A Combined Systemic Strategy for Overcoming Cisplatin Resistance in Head and Neck Cancer: From Target Identification to Drug Discovery

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Establishment of Cisplatin-Resistant (CR) Sublines of HNC Cells
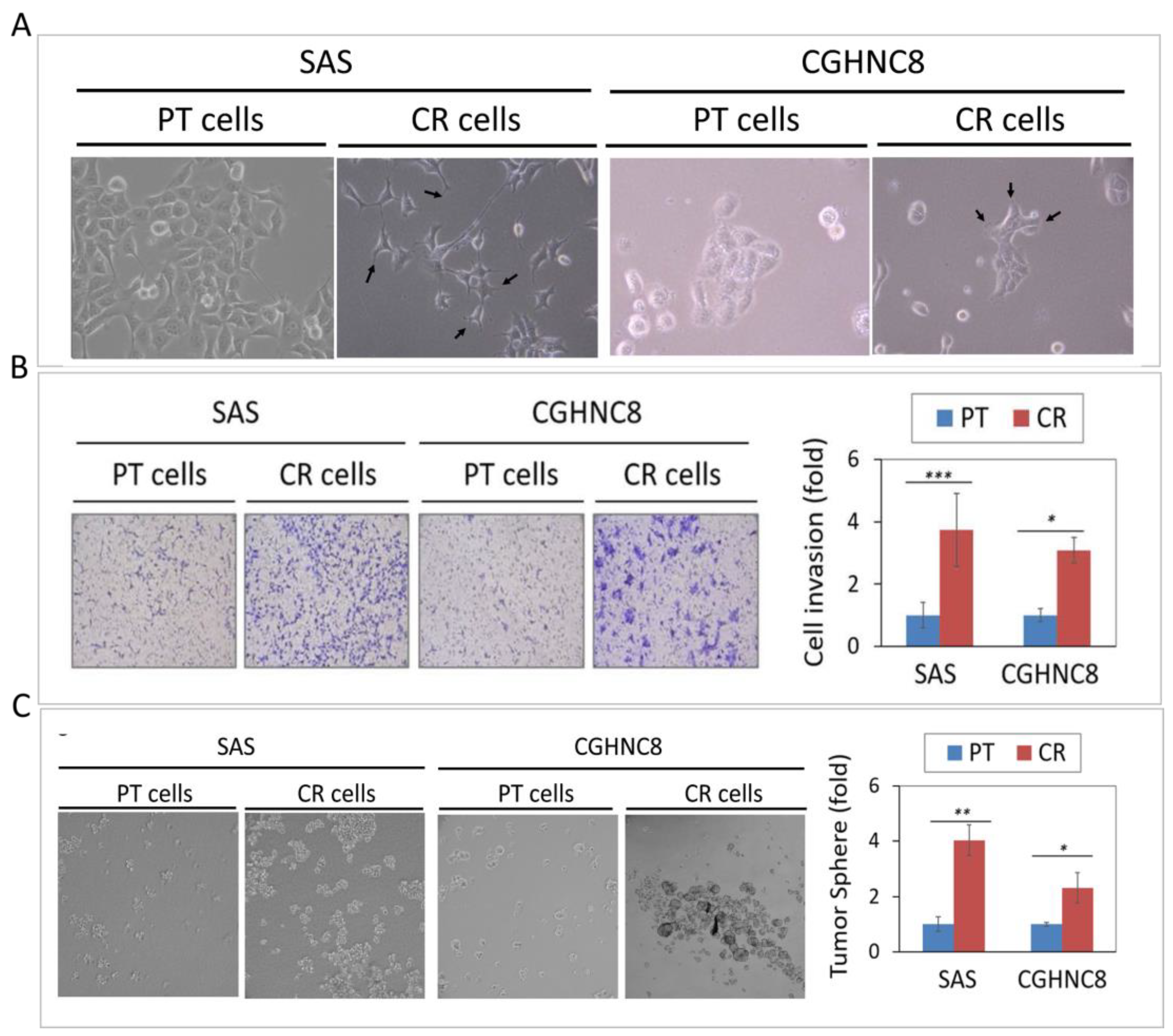
2.2. The CR Sublines Exhibit Highly Invasive and Cancer Stemness Properties
2.3. Transcriptomic Profile and Functional Pathways Associated with CR in HNC
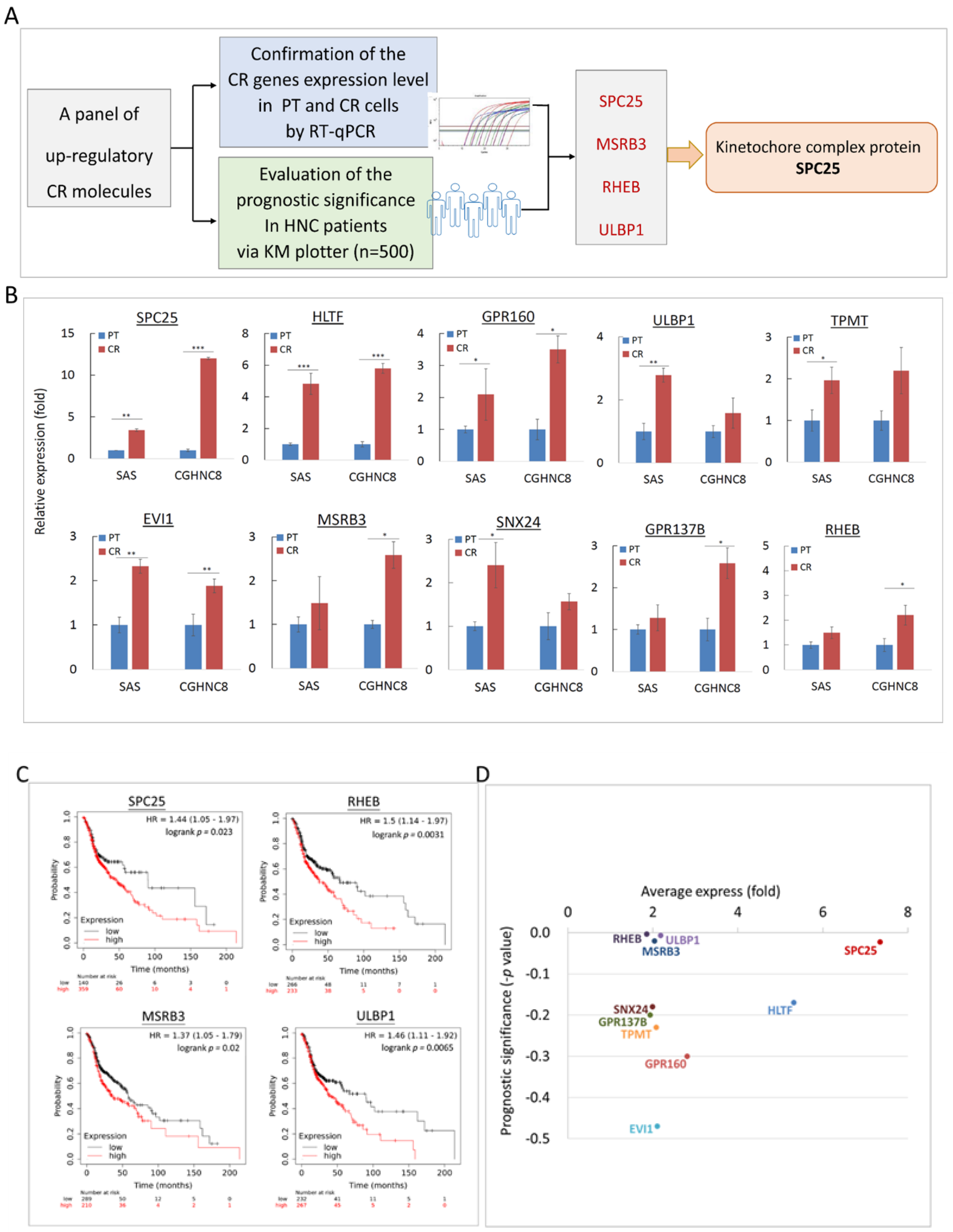
2.4. Panel Molecules that Were Upregulated in CR Cells and Led to Poor Prognosis in HNC Patients
2.5. Cmap Analysis Identified Celastrol as a Potent Compound to Overcome CR
2.6. Silencing Kinetochore Protein SPC25 Increased Cisplatin Sensitivity in HNC Cells
3. Discussion
4. Materials and Methods
4.1. Establishment of Cisplatin-Resistant (CR) Sublines of HNC
4.2. Transcriptional Profiling and Validation of CR Genes in HNC
4.3. Analysis of Functional Pathways and Prognostic Significance in HNC
4.4. Cellular Protein Extraction and Immunoblot Analysis
4.5. Plasmid Construction and Cellular Transfection
4.6. Cell Survival, Colony Formation, and Chemosensitivity Analyses
4.7. Cell Invasion and Tumorsphere Assays
4.8. Connectivity Map (Cmap) Analysis to Search Potential CR Sensitizers
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastman, A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol. Ther. 1987, 34, 155–166. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, e102925. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.J.; Ahn, M.J.; Kim, H.S.; Yi, S.Y.; Han, J.; Lee, S.K.; Ahn, Y.C.; Jeong, H.S.; Son, Y.I.; Baek, J.H.; et al. ERCC1 expression as a predictive marker of squamous cell carcinoma of the head and neck treated with cisplatin-based concurrent chemoradiation. Br. J. Cancer 2008, 99, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.J.; Chen, C.H.; Chien, C.Y.; Li, S.H.; Tsai, H.T.; Chen, Y.J. High ERCC1 expression predicts cisplatin-based chemotherapy resistance and poor outcome in unresectable squamous cell carcinoma of head and neck in a betel-chewing area. J. Transl. Med. 2011, 9, e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuelei, M.; Jingwen, H.; Wei, D.; Hongyu, Z.; Jing, Z.; Changle, S.; Lei, L. ERCC1 plays an important role in predicting survival outcomes and treatment response for patients with HNSCC: A meta-analysis. Oral Oncol. 2015, 51, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Warta, R.; Theile, D.; Mogler, C.; Herpel, E.; Grabe, N.; Lahrmann, B.; Plinkert, P.K.; Herold-Mende, C.; Weiss, J.; Dyckhoff, G. Association of drug transporter expression with mortality and progression-free survival in stage IV head and neck squamous cell carcinoma. PLoS ONE 2014, 9, e108908. [Google Scholar] [CrossRef]
- Tanimoto, T.; Tsuda, H.; Imazeki, N.; Ohno, Y.; Imoto, I.; Inazawa, J.; Matsubara, O. Nuclear expression of cIAP-1, an apoptosis inhibiting protein, predicts lymph node metastasis and poor patient prognosis in head and neck squamous cell carcinomas. Cancer Lett. 2005, 224, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Feng, Z.E.; Yan, M.; Hanada, S.; Zuo, H.; Yang, C.Z.; Han, Z.G.; Guo, W.; Chen, W.T.; Zhang, P. XIAP is a predictor of cisplatin-based chemotherapy response and prognosis for patients with advanced head and neck cancer. PLoS ONE 2012, 7, e31601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chen, B.L.; Zhou, Y.W.; Guo, R.W.; Shuai, M.T.; Zeng, J.X.; Leng, A.M. Expression and clinical significance of Apollon in esophageal squamous cell carcinoma. Mol. Med. Rep. 2016, 14, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef]
- Peitzsch, C.; Nathansen, J.; Schniewind, S.I.; Schwarz, F.; Dubrovska, A. Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma: Identification, Characterization and Clinical Implications. Cancers 2019, 11, 616. [Google Scholar] [CrossRef] [Green Version]
- Xia, P. Surface markers of cancer stem cells in solid tumors. Curr. Stem Cell Res. 2014, 9, 102–111. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharm. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Gyorffy, B.; Benke, Z.; Lanczky, A.; Balazs, B.; Szallasi, Z.; Timar, J.; Schafer, R. RecurrenceOnline: An online analysis tool to determine breast cancer recurrence and hormone receptor status using microarray data. Breast Cancer Res. Treat. 2012, 132, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szasz, A.M.; Lanczky, A.; Nagy, A.; Forster, S.; Hark, K.; Green, J.E.; Boussioutas, A.; Busuttil, R.; Szabo, A.; Gyorffy, B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 2016, 7, 49322–49333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiato, H.; DeLuca, J.; Salmon, E.D.; Earnshaw, W.C. The dynamic kinetochore-microtubule interface. J. Cell Sci. 2004, 117, 5461–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheeseman, I.M.; Desai, A. Molecular architecture of the kinetochore-microtubule interface. Nat. Rev. Mol. Cell Biol. 2008, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Pasqualato, S.; Screpanti, E.; Varetti, G.; Santaguida, S.; Dos Reis, G.; Maiolica, A.; Polka, J.; De Luca, J.G.; De Wulf, P.; et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell 2008, 133, 427–439. [Google Scholar] [CrossRef] [Green Version]
- DeLuca, J.G.; Musacchio, A. Structural organization of the kinetochore-microtubule interface. Curr. Opin. Cell Biol. 2012, 24, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Dai, X.; Chen, Y.; Shen, Y.; Lei, S.; Xiao, T.; Bartfai, T.; Ding, J.; Wang, M.W. G protein-coupled receptor GPR160 is associated with apoptosis and cell cycle arrest of prostate cancer cells. Oncotarget 2016, 7, 12823–12839. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wang, H.; Deng, J.; Zheng, X.; Ling, Y.; Gong, Y. Prognostic significance of the EVI1 gene expression in patients with acute myeloid leukemia: A meta-analysis. Ann. Hematol. 2019, 98, 2485–2496. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Zhao, T.; Zhu, H.H.; Wang, J.; Jia, J.S.; Lai, Y.Y.; Zhao, X.S.; Shi, H.X.; Liu, Y.R.; Jiang, H.; et al. High EVI1 Expression Predicts Poor Outcomes in Adult Acute Myeloid Leukemia Patients with Intermediate Cytogenetic Risk Receiving Chemotherapy. Med. Sci. Monit. 2018, 24, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debauve, G.; Capouillez, A.; Belayew, A.; Saussez, S. The helicase-like transcription factor and its implication in cancer progression. Cell Mol. Life Sci. 2008, 65, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Brown, G.W. Post-replication repair: Rad5/HLTF regulation, activity on undamaged templates, and relationship to cancer. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 301–332. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Jiang, L.; Luo, S.; Zhao, X.; Hu, H.; Zhao, G.; Tang, W. Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J. Transl. Med. 2020, 18, e160. [Google Scholar] [CrossRef] [Green Version]
- Sohretoglu, D.; Zhang, C.; Luo, J.; Huang, S. ReishiMax inhibits mTORC1/2 by activating AMPK and inhibiting IGFR/PI3K/Rheb in tumor cells. Signal Transduct. Target. Ther. 2019, 4, e21. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Shen, L.; Li, F.; Yang, J.; Wan, X.; Ouyang, M. Silencing of RHEB inhibits cell proliferation and promotes apoptosis in colorectal cancer cells via inhibition of the mTOR signaling pathway. J. Cell Physiol. 2020, 235, 442–453. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Zhao, M.; Huang, H.; Wu, J. Increased expression of methionine sulfoxide reductases B3 is associated with poor prognosis in gastric cancer. Oncol. Lett. 2019, 18, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Kwak, G.H.; Kim, T.H.; Kim, H.Y. Down-regulation of MsrB3 induces cancer cell apoptosis through reactive oxygen species production and intrinsic mitochondrial pathway activation. Biochem. Biophys. Res. Commun. 2017, 483, 468–474. [Google Scholar] [CrossRef]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, e2040. [Google Scholar] [CrossRef] [Green Version]
- Martin-Lluesma, S.; Stucke, V.M.; Nigg, E.A. Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 2002, 297, 2267–2270. [Google Scholar] [CrossRef]
- McCleland, M.L.; Kallio, M.J.; Barrett-Wilt, G.A.; Kestner, C.A.; Shabanowitz, J.; Hunt, D.F.; Gorbsky, G.J.; Stukenberg, P.T. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr. Biol. 2004, 14, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Musacchio, A.; Petrovic, A. The Ndc80 complex: Hub of kinetochore activity. Febs. Lett. 2007, 581, 2862–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.C.; Lee, S.E.; Xu, Y.N.; Kim, N.H. Perturbation of Spc25 expression affects meiotic spindle organization, chromosome alignment and spindle assembly checkpoint in mouse oocytes. Cell Cycle 2010, 9, 4552–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhl, L.M.; Vader, G. Kinetochores, cohesin, and DNA breaks: Controlling meiotic recombination within pericentromeres. Yeast 2019, 36, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, H.; Yang, H.; Dai, H. SPC25 upregulation increases cancer stem cell properties in non-small cell lung adenocarcinoma cells and independently predicts poor survival. Biomed. Pharm. 2018, 100, 233–239. [Google Scholar] [CrossRef]
- Cui, F.; Hu, J.; Fan, Y.; Tan, J.; Tang, H. Knockdown of spindle pole body component 25 homolog inhibits cell proliferation and cycle progression in prostate cancer. Oncol. Lett. 2018, 15, 5712–5720. [Google Scholar] [CrossRef] [Green Version]
- Cui, F.; Tang, H.; Tan, J.; Hu, J. Spindle pole body component 25 regulates stemness of prostate cancer cells. Aging 2018, 10, 3273–3282. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, Y.; Li, Z.; Bu, Q.; Sun, T.; Wang, H.; Sun, H.; Cao, X. Up-regulation of SPC25 promotes breast cancer. Aging 2019, 11, 5689–5704. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, K.; Huang, Y.; Luo, F.; Hu, K.; Cai, Q. SPC25 may promote proliferation and metastasis of hepatocellular carcinoma via p53. Febs. Open Bio. 2020, 10, 1261–1275. [Google Scholar] [CrossRef]
- Sun, Z.Y.; Wang, W.; Gao, H.; Chen, Q.F. Potential therapeutic targets of the nuclear division cycle 80 (NDC80) complexes genes in lung adenocarcinoma. J. Cancer 2020, 11, 2921–29344. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ma, L.; Zhou, G.B. The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules 2011, 16, 5283–5297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.N.; Zhao, N.; Chen, J.Y.; Ye, P.P.; Nan, X.W.; Zhou, H.H.; Jiang, Q.W.; Yang, Y.; Huang, J.R.; Yuan, M.L.; et al. Celastrol Inhibits the Growth of Ovarian Cancer Cells in vitro and in vivo. Front. Oncol. 2019, 9, e2. [Google Scholar] [CrossRef] [PubMed]
- Mi, C.; Shi, H.; Ma, J.; Han, L.Z.; Lee, J.J.; Jin, X. Celastrol induces the apoptosis of breast cancer cells and inhibits their invasion via downregulation of MMP-9. Oncol. Rep. 2014, 32, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.; Zhao, W.; Kong, X.; Li, H.; Ouyang, J. Celastrol inhibits lipopolysaccharide-induced angiogenesis by suppressing TLR4-triggered nuclear factor-kappa B activation. Acta. Haematol. 2014, 131, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.X.; Li, N.; Wu, J.L.; Zhou, Y.L.; He, J.X.; Liu, L.; Leung, E.L. Celastrol induces apoptosis in gefitinib-resistant non-small cell lung cancer cells via caspases-dependent pathways and Hsp90 client protein degradation. Molecules 2014, 19, 3508–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzanotte, L.; An, N.; Mol, I.M.; Lowik, C.W.; Kaijzel, E.L. A new multicolor bioluminescence imaging platform to investigate NF-kappaB activity and apoptosis in human breast cancer cells. PLoS ONE 2014, 9, e85550. [Google Scholar] [CrossRef]
- Sha, M.; Ye, J.; Zhang, L.X.; Luan, Z.Y.; Chen, Y.B.; Huang, J.X. Celastrol induces apoptosis of gastric cancer cells by miR-21 inhibiting PI3K/Akt-NF-kappaB signaling pathway. Pharmacology 2014, 93, 39–46. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, S.; Ren, H.; Huang, H.; Ji, W.; Hao, J. Inhibition of autophagy strengthens celastrol-induced apoptosis in human pancreatic cancer in vitro and in vivo models. Curr. Mol. Med. 2014, 14, 555–563. [Google Scholar] [CrossRef]
- Zheng, L.; Fu, Y.; Zhuang, L.; Gai, R.; Ma, J.; Lou, J.; Zhu, H.; He, Q.; Yang, B. Simultaneous NF-kappaB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo. Int. J. Cancer 2014, 135, 1721–1732. [Google Scholar] [CrossRef]
- Boridy, S.; Le, P.U.; Petrecca, K.; Maysinger, D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014, 5, e1216. [Google Scholar] [CrossRef]
- Zanphorlin, L.M.; Alves, F.R.; Ramos, C.H. The effect of celastrol, a triterpene with antitumorigenic activity, on conformational and functional aspects of the human 90kDa heat shock protein Hsp90alpha, a chaperone implicated in the stabilization of the tumor phenotype. Biochim. Biophys. Acta 2014, 1840, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Fribley, A.M.; Miller, J.R.; Brownell, A.L.; Garshott, D.M.; Zeng, Q.; Reist, T.E.; Narula, N.; Cai, P.; Xi, Y.; Callaghan, M.U.; et al. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp. Cell Res. 2015, 330, 412–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.Y.; Guo, Y.; Zhang, W.; Ma, C.G.; Zhang, Y.X.; Wang, C.; Wang, H.X. Celastrol enhanced the anticancer effect of lapatinib in human hepatocellular carcinoma cells in vitro. J. Buon. 2014, 19, 412–418. [Google Scholar] [PubMed]
- Moreira, H.; Szyjka, A.; Gasiorowski, K. Chemopreventive activity of celastrol in drug-resistant human colon carcinoma cell cultures. Oncotarget 2018, 9, 21211–21223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.Y.; Chen, Y.J.; Lu, Y.C.; Liao, C.T.; Chen, I.H.; Chang, J.T.; Huang, Y.C.; Chen, W.H.; Huang, C.C.; Tsai, C.Y.; et al. Fascin is a circulating tumor marker for head and neck cancer as determined by a proteomic analysis of interstitial fluid from the tumor microenvironment. Clin. Chem. Lab. Med. 2015, 53, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Chen, Y.J.; Wang, H.M.; Tsai, C.Y.; Chen, W.H.; Huang, Y.C.; Fan, K.H.; Tsai, C.N.; Huang, S.F.; Kang, C.J.; et al. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer Prev. Res. 2012, 5, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Lin, C.Y.; Lee, L.Y.; Chen, Y.J.; Lu, Y.C.; Wang, H.M.; Liao, C.T.; Chang, J.T.; Cheng, A.J. Molecular chaperones as a common set of proteins that regulate the invasion phenotype of head and neck cancer. Clin. Cancer Res. 2011, 17, 4629–4641. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Chang, J.T.; Chiu, C.; Lu, Y.C.; Li, Y.L.; Chiang, C.H.; You, G.R.; Lee, L.Y.; Cheng, A.J. Areca nut contributes to oral malignancy through facilitating the conversion of cancer stem cells. Mol. Carcinog. 2016, 55, 1012–1023. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, e9227. [Google Scholar] [CrossRef]
- Chen, Y.J.; Lee, L.Y.; Chao, Y.K.; Chang, J.T.; Lu, Y.C.; Li, H.F.; Chiu, C.C.; Li, Y.C.; Li, Y.L.; Chiou, J.F.; et al. DSG3 facilitates cancer cell growth and invasion through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP signaling pathway. PLoS ONE 2013, 8, e64088. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Lin, Y.S.; Chen, C.L.; Chao, P.Z.; Chiou, J.F.; Kuo, C.C.; Lee, F.P.; Lin, Y.F.; Sung, Y.H.; Lin, Y.T.; et al. Targeting annexin A2 reduces tumorigenesis and therapeutic resistance of nasopharyngeal carcinoma. Oncotarget 2015, 6, 26946–26959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.C.; Lee, L.Y.; Li, Y.C.; Chen, Y.J.; Lu, Y.C.; Li, Y.L.; Wang, H.M.; Chang, J.T.; Cheng, A.J. Grp78 as a therapeutic target for refractory head-neck cancer with CD24(-)CD44(+) stemness phenotype. Cancer Gene 2013, 20, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.A.; Rajpal, D.K. Applications of Connectivity Map in drug discovery and development. Drug Discov. Today 2012, 17, 1289–1298. [Google Scholar] [CrossRef]
- Chung, F.H.; Chiang, Y.R.; Tseng, A.L.; Sung, Y.C.; Lu, J.; Huang, M.C.; Ma, N.; Lee, H.C. Functional Module Connectivity Map (FMCM): A framework for searching repurposed drug compounds for systems treatment of cancer and an application to colorectal adenocarcinoma. PLoS ONE 2014, 9, e86299. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; You, G.-R.; Lai, M.-Y.; Lu, L.-S.; Chen, C.-Y.; Ting, L.-L.; Lee, H.-L.; Kanno, Y.; Chiou, J.-F.; Cheng, A.-J. A Combined Systemic Strategy for Overcoming Cisplatin Resistance in Head and Neck Cancer: From Target Identification to Drug Discovery. Cancers 2020, 12, 3482. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113482
Chen Y-J, You G-R, Lai M-Y, Lu L-S, Chen C-Y, Ting L-L, Lee H-L, Kanno Y, Chiou J-F, Cheng A-J. A Combined Systemic Strategy for Overcoming Cisplatin Resistance in Head and Neck Cancer: From Target Identification to Drug Discovery. Cancers. 2020; 12(11):3482. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113482
Chicago/Turabian StyleChen, Yin-Ju, Guo-Rung You, Meng-Yu Lai, Long-Sheng Lu, Chang-Yu Chen, Lai-Lei Ting, Hsin-Lun Lee, Yuzuka Kanno, Jeng-Fong Chiou, and Ann-Joy Cheng. 2020. "A Combined Systemic Strategy for Overcoming Cisplatin Resistance in Head and Neck Cancer: From Target Identification to Drug Discovery" Cancers 12, no. 11: 3482. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113482