population cells in human cancers. Cancer Lett. 2008, 268, 1–9. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ring, A.; Nguyen, C.; Smbatyan, G.; Tripathy, D.; Yu, M.; Press, M.; Kahn, M.; Lang, J.E. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers 2018, 10, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10120525
Ring A, Nguyen C, Smbatyan G, Tripathy D, Yu M, Press M, Kahn M, Lang JE. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers. 2018; 10(12):525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10120525
Chicago/Turabian StyleRing, Alexander, Cu Nguyen, Goar Smbatyan, Debu Tripathy, Min Yu, Michael Press, Michael Kahn, and Julie E. Lang. 2018. "CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer" Cancers 10, no. 12: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10120525