Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair

 and
and
Abstract
:1. Introduction
2. Results
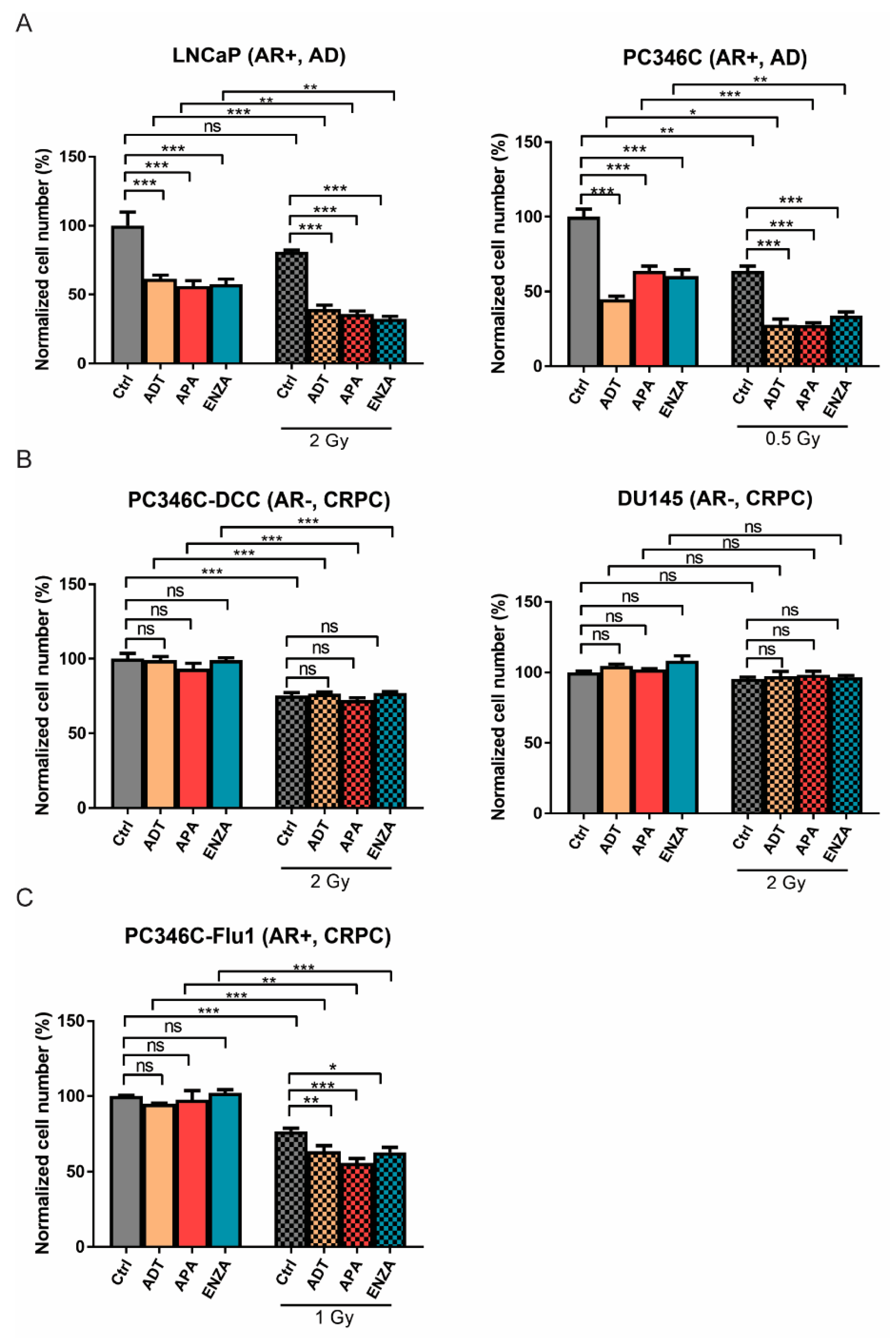
2.1. AR Suppression Enhances IR-Induced Cell Killing
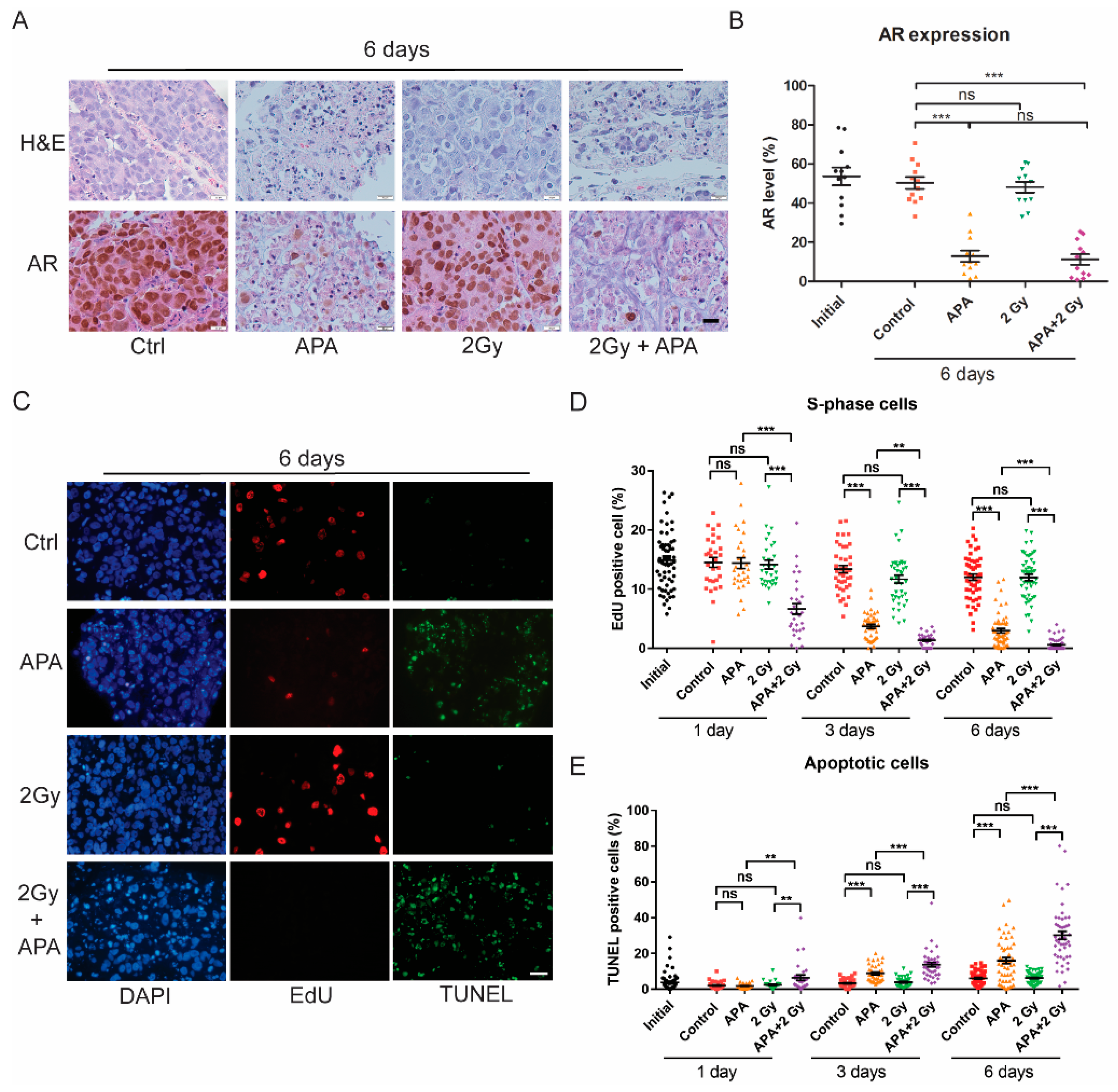
2.2. Apalutamide Enhances PCa Tissue Slice IR Sensitivity
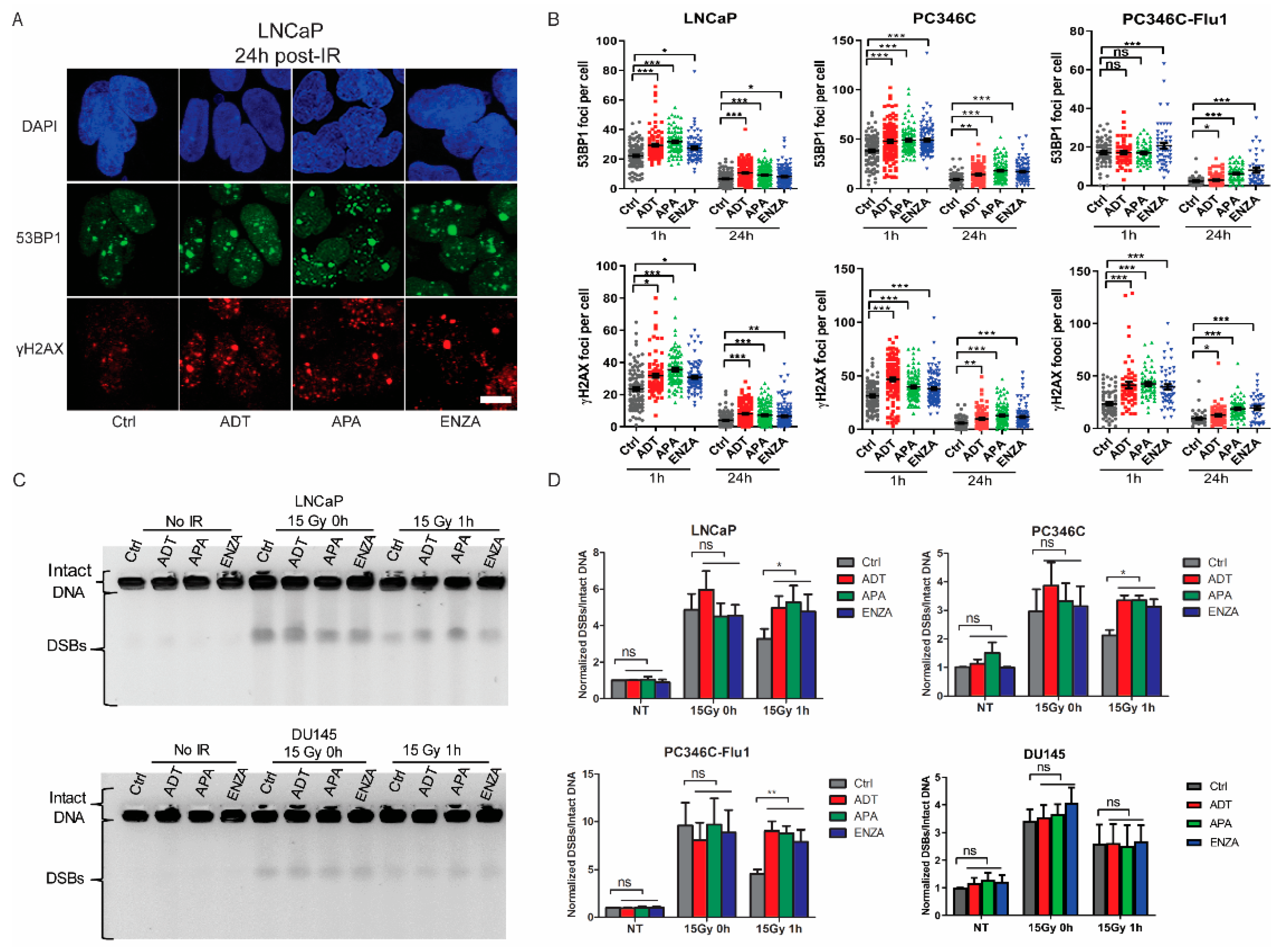
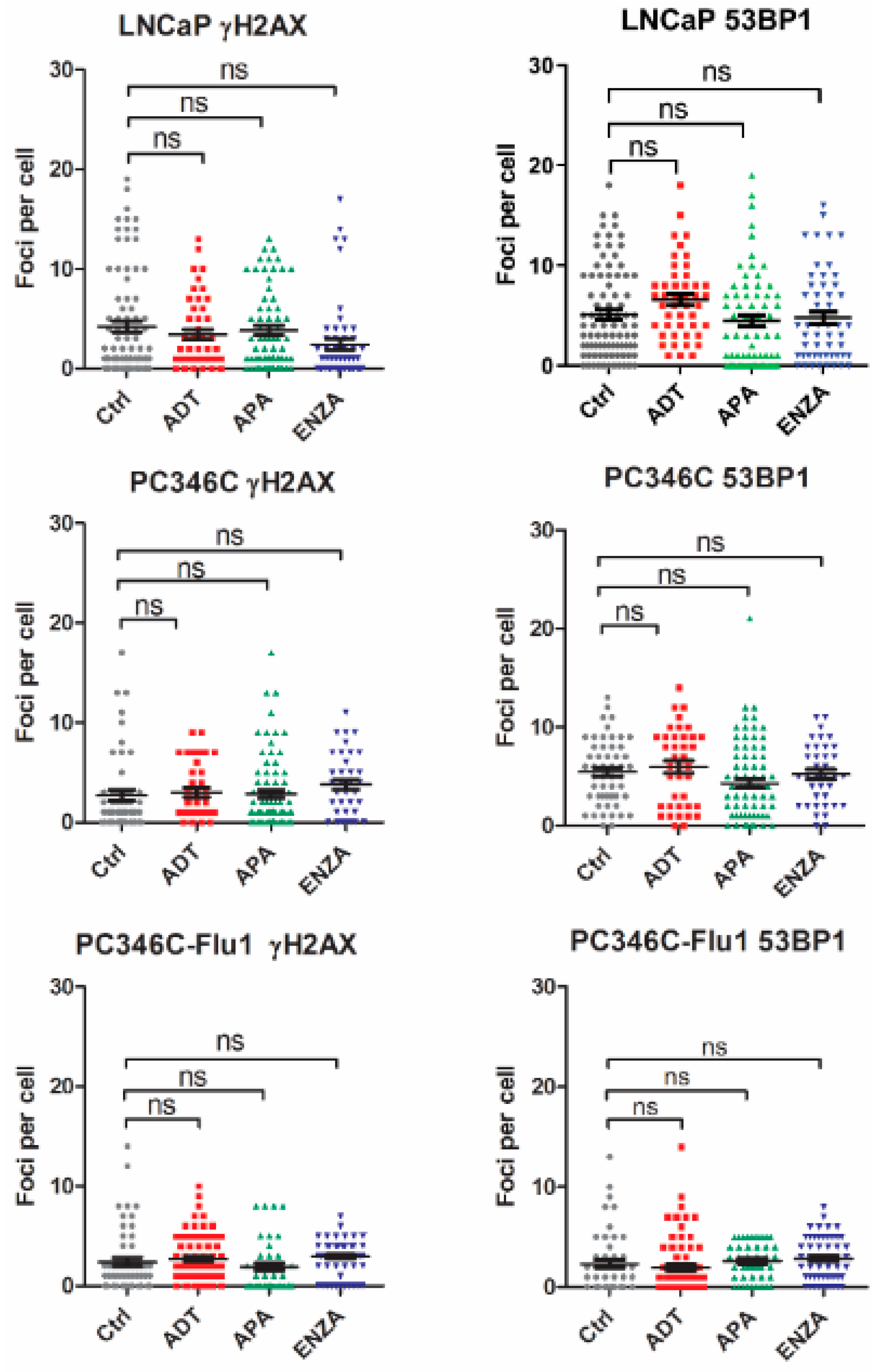
2.3. AR Suppression Inhibits DNA Damage Repair
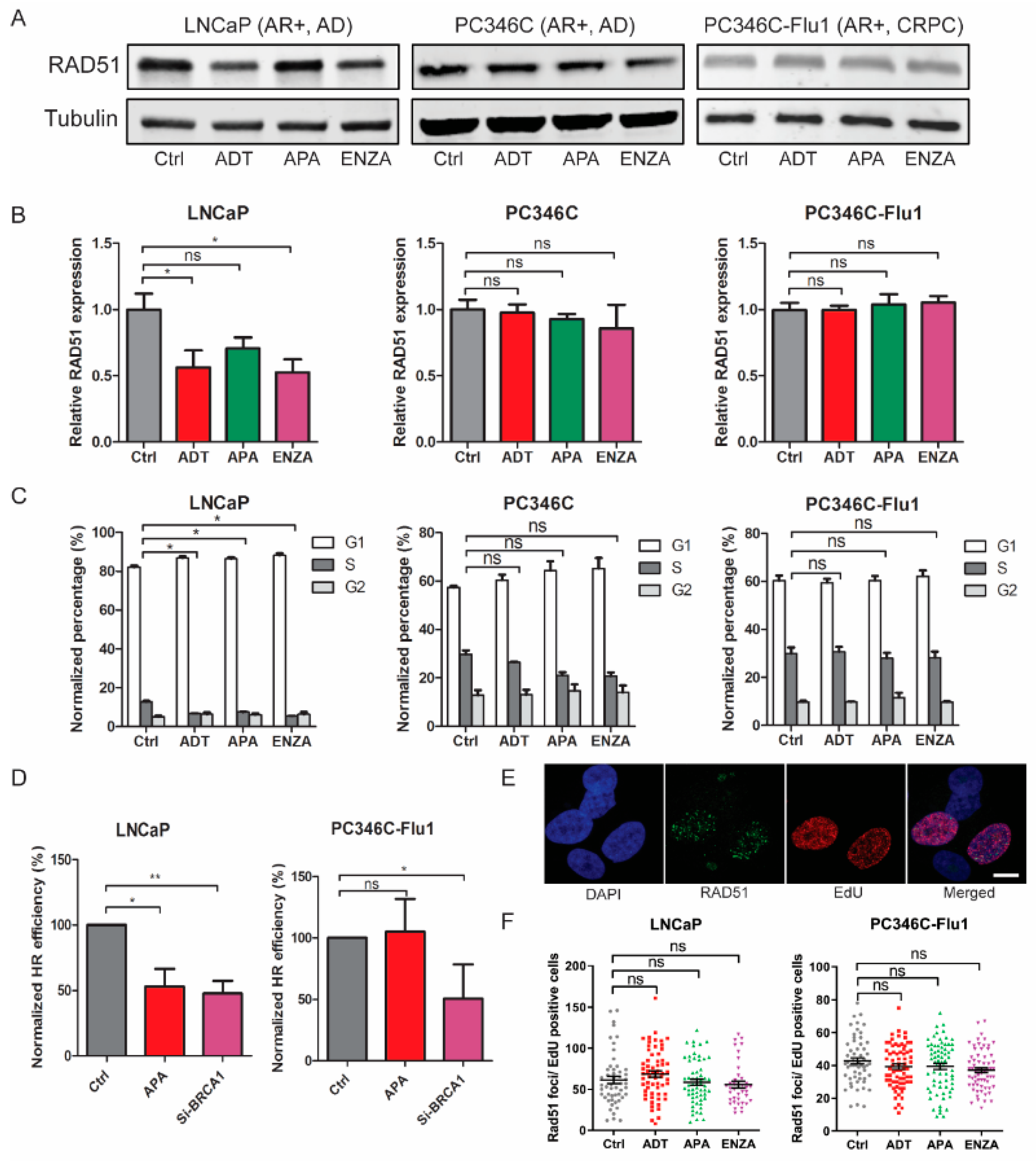
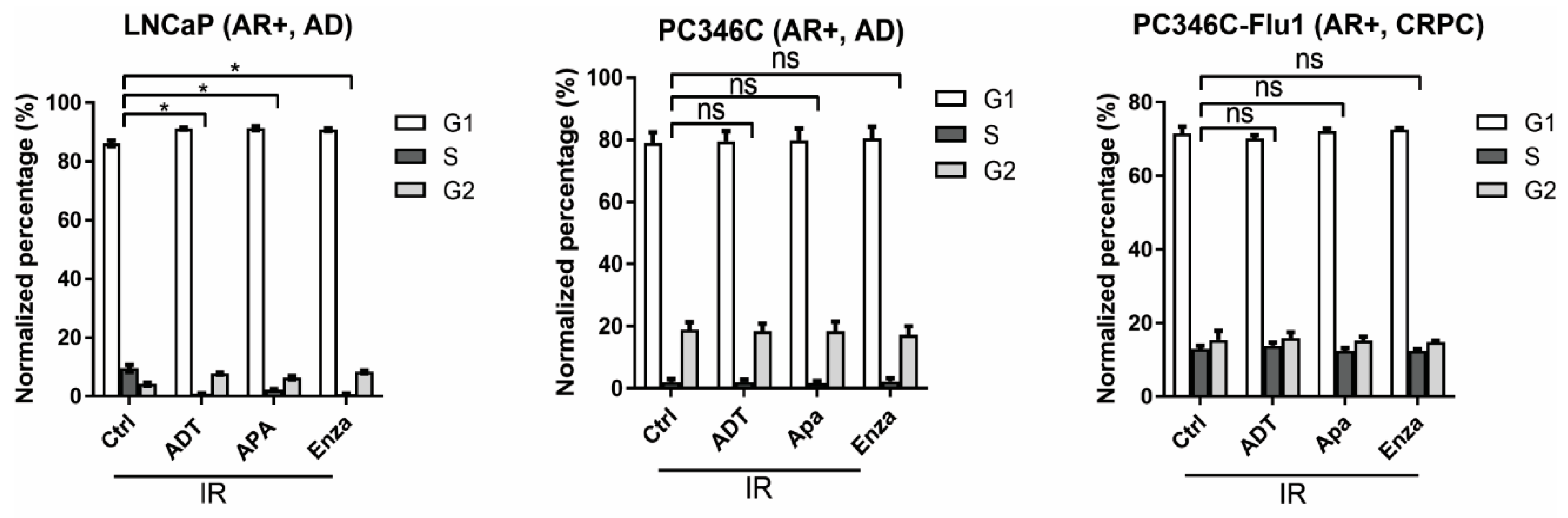
2.4. Reduced HR Efficiency Provoked by AR Suppression Results from Cell-Cycle Alterations
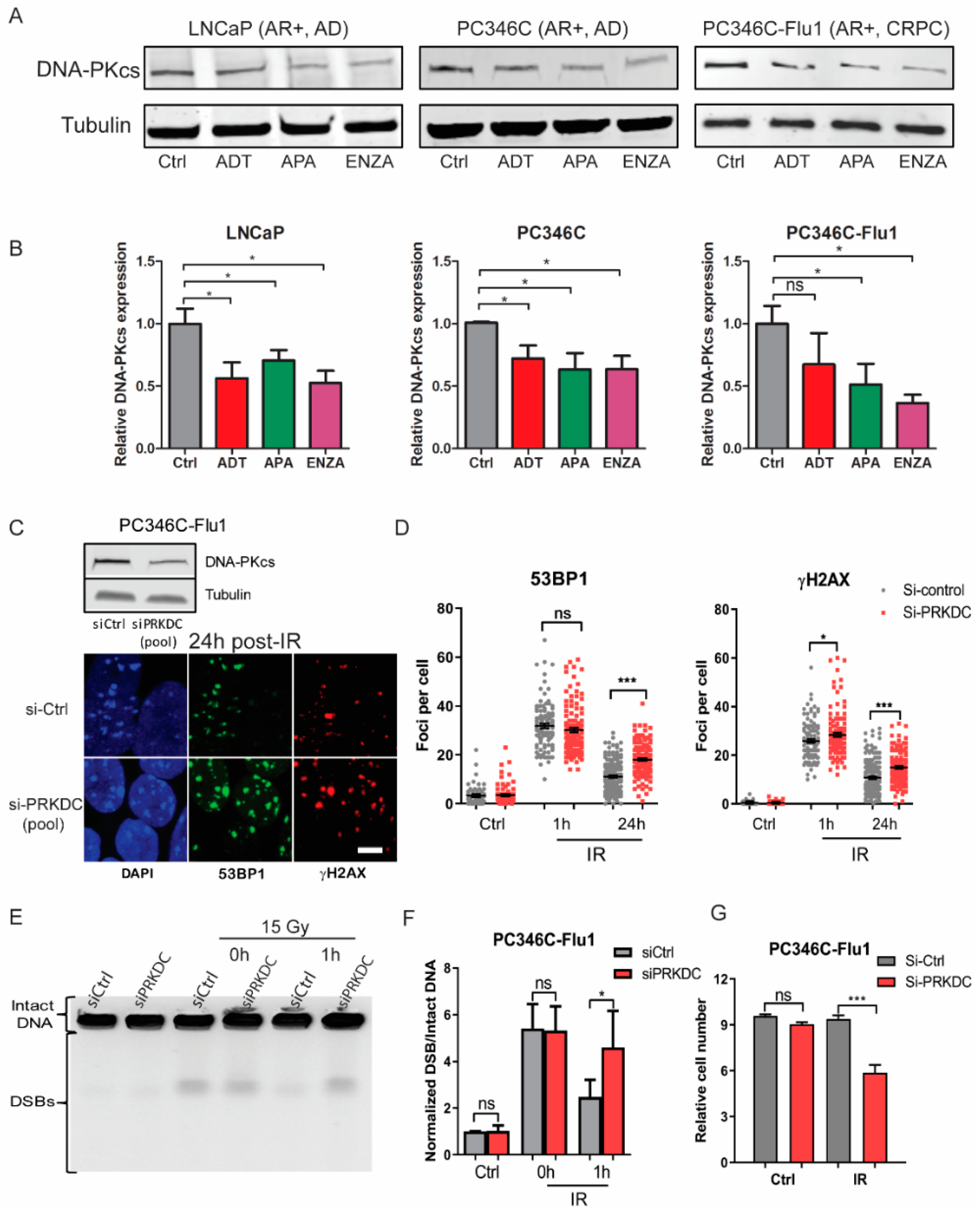
2.5. NHEJ Inhibition by AR Suppression Contributed to Radiosensitization
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Irradiation (IR)
4.4. Cell Growth Assay
4.5. Immunofluorescent Staining on Cells
4.6. Cell-Cycle Analysis
4.7. Tissue Slice Culture
4.8. Tissue Section Terminal Deoxynucleotidyl Transferase dUTP Nick end Labeling (TUNEL) and EdU Click-IT Assays
4.9. Tissue Section Hematoxylin and Eosin (H&E)
4.10. Tissue Section Immunohistochemical Staining
4.11. Pulsed-Field Gel Electrophoresis
4.12. HR Assay
4.13. Western Blot
4.14. RNA Interference
4.15. Image Acquisition and Quantification
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | DU145 | LNCaP | PC346C | PC346C-DCC | PC346C-Flu1 |
|---|---|---|---|---|---|
| Culture Medium | RPMI 1640 FCS (10%) PS | RPMI 1640 FCS (10%) PS | DMEM/Ham’s F12 BSA (0.01%) FCS (2%) Epidermal growth factor (10 ng/mL) Insulin–transferrin–selenium (1%) Hydrocortisone (0.5 µg/mL) Triiodothyronine (1 nM) Phosphoethanolamine (0.1 mM) Cholera toxin (50 ng/mL) Fibronectin (100 ng/mL) Fetuine (20 µg/mL) R1881 (0.1 nM) PS | DMEM/Ham’s F12 BSA (0.01%) DCC (2%) Epidermal growth factor (10 ng/mL) Insulin–transferrin–selenium (1%) Hydrocortisone (0.5 µg/mL) Triiodothyronine (1 nM) Phosphoethanolamine (0.1 mM) Cholera toxin (50 ng/mL) Fibronectin (100 ng/mL) Fetuine (20 µg/mL) R1881 (0.1 nM) PS | PC346C-DCC medium plus OH-flutamide (1 µM) |
| AR | – | + | + | − | ++ |
| p53 | Missense Mutation | Silent | WT | WT | WT |
| BRCA2 | Missense Mutation | WT | WT | WT | WT |
| PTEN | WT | Frame shift deletion | Nonsense mutation | Nonsense mutation | Nonsense mutation |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Djavan, B.; Moul, J.W.; Zlotta, A.; Remzi, M.; Ravery, V. Psa progression following radical prostatectomy and radiation therapy: New standards in the new millennium. Eur. Urol. 2003, 43, 12–27. [Google Scholar] [CrossRef]
- Khan, M.A.; Han, M.; Partin, A.W.; Epstein, J.I.; Walsh, P.C. Long-term cancer control of radical prostatectomy in men younger than 50 years of age: Update 2003. Urology 2003, 62, 86–91. [Google Scholar] [CrossRef]
- Schmidt-Hansen, M.; Hoskin, P.; Kirkbride, P.; Hasler, E.; Bromham, N. Hormone and radiotherapy versus hormone or radiotherapy alone for non-metastatic prostate cancer: A systematic review with meta-analyses. Clin. Oncol. 2014, 26, 21–46. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.U.; Hunt, D.; McGowan, D.G.; Amin, M.B.; Chetner, M.P.; Bruner, D.W.; Leibenhaut, M.H.; Husain, S.M.; Rotman, M.; Souhami, L.; et al. Radiotherapy and short-term androgen deprivation for localized prostate cancer. N. Engl. J. Med. 2011, 365, 107–118. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Manola, J.; Loffredo, M.; Renshaw, A.A.; DellaCroce, A.; Kantoff, P.W. 6-month androgen suppression plus radiation therapy vs radiation therapy alone for patients with clinically localized prostate cancera randomized controlled trial. JAMA 2004, 292, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “brcaness” and parp inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, e7479. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the parp pathway in prostate cancer. Nat. Commun. 2017, 8, e374. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. Arn-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef]
- Ghashghaei, M.; Niazi, T.M.; Heravi, M.; Bekerat, H.; Trifiro, M.; Paliouras, M.; Muanza, T. Enhanced radiosensitization of enzalutamide via schedule dependent administration to androgen-sensitive prostate cancer cells. Prostate 2018, 78, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; van Weerden, W.M.; de Ridder, C.M.A.; Erkens-Schulze, S.; Schonfeld, E.; Meijer, T.G.; Kanaar, R.; van Gent, D.C.; Nonnekens, J. Ex vivo treatment of prostate tumor tissue recapitulates in vivo therapy response. Prostate 2019, 79, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Wang, J.; Freeman, M.L.; Kirschner, A.N. Radiosensitization by enzalutamide for human prostate cancer is mediated through the DNA damage repair pathway. PLoS ONE 2019, 14, e0214670. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.-J.; Chen, Y.; Chen, D.; Niu, Y.; Li, G.; Keng, P.; Yeh, S.; Chang, C. Preclinical study using androgen receptor (ar) degradation enhancer to increase radiotherapy efficacy via targeting radiation-increased ar to better suppress prostate cancer progression. EBioMedicine 2019, 40, 504–516. [Google Scholar] [CrossRef]
- van Oijen, M.G.; Medema, R.H.; Slootweg, P.J.; Rijksen, G. Positivity of the proliferation marker ki-67 in noncycling cells. Am. J. Clin. Pathol. 1998, 110, 24–31. [Google Scholar] [CrossRef]
- Tarish, F.L.; Schultz, N.; Tanoglidi, A.; Hamberg, H.; Letocha, H.; Karaszi, K.; Hamdy, F.C.; Granfors, T.; Helleday, T. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci. Transl. Med. 2015, 7, e312. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: Results from nci 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly (adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Van der Steen, T.; Tindall, D.J. Are androgen receptor variants a substitute for the full-length receptor? Nat. Rev. Urol. 2015, 12, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. Ar-v7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, R.; Xu, K.; Ding, S.; Li, J.; Baek, G.; Ramanand, S.G.; Ding, S.; Liu, Z.; Gao, Y.; et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res. 2017, 77, 4745–4754. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Babich, J.W.; Kratochwil, C.; Giesel, F.L.; Eisenhut, M.; Kopka, K.; Haberkorn, U. The rise of psma ligands for diagnosis and therapy of prostate cancer. J. Nucl. Med. 2016, 57, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Essers, J.; Hendriks, R.W.; Wesoly, J.; Beerens, C.E.; Smit, B.; Hoeijmakers, J.H.; Wyman, C.; Dronkert, M.L.; Kanaar, R. Analysis of mouse rad54 expression and its implications for homologous recombination. DNA Repair 2002, 1, 779–793. [Google Scholar] [CrossRef]
- Weterings, E.; Verkaik, N.S.; Bruggenwirth, H.T.; Hoeijmakers, J.H.; van Gent, D.C. The role of DNA dependent protein kinase in synapsis of DNA ends. Nucleic Acids Res. 2003, 31, 7238–7246. [Google Scholar] [CrossRef] [Green Version]
- Moll, J.M.; Kumagai, J.; van Royen, M.E.; Teubel, W.J.; van Soest, R.J.; French, P.J.; Homma, Y.; Jenster, G.; de Wit, R.; van Weerden, W.M. A bypass mechanism of abiraterone-resistant prostate cancer: Accumulating cyp17a1 substrates activate androgen receptor signaling. Prostate 2019, 79, 937–948. [Google Scholar] [CrossRef]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (du 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef]
- Marques, R.B.; Erkens-Schulze, S.; de Ridder, C.M.; Hermans, K.G.; Waltering, K.; Visakorpi, T.; Trapman, J.; Romijn, J.C.; van Weerden, W.M.; Jenster, G. Androgen receptor modifications in prostate cancer cells upon long-termandrogen ablation and antiandrogen treatment. Int. J. Cancer 2005, 117, 221–229. [Google Scholar] [CrossRef]
- Nonnekens, J.; van Kranenburg, M.; Beerens, C.E.; Suker, M.; Doukas, M.; van Eijck, C.H.; de Jong, M.; van Gent, D.C. Potentiation of peptide receptor radionuclide therapy by the parp inhibitor olaparib. Theranostics 2016, 6, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- van den Tempel, N.; Laffeber, C.; Odijk, H.; van Cappellen, W.A.; van Rhoon, G.C.; Franckena, M.; Kanaar, R. The effect of thermal dose on hyperthermia-mediated inhibition of DNA repair through homologous recombination. Oncotarget 2017, 8, e44593. [Google Scholar]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in brca-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- van Weerden, W.M.; de Ridder, C.M.; Verdaasdonk, C.L.; Romijn, J.C.; van der Kwast, T.H.; Schroder, F.H.; van Steenbrugge, G.J. Development of seven new human prostate tumor xenograft models and their histopathological characterization. Am. J. Pathol. 1996, 149, 1055–1062. [Google Scholar]
- Naipal, K.A.T.; Verkaik, N.S.; Sánchez, H.; van Deurzen, C.H.M.; den Bakker, M.A.; Hoeijmakers, J.H.J.; Kanaar, R.; Vreeswijk, M.P.G.; Jager, A.; van Gent, D.C. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 2016, 16, e78. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase Ⅰ poisoning results in parp-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, e417. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Liao, C.-Y.; Chtatou, H.; Incrocci, L.; van Gent, D.C.; van Weerden, W.M.; Nonnekens, J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers 2019, 11, 1593. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101593
Zhang W, Liao C-Y, Chtatou H, Incrocci L, van Gent DC, van Weerden WM, Nonnekens J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers. 2019; 11(10):1593. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101593
Chicago/Turabian StyleZhang, Wenhao, Chen-Yi Liao, Hajar Chtatou, Luca Incrocci, Dik C. van Gent, Wytske M. van Weerden, and Julie Nonnekens. 2019. "Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair" Cancers 11, no. 10: 1593. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101593