G1 Cell Cycle Arrest and Extrinsic Apoptotic Mechanisms Underlying the Anti-Leukemic Activity of CDK7 Inhibitor BS-181


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
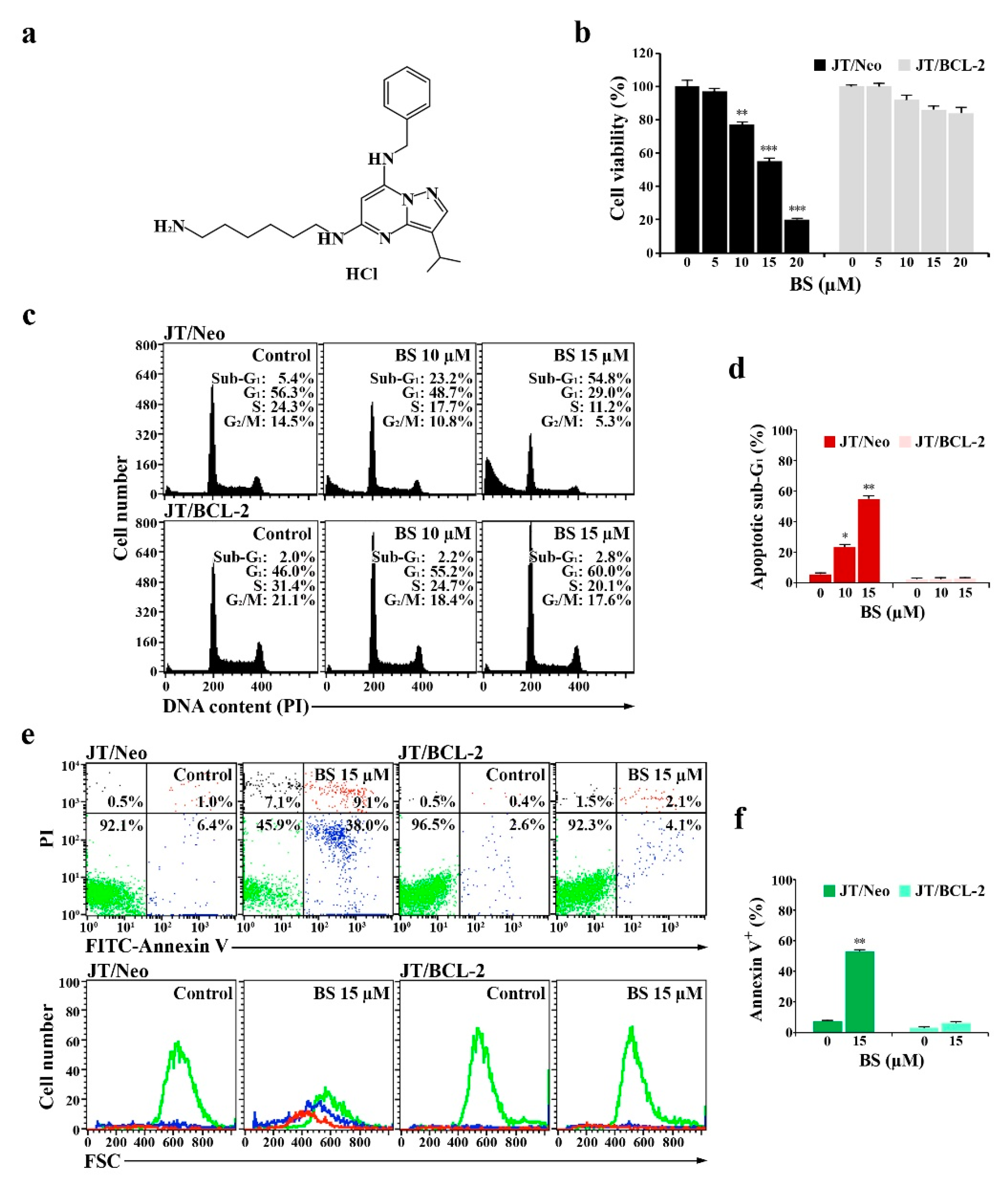
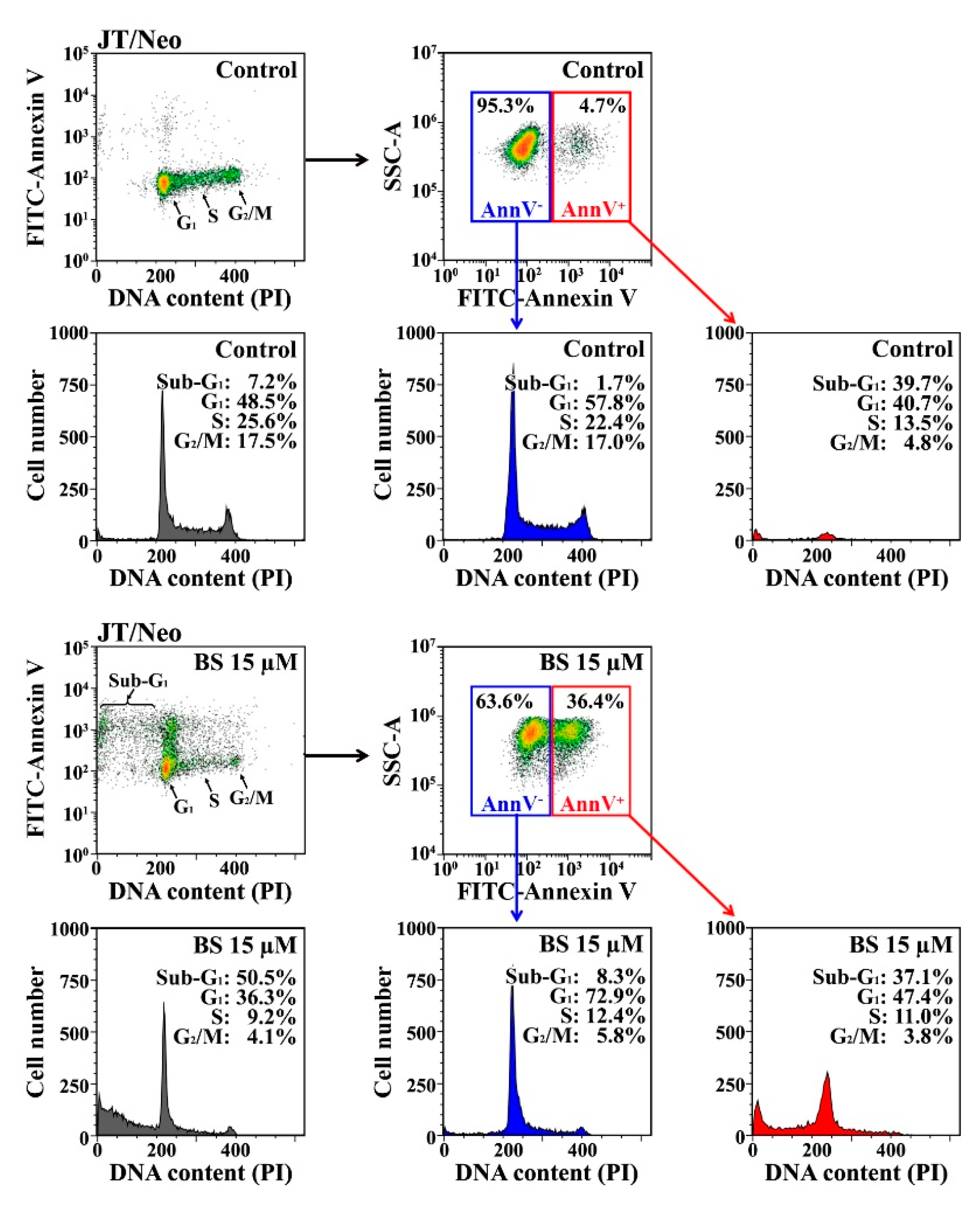
2.1. BS-181 Cytotoxicity Is Exerted by G1 Cell Cycle Arrest and BCL-2-Sensitive Apoptosis in Jurkat T Cells
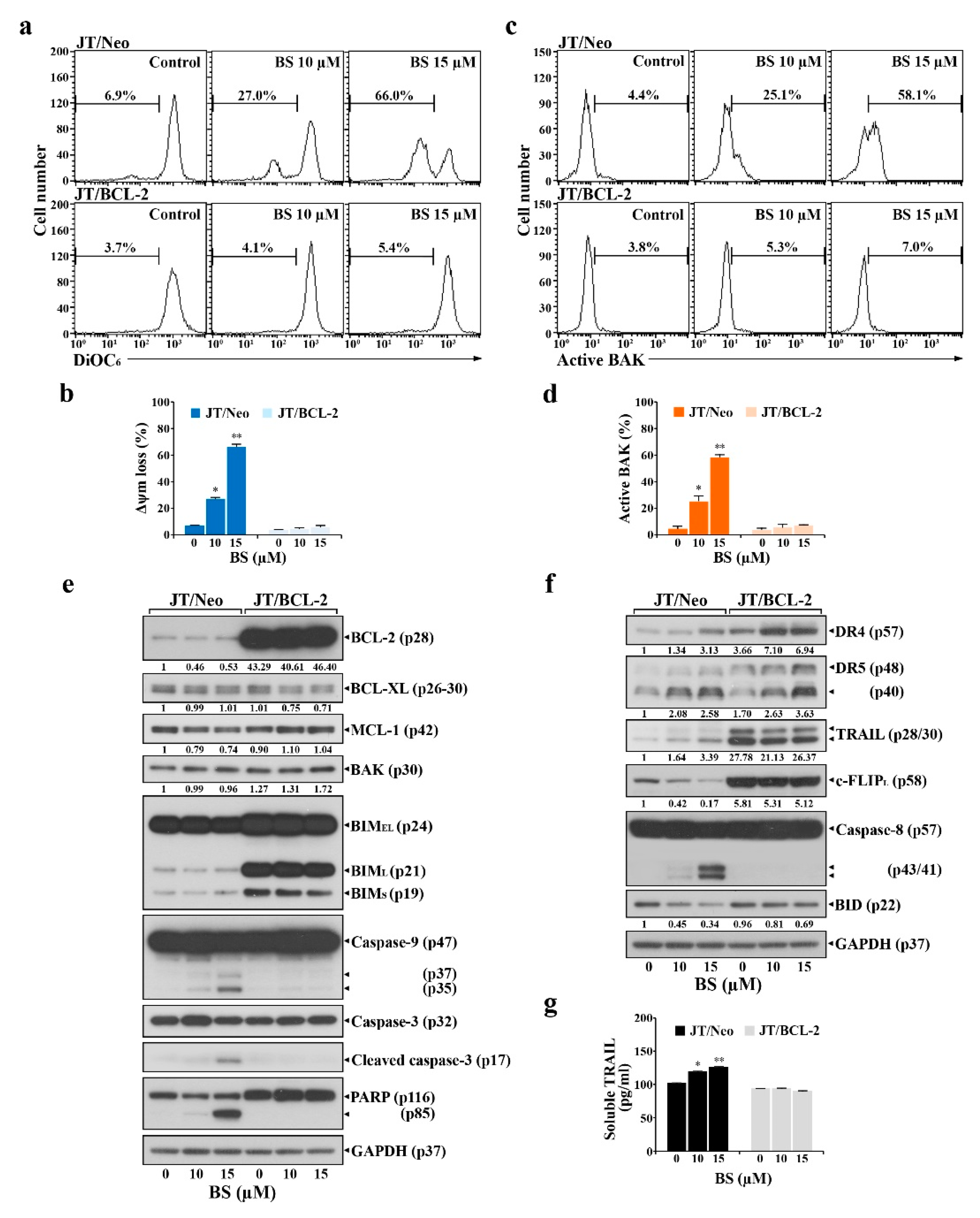
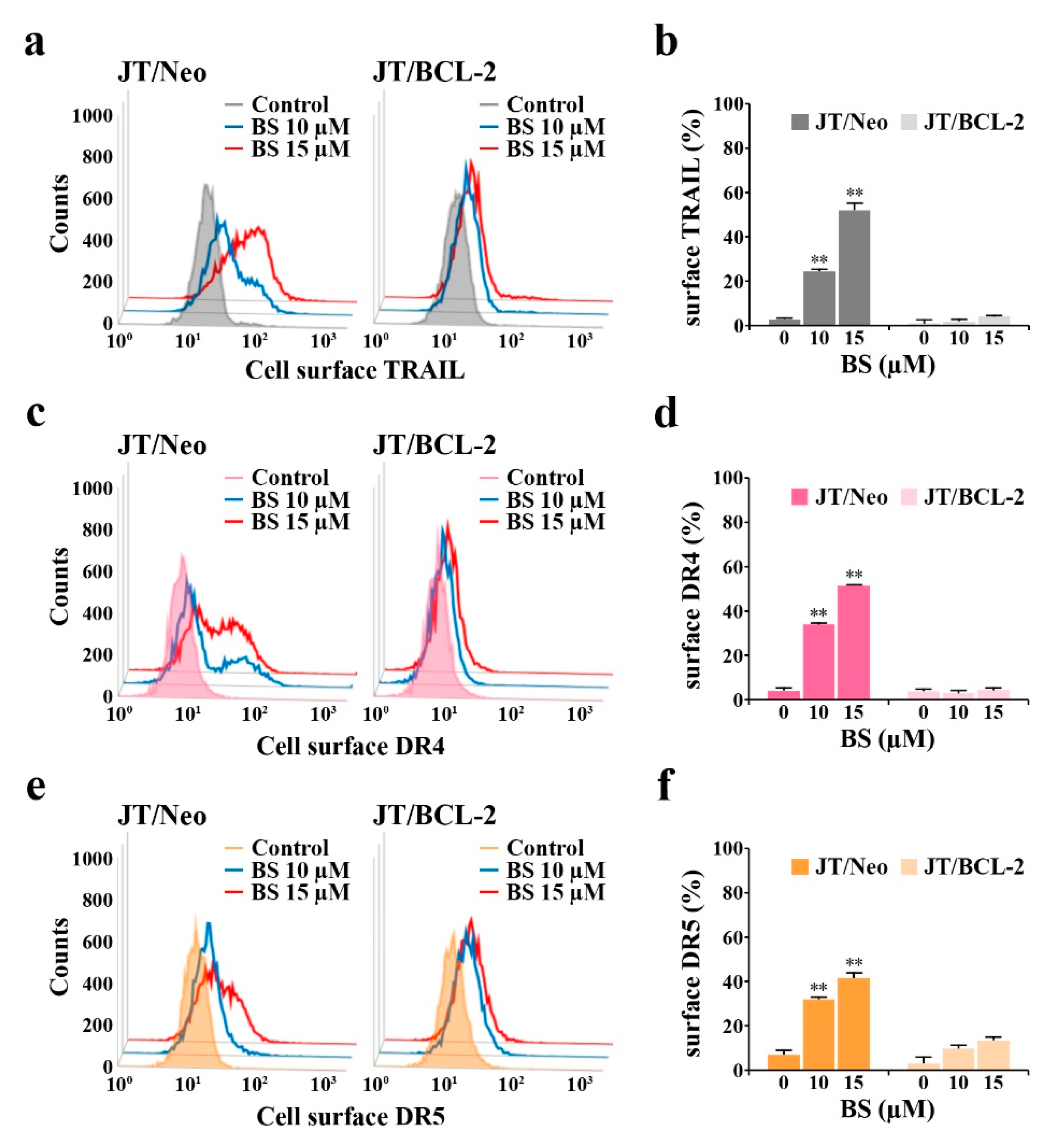
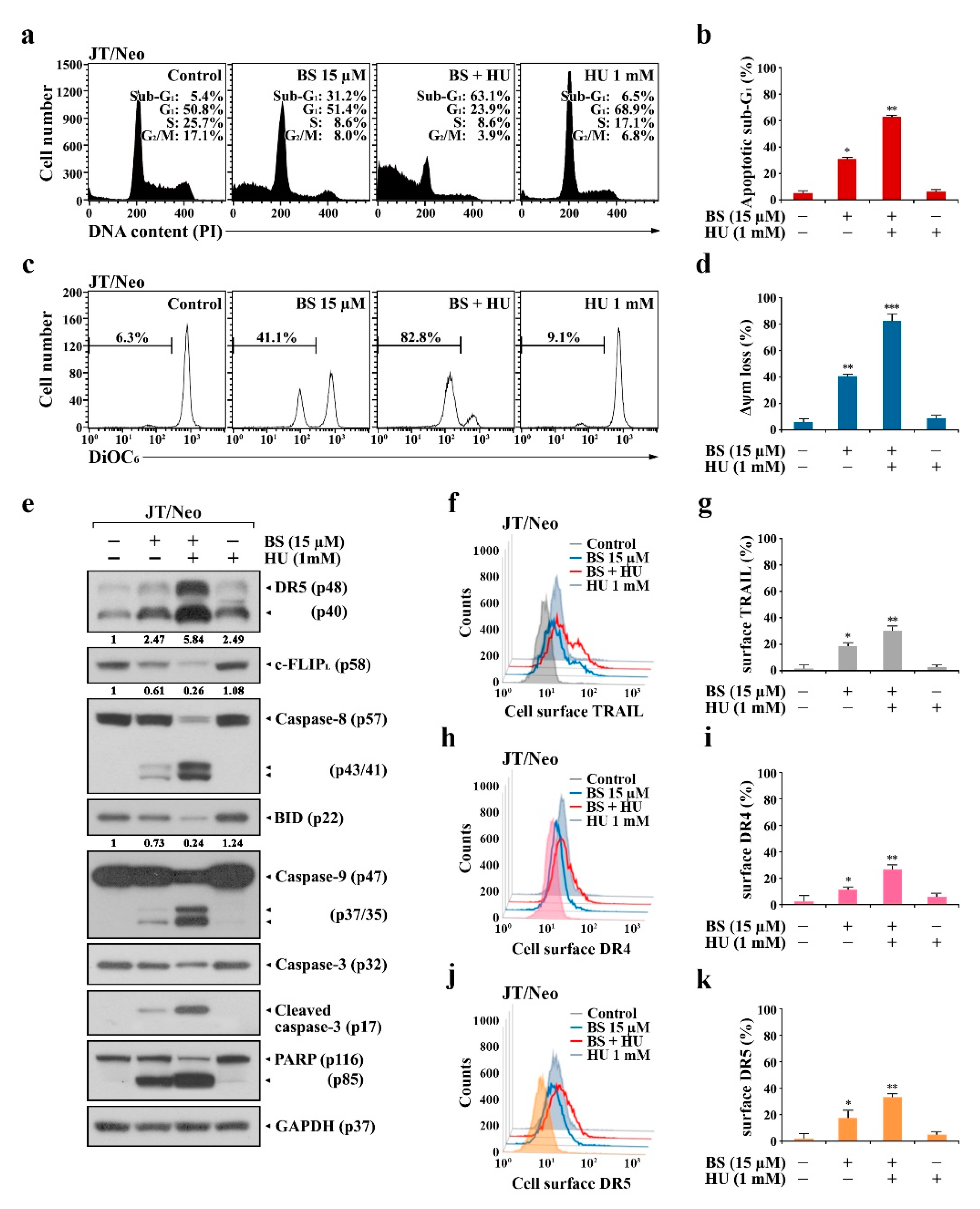
2.2. BCL-2 Overexpression Abrogates Extrinsic TRAIL/DR5 Upregulation-Mediated Apoptosis and Subsequent Mitochondrial Damage-Mediated Apoptosis in BS-181-Treated JT/Neo Cells
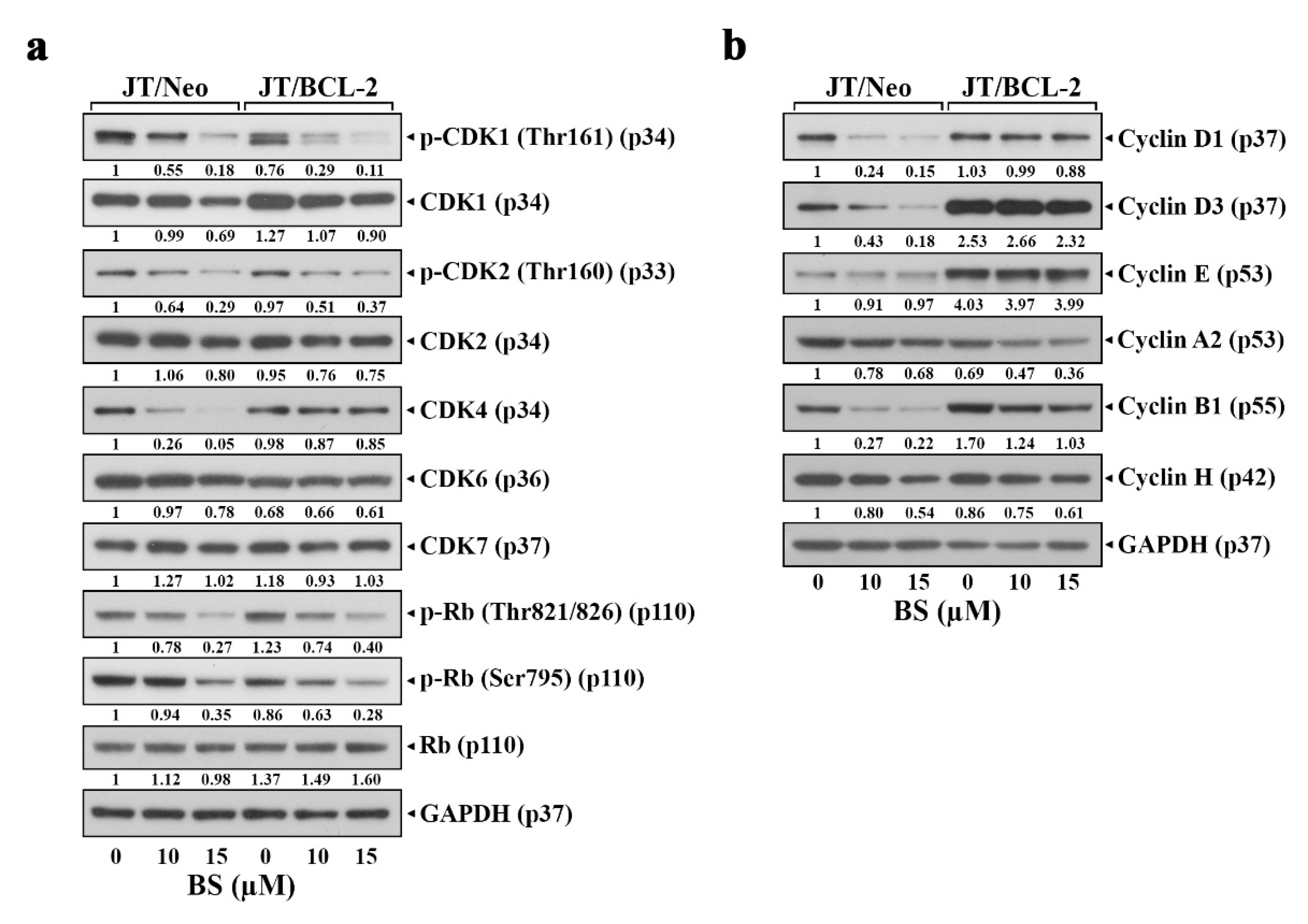
2.3. BS-181 Preferentially Induces Apoptosis in G1–Arrested JT/Neo Cells after Attenuation of CDK7-Mediated Retinoblastoma (Rb) Inactivation and Cell Cycle Progression
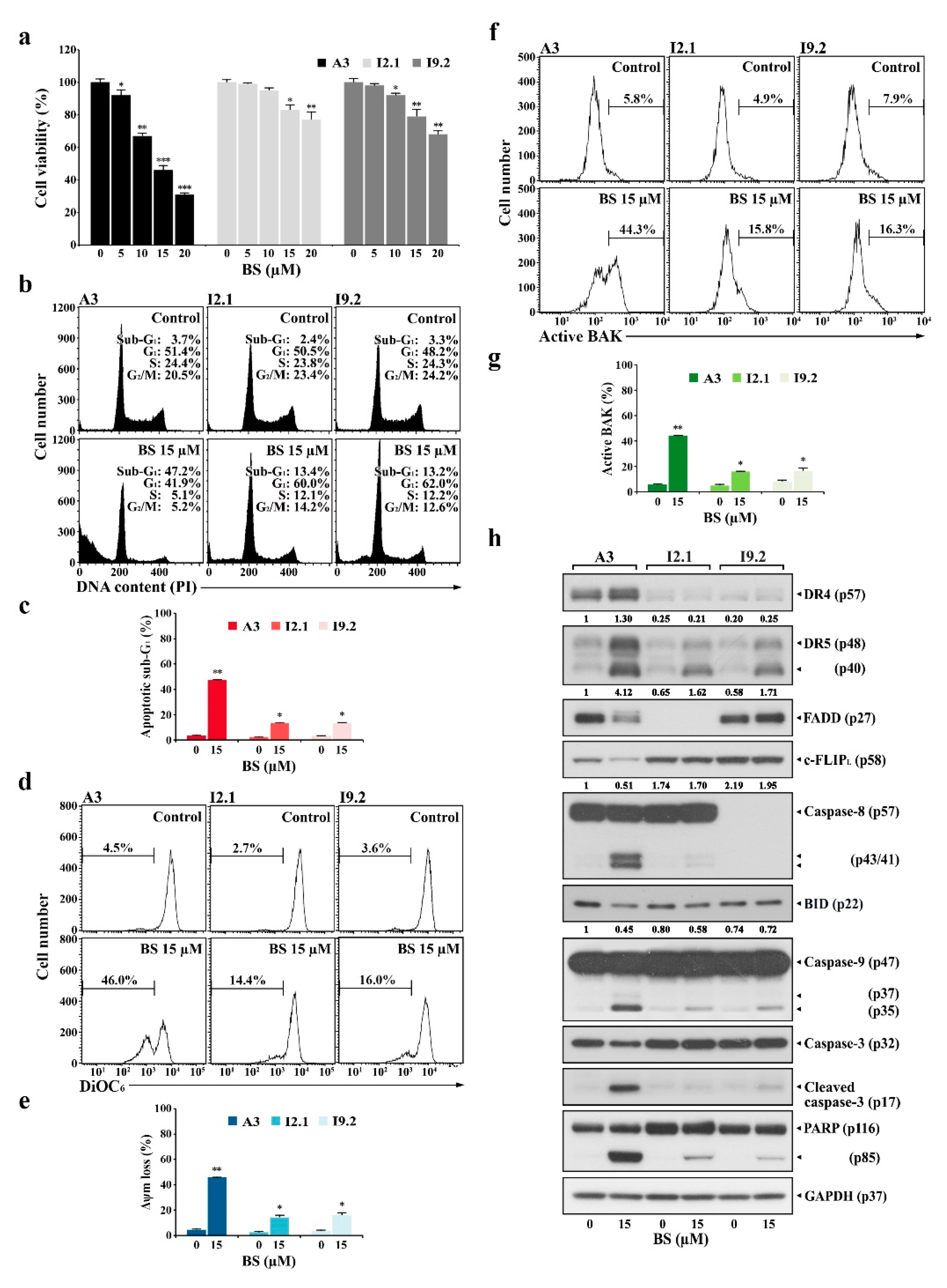
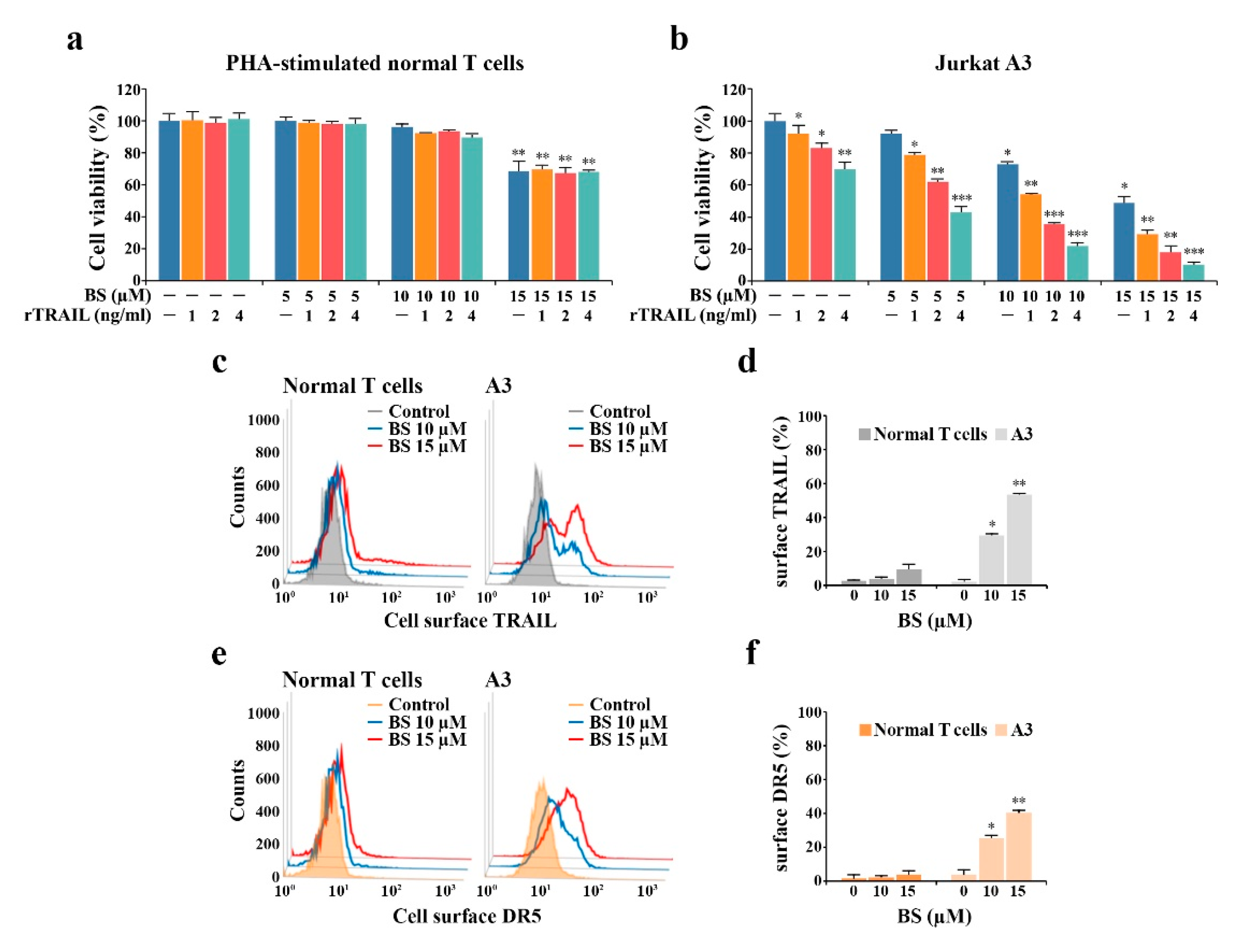
2.4. BS-181-Induced Apoptosis Occurs Primarily through Extrinsic TRAIL/DR5-Mediated Apoptotic Signaling and Subsequent BAK-Dependent Mitochondrial Apoptosis Induction
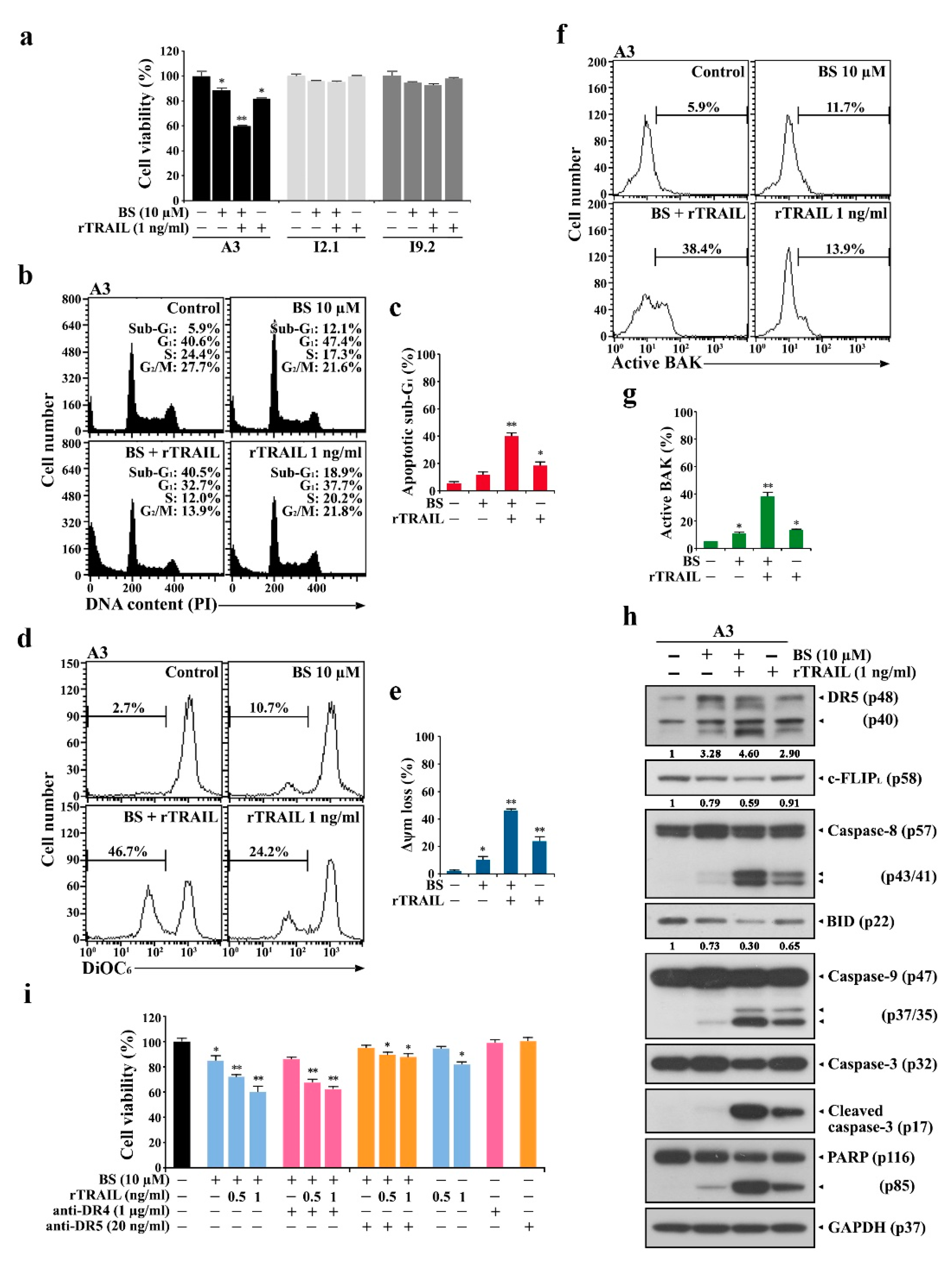
2.5. rTRAIL Exhibits No Synergistic Effects on BS-181 Cytotoxicity in Normal Human T Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Normal Human Peripheral Blood T Lymphocytes
4.4. Cytotoxicity Assay
4.5. Flow Cytometric Analysis
4.6. Preparation of Cell Lysates and Western Blot Analysis
4.7. ELISA for the Detection of Extracellular Soluble TRAIL
4.8. Statistical Analysis
4.9. Study Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3398–3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.M.; Silverman, L.B.; Levy, D.E.; Dalton, V.K.; Gelber, R.D.; Lehmann, L.; Cohen, H.J.; Sallan, S.E.; Asselin, B.L. Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J. Clin. Oncol. 2003, 21, 3616–3622. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Gillet, J.-P.; Sauerbrey, A.; Zintl, F.; Bertholet, V.; de Longueville, F.; Jose Remacle, J.; Steinbach, D. Expression profiling of ATP-binding cassette transporters in childhood T-cell acute lymphoblastic leukemia. Mol. Cancer Ther. 2006, 5, 1986–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavese, M.; Santo, L.; Raje, N. Cyclin dependent kinases in cancer: Potential for therapeutic intervention. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Gabrielli, B.; Brooks, K.; Pavey, S. Defective cell cycle checkpoints as targets for anti-cancer therapies. Front. Pharmacol. 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Cordon-Cardo, C. Mutations of cell cycle regulators. Biological and clinical implications for human neoplasia. Am. J. Pathol. 1995, 147, 545–560. [Google Scholar] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Corona, S.P.; Ravelli, A.; Cretella, D.; Cappelletti, M.R.; Zanotti, L.; Dester, M.; Gobbi, A.; Petronini, P.G.; Generali, D. CDK4/6 inhibitors in HER2-positive breast cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 208–214. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.F.; Kuijpers, C.J.; Kroep, J.R. CDK4/6 inhibition in early and metastatic breast cancer: A review. Cancer Treat. Rev. 2017, 60, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette-Egly, C.; Adam, S.; Rossignol, M.; Egly, J.M.; Chambon, P. Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell 1997, 90, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Riedl, T.; Washbrook, E.; Pace, P.E.; Coombes, R.C.; Egly, J.M.; Ali, S. Activation of estrogen receptor a by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 2000, 6, 127–137. [Google Scholar] [CrossRef]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.B.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.J.; Periyasamy, M.; et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.Y.; Liu, Q.Y.; Cao, J.; Chen, J.W.; Liu, Z.S. Selective CDK7 inhibition with BS-181suppresses cell proliferation and induces cell cycle arrest and apoptosis in gastric cancer. Drug Des. Devel. Ther. 2016, 10, 1181–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.R.; Jun, D.Y.; Kim, Y.H.; Lee, J.Y.; Kim, Y.H. Prometaphase arrest-dependent phosphorylation of Bcl-2 family proteins and activation of mitochondrial apoptotic pathway are associated with 17α-estradiol-induced apoptosis in human Jurkat T cells. Biochim. Biophys. Acta 2013, 1833, 2220–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada-Oikawa, S.; Oikawa, S.; Kawanishi, S. Role of ultraviolet A-induced oxidative DNA damage in apoptosis via loss of mitochondrial membrane potential and caspase-3 activation. Biochem. Biophys. Res. Commun. 1998, 247, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Shankar, S.; Srivastava, R.K. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: Mechanisms and clinical implications. Drug Resist. Update. 2004, 7, 139–156. [Google Scholar] [CrossRef]
- Kruyt, F.A. TRAIL and cancer therapy. Cancer Lett. 2008, 263, 14–25. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of apoptosis and necroptosis by c-FLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [Green Version]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef] [Green Version]
- Fesquet, D.; Labbé, J.C.; Derancourt, J.; Capony, J.P.; Galas, S.; Girard, F.; Lorca, T.; Shuttleworth, J.; Doree, M.; Cavadore, J.C. The MO15 gene encodes the catalytic subunit of a protein kinase that activates cdc2 and other cyclin-dependent kinases (CDKs) through phosphorylation of Thr161 and its homologues. EMBO J. 1993, 12, 3111–3121. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Wang, J.Y. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J. Biol. Chem. 1996, 271, 8313–8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim. Biophys. Acta 2001, 1471, M123–M133. [Google Scholar] [CrossRef]
- Kim, Y.H.; Proust, J.J.; Buchholz, M.J.; Chrest, F.J.; Nordin, A.A. Expression of the murine homologue of the cell cycle control protein p34cdc2 in T lymphocytes. J. Immunol. 1992, 149, 17–23. [Google Scholar] [PubMed]
- Koc, A.; Wheeler, L.J.; Mathews, C.K.; Merrill, G.F. Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J. Biol. Chem. 2004, 279, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juo, P.; Woo, M.S.; Kuo, C.J.; Signorelli, P.; Biemann, H.P.; Hannun, Y.A.; Blenis, J. FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ. 1999, 10, 797–804. [Google Scholar] [PubMed]
- Shetty, S.; Gladden, J.B.; Henson, E.S.; Hu, X.; Villanueva, J.; Haney, N.; Gibson, S.B. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NF-kB activation in epithelial derived cell lines. Apoptosis 2002, 7, 413–420. [Google Scholar] [CrossRef]
- Lange, C.A.; Yee, D. Killing the second messenger: Targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr. Relat. Cancer 2011, 18, C19–C24. [Google Scholar] [CrossRef]
- Guillouf, C.; Grana, X.; Selvakumaran, M.; De Luca, A.; Giordano, A.; Hoffman, B.; Liebermann, D.A. Dissection of the genetic programs of p53-mediated G1 growth arrest and apoptosis: Blocking p53-induced apoptosis unmasks G1 arrest. Blood 1995, 85, 2691–2698. [Google Scholar] [CrossRef] [Green Version]
- Laukens, B.; Jennewein, C.; Schenk, B.; Vanlangenakker, N.; Schier, A.; Cristofanon, A.; Zobel, K.; Deshayes, K.; Vucic, D.; Jeremias, I.; et al. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor α-induced necroptosis. Neoplasia 2011, 13, 971–979. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.-P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metast. Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Periyasamy, M.; Sava, G.P.; Bondke, A.; Slafer, B.W.; Kroll, S.H.B.; Barbazanges, M.; Starkey, R.; Ottaviani, S.; Harrod, A.; et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol. Cancer Ther. 2018, 17, 1156–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.T.; Lee, J.Y.; Han, C.R.; Kim, Y.H.; Jun, D.Y.; Taub, D.; Kim, Y.H. Dependency of 2-methoxyestradiol-induced mitochondrial apoptosis on mitotic spindle network impairment and prometaphase arrest in human Jurkat T cells. Biochem. Pharmacol. 2015, 94, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.E.; Jun, D.Y.; Kim, J.S.; Kim, Y.H. Cis-trimethoxy resveratrol induces intrinsic apoptosis via prometaphase arrest and prolonged CDK1 activation pathway in human Jurkat T cells. Oncotarget 2018, 9, 4969–4984. [Google Scholar] [CrossRef] [Green Version]
- Jun, D.Y.; Kim, J.S.; Park, H.S.; Han, C.R.; Fang, Z.; Woo, M.H.; Rhee, I.K.; Kim, Y.H. Apoptogenic activity of auraptene of Zanthoxylum schinifolium toward human acute leukemia Jurkat T cells is associated with ER stress-mediated caspase-8 activation that stimulates mitochondria-dependent or -independent caspase cascade. Carcinogenesis 2007, 8, 1303–1313. [Google Scholar] [CrossRef]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Zanin, C.; Vayssiere, J.L.; Petit, P.X.; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995, 181, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Jun, D.Y.; Han, C.R.; Woo, H.J.; Kim, Y.H. Proteasome inhibitor MG132-induced apoptosis via ER stress-mediated apoptotic pathway and its potentiation by protein tyrosine kinase p56lck in human Jurkat T cells. Biochem. Pharmacol. 2011, 82, 1110–1125. [Google Scholar] [CrossRef]
- Samraj, A.K.; Stroh, C.; Fischer, U.; Schulze-Osthoff, K. The tyrosine kinase Lck is positive regulator of the mitochondrial apoptosis pathway by controlling Bak expression. Oncogene 2005, 25, 186–197. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor and Normal Cells | IC50 (μM) * |
|---|---|
| Unstimulated human peripheral T cells | 43.9 |
| PHA-stimulated human peripheral T cells ** | 18.9 |
| Human acute T cell leukemia Jurkat (JT/Neo) cells | 15.0 |
| Human acute T cell leukemia Jurkat (JT/BCL-2) cells | 50.8 |
| Human acute T cell leukemia Jurkat (J/Neo) cells | 14.2 |
| Human acute T cell leukemia Jurkat (J/BCL-XL) cells | 48.1 |
| Human acute T cell leukemia Jurkat A3 cells | 14.5 |
| Human monoblastoid U937 cells | 16.4 |
| Human cervical carcinoma HeLa cells | 17.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.Y.; Kim, K.Y.; Jun, D.Y.; Hwang, S.-K.; Kim, Y.H. G1 Cell Cycle Arrest and Extrinsic Apoptotic Mechanisms Underlying the Anti-Leukemic Activity of CDK7 Inhibitor BS-181. Cancers 2020, 12, 3845. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123845
Park SY, Kim KY, Jun DY, Hwang S-K, Kim YH. G1 Cell Cycle Arrest and Extrinsic Apoptotic Mechanisms Underlying the Anti-Leukemic Activity of CDK7 Inhibitor BS-181. Cancers. 2020; 12(12):3845. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123845
Chicago/Turabian StylePark, Shin Young, Ki Yun Kim, Do Youn Jun, Su-Kyeong Hwang, and Young Ho Kim. 2020. "G1 Cell Cycle Arrest and Extrinsic Apoptotic Mechanisms Underlying the Anti-Leukemic Activity of CDK7 Inhibitor BS-181" Cancers 12, no. 12: 3845. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123845