ABCG2 Overexpression Contributes to Pevonedistat Resistance

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
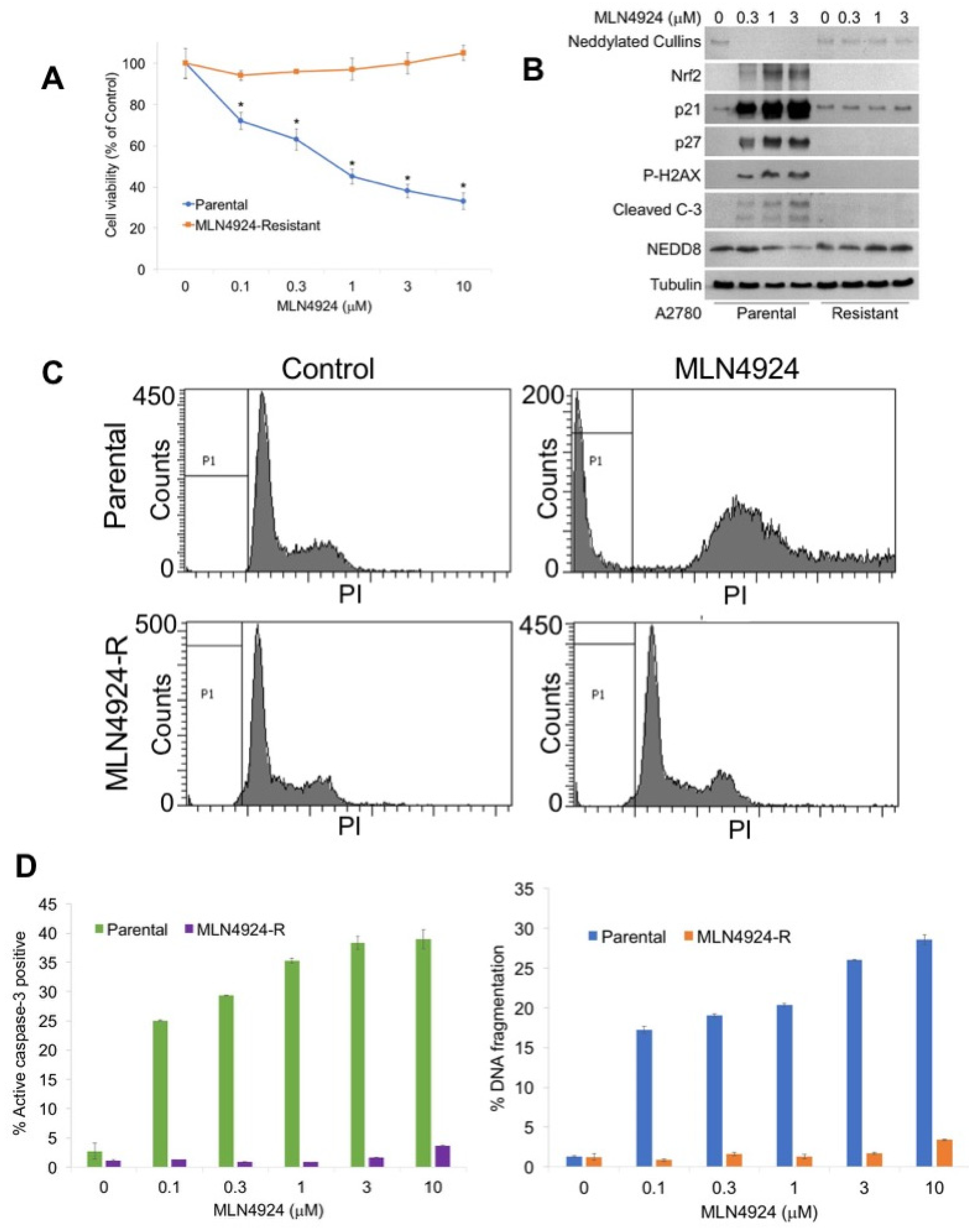
2.1. Generation of a Novel MLN4924 Resistant Ovarian Cancer Model System
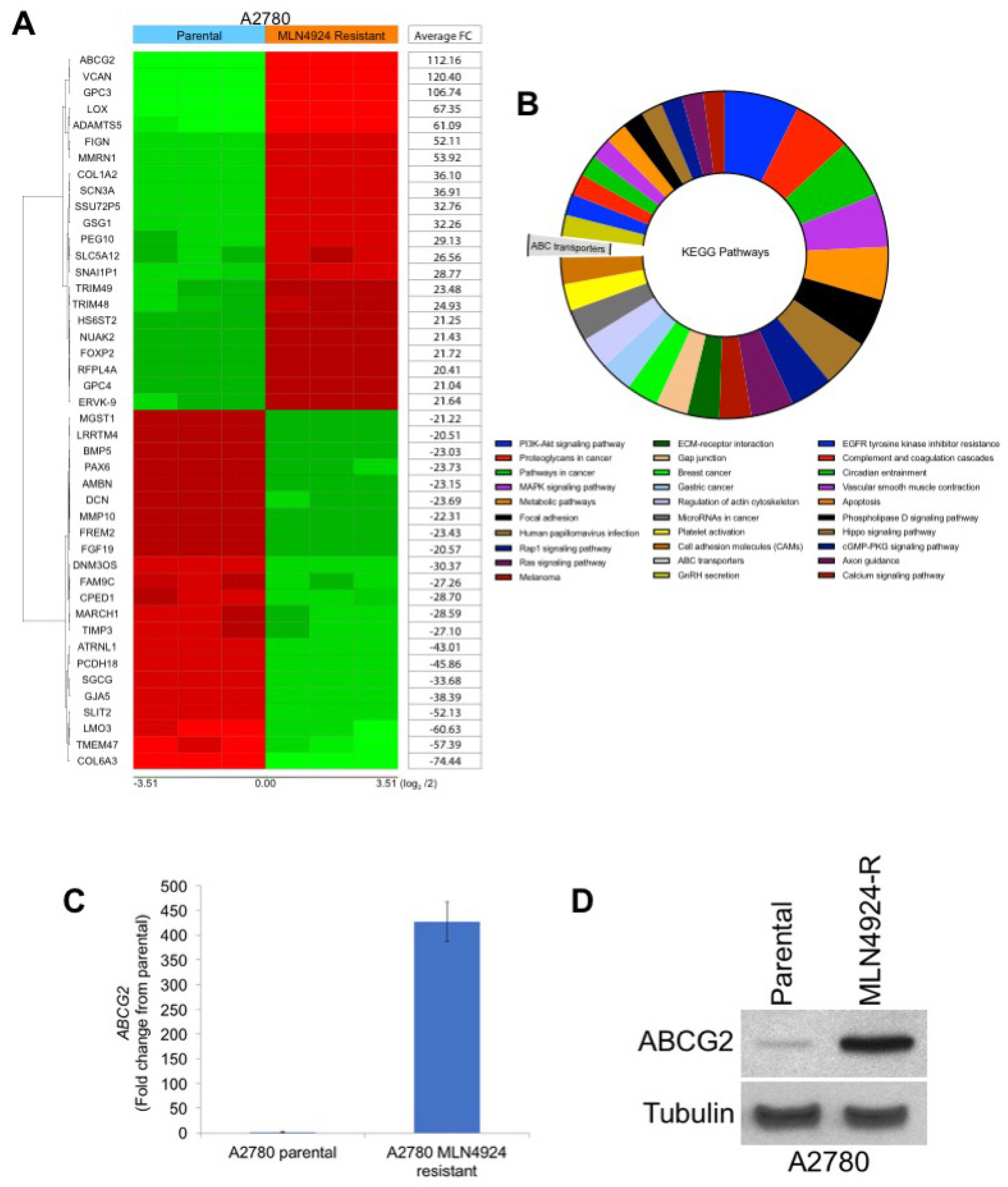
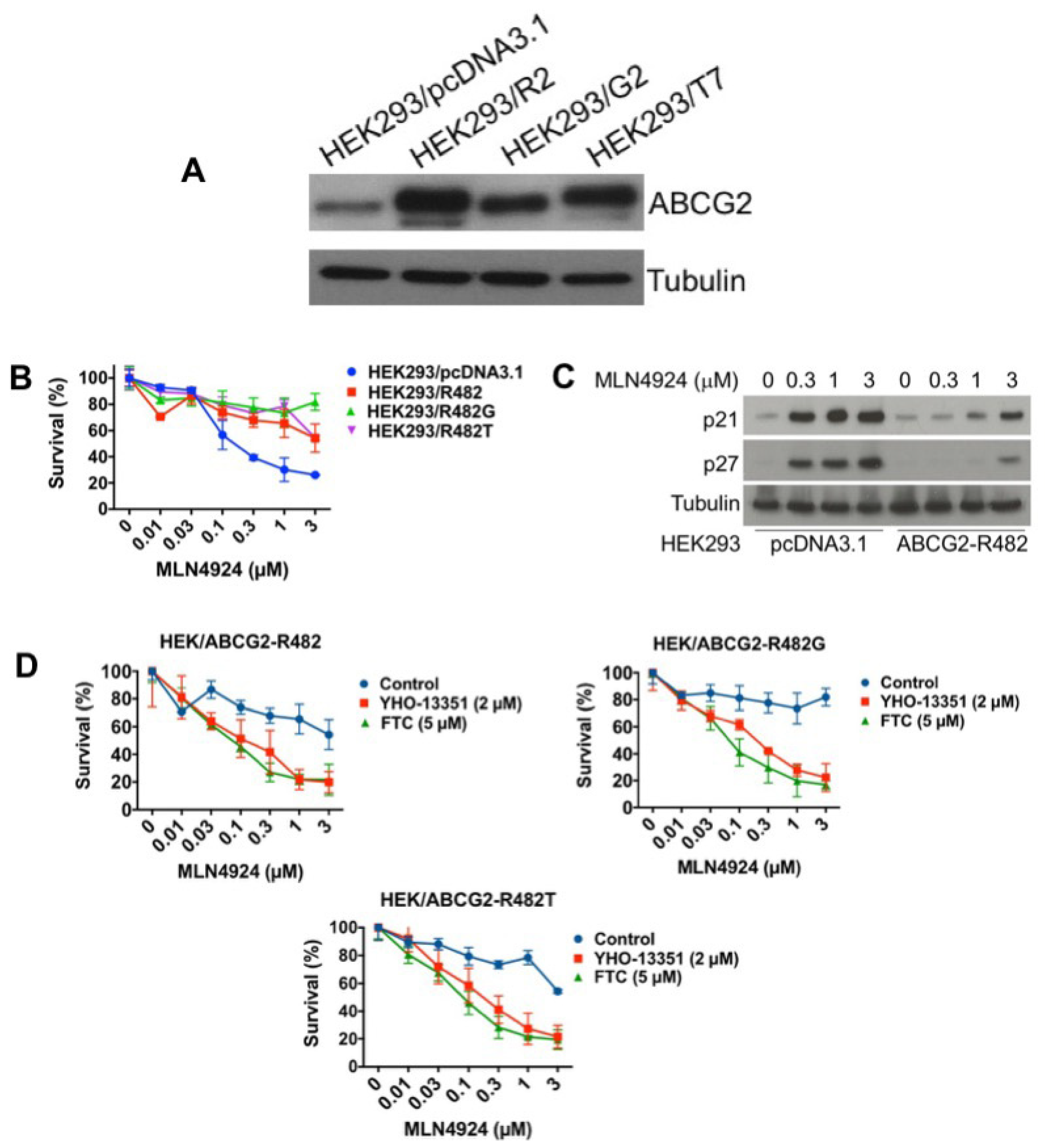
2.2. ABCG2 is Highly Upregulated in MLN4924-Resistant Cells
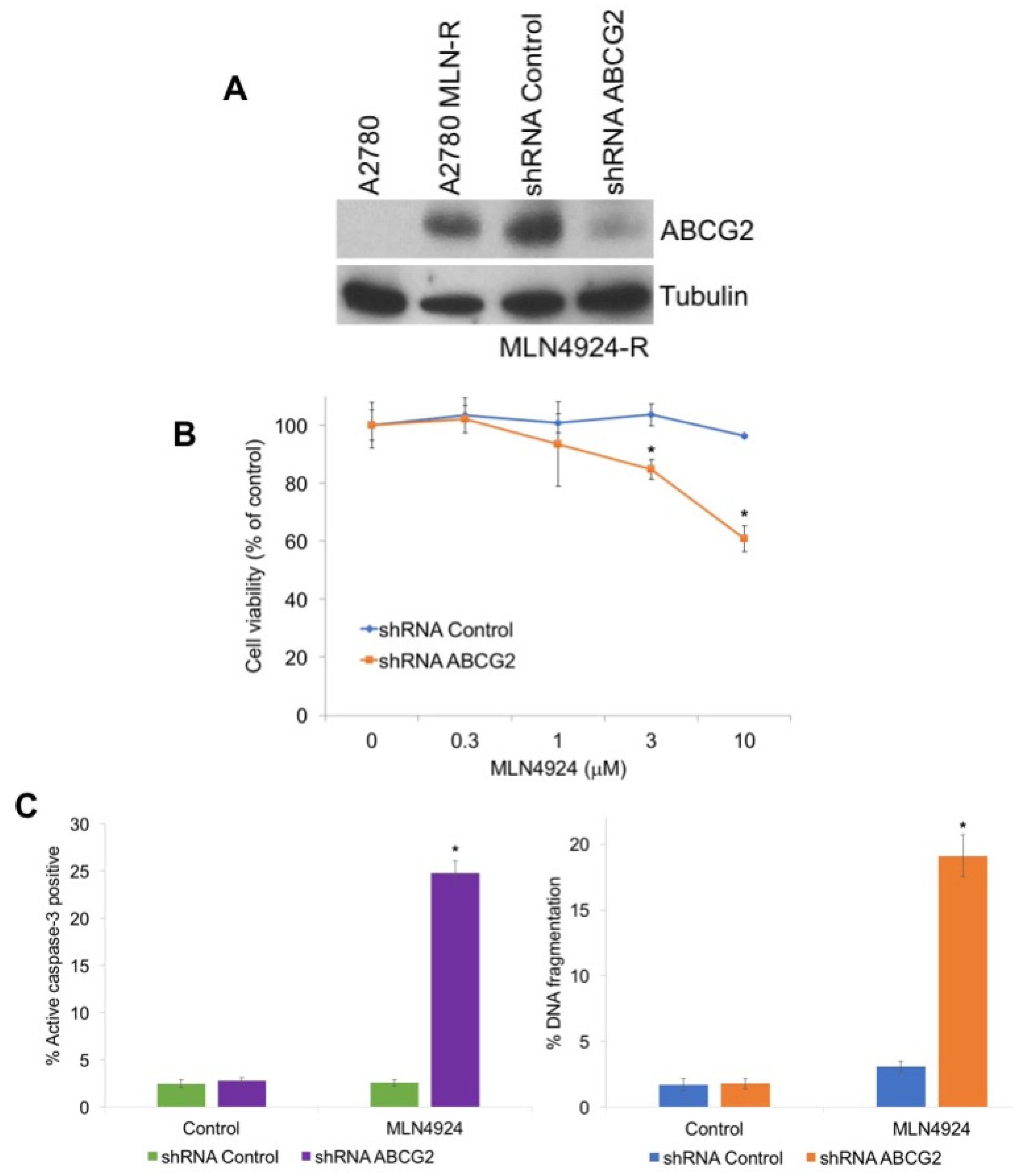
2.3. Targeting ABCG2 Overexpression Diminishes Resistance to MLN4924
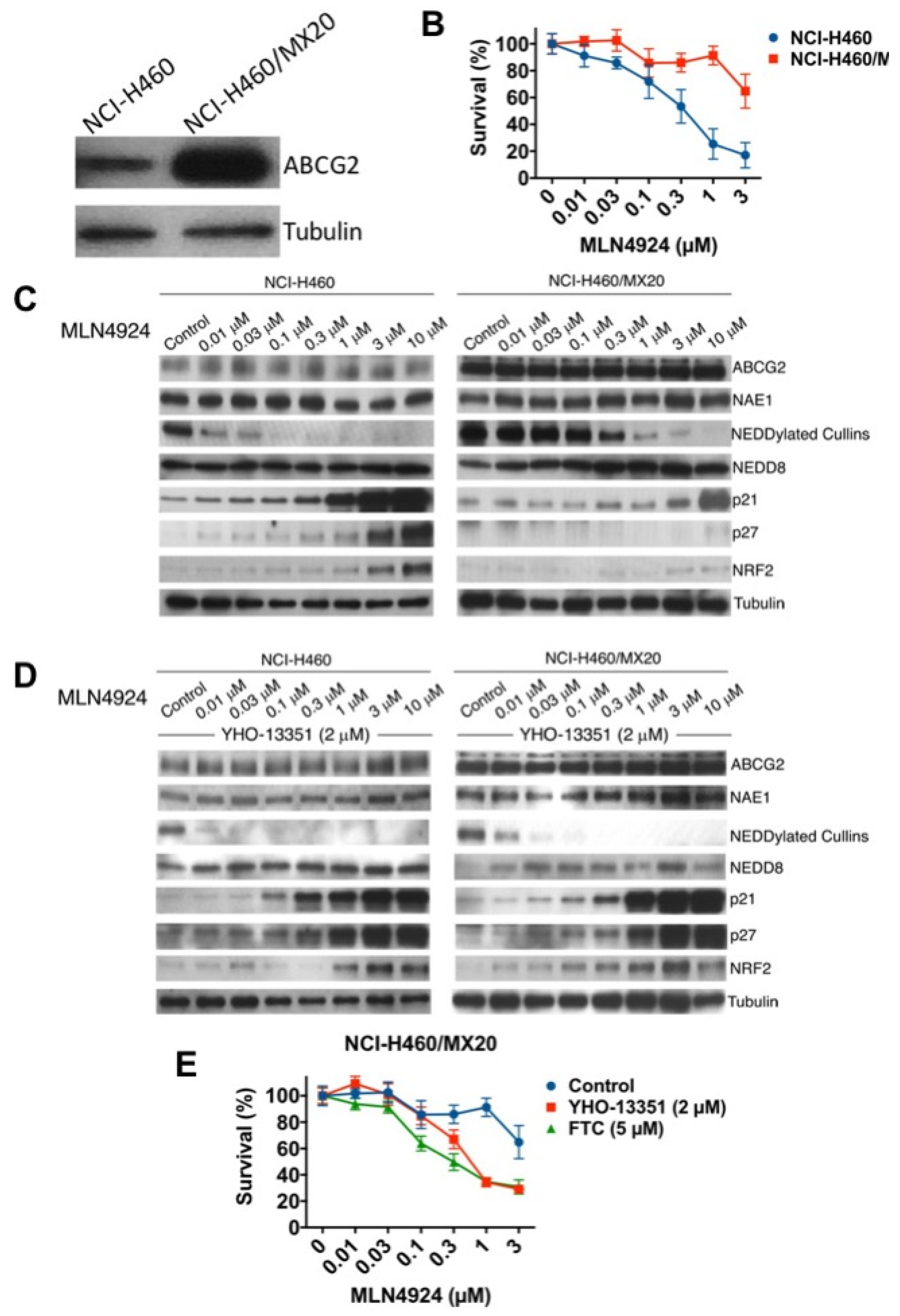
2.4. Mitoxantrone-Selected ABCG2-Overexpressing Cells are Resistant to MLN4924
2.5. ABCG2 Overexpression is Sufficient to Confer Resistance to MLN4924
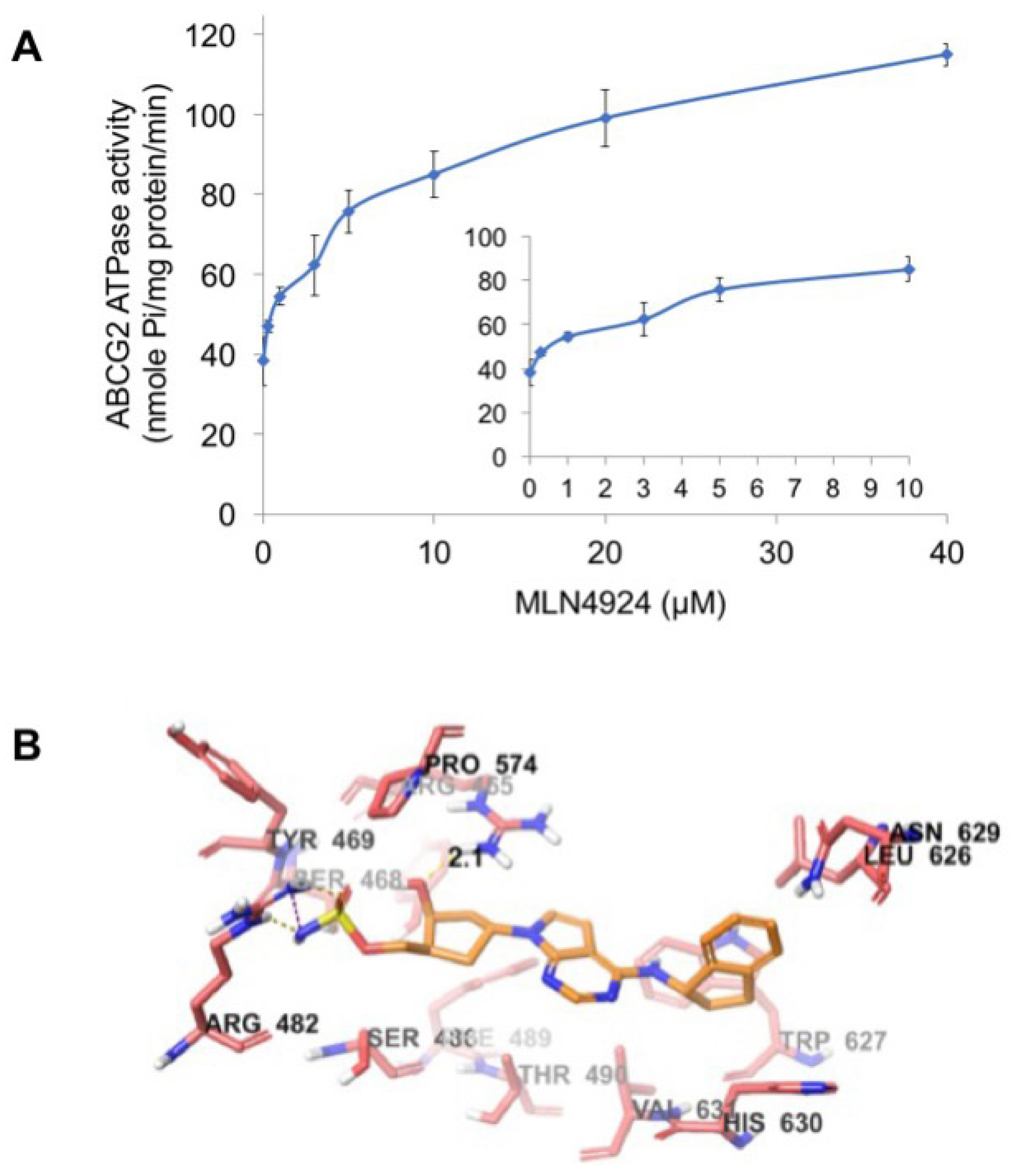
2.6. MLN4924 Stimulates ABCG2 ATPase Activity
2.7. Docking Analysis of MLN4924 with Human ABCG2
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Antibodies and Reagents
4.3. Quantification of Drug-Induced Cytotoxicity
4.4. Mutation Analysis of NAEβ (UBA3) in A2780 and A2780/MLN-R Cells
4.5. Transcriptome Profiling
4.6. Immunoblotting
4.7. qRT-PCR
4.8. shRNA Knockdown of ABCG2
4.9. ATPase Activity of ABCG2
4.10. Molecular Modeling of Human ABCG2 and Docking of MLN4924
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Podust, V.N.; Brownell, J.E.; Gladysheva, T.B.; Luo, R.S.; Wang, C.; Coggins, M.B.; Pierce, J.W.; Lightcap, E.S.; Chau, V. A nedd8 conjugation pathway is essential for proteolytic targeting of p27kip1 by ubiquitination. Proc. Natl. Acad. Sci. USA 2000, 97, 4579–4584. [Google Scholar] [CrossRef] [Green Version]
- Milhollen, M.A.; Thomas, M.P.; Narayanan, U.; Traore, T.; Riceberg, J.; Amidon, B.S.; Bence, N.F.; Bolen, J.B.; Brownell, J.; Dick, L.R.; et al. Treatment-emergent mutations in naebeta confer resistance to the nedd8-activating enzyme inhibitor mln4924. Cancer Cell 2012, 21, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. Mln4924: A novel first-in-class inhibitor of nedd8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Espitia, C.M.; Possemato, A.; Beausoleil, S.A.; Milhollen, M.; Blakemore, S.; Thomas, M.; Berger, A.; et al. Disrupting protein neddylation with mln4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin. Cancer Res. 2013, 19, 3577–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarantopoulos, J.; Shapiro, G.I.; Cohen, R.B.; Clark, J.W.; Kauh, J.S.; Weiss, G.J.; Cleary, J.M.; Mahalingam, D.; Pickard, M.D.; Faessel, H.M.; et al. Phase i study of the investigational nedd8-activating enzyme inhibitor pevonedistat (tak-924/mln4924) in patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase i study of the novel investigational nedd8-activating enzyme inhibitor pevonedistat (mln4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of nedd8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [Green Version]
- Le-Trilling, V.T.; Megger, D.A.; Katschinski, B.; Landsberg, C.D.; Ruckborn, M.U.; Tao, S.; Krawczyk, A.; Bayer, W.; Drexler, I.; Tenbusch, M.; et al. Broad and potent antiviral activity of the nae inhibitor mln4924. Sci. Rep. 2016, 6, 19977. [Google Scholar] [CrossRef]
- Nekorchuk, M.D.; Sharifi, H.J.; Furuya, A.K.; Jellinger, R.; de Noronha, C.M. Hiv relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology 2013, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Yao, W.; Wang, K.; Qian, Y.; Chen, H.; Jung, Y.S. Inhibition of neddylation pathway represses influenza virus replication and pro-inflammatory responses. Virology 2018, 514, 230–239. [Google Scholar] [CrossRef]
- Toth, J.I.; Yang, L.; Dahl, R.; Petroski, M.D. A gatekeeper residue for nedd8-activating enzyme inhibition by mln4924. Cell Rep. 2012, 1, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.W.; Toth, J.I.; da Silva, S.R.; Paiva, S.L.; Lukkarila, J.L.; Hurren, R.; Maclean, N.; Sukhai, M.A.; Bhattacharjee, R.N.; Goard, C.A.; et al. Mutations in uba3 confer resistance to the nedd8-activating enzyme inhibitor mln4924 in human leukemic cells. PLoS One 2014, 9, e93530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Keaton, M.; Carraway, H.; Sekeres, M.A.; Maciejewski, J.P.; Carew, J.S. The nedd8-activating enzyme inhibitor mln4924 disrupts nucleotide metabolism and augments the efficacy of cytarabine. Clin. Cancer Res. 2015, 21, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconte, V.; Nawrocki, S.T.; Espitia, C.M.; Kelly, K.R.; Possemato, A.; Beausoleil, S.A.; Han, Y.; Carraway, H.E.; Nazha, A.; Advani, A.S.; et al. Comprehensive quantitative proteomic profiling of the pharmacodynamic changes induced by mln4924 in acute myeloid leukemia cells establishes rationale for its combination with azacitidine. Leukemia 2016, 30, 1190–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Li, S.; Li, W.; Xie, M.; Wei, Z.; Guo, H.; Wei, W.; Zhang, S. Disruption of protein neddylation with mln4924 attenuates paclitaxel-induced apoptosis and microtubule polymerization in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2019, 508, 986–990. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Shibata, E.; Park, J.; Bryant, J.L.; Conaway, M.R.; Modesitt, S.C.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Dutta, A. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor mln4924. Mol. Cancer Ther. 2013, 12, 1958–1967. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.W.; Zhou, J.J.; Yu, C.; Xu, Y.; Guo, L.J.; Zhang, H.Y.; Zhou, D.; Song, F.Z.; Fan, H.Y. Ubiquitin e3 ligase crl4(cdt2/dcaf2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J. Biol. Chem. 2013, 288, 29680–29691. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase ib study of pevonedistat, a nedd8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest. New Drugs 2019, 37, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Ji, N.; Yang, Y.; Cai, C.Y.; Lei, Z.N.; Wang, J.Q.; Gupta, P.; Shukla, S.; Ambudkar, S.V.; Kong, D.; Chen, Z.S. Selonsertib (gs-4997), an ask1 inhibitor, antagonizes multidrug resistance in abcb1- and abcg2-overexpressing cancer cells. Cancer Lett. 2019, 440–441, 82–93. [Google Scholar] [CrossRef]
- Swords, R.T.; Erba, H.P.; DeAngelo, D.J.; Bixby, D.L.; Altman, J.K.; Maris, M.; Hua, Z.; Blakemore, S.J.; Faessel, H.; Sedarati, F.; et al. Pevonedistat (mln4924), a first-in-class nedd8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: A phase 1 study. Br. J. Haematol. 2015, 169, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Nicolle, E.; Boumendjel, A.; Macalou, S.; Genoux, E.; Ahmed-Belkacem, A.; Carrupt, P.A.; Di Pietro, A. Qsar analysis and molecular modeling of abcg2-specific inhibitors. Adv. Drug Deliv. Rev. 2009, 61, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Peng, H.; Zhang, J.T. Human multidrug transporter abcg2, a target for sensitizing drug resistance in cancer chemotherapy. Curr. Med. Chem. 2007, 14, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Shiozawa, K.; Hassel, B.A.; Ross, D.D. Complex interaction of bcrp/abcg2 and imatinib in bcr-abl-expressing cells: Bcrp-mediated resistance to imatinib is attenuated by imatinib-induced reduction of bcrp expression. Blood 2006, 108, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with abcb1 and abcg2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of abc transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Hang, D.; Dong, H.C.; Ning, T.; Dong, B.; Hou, D.L.; Xu, W.G. Prognostic value of the stem cell markers cd133 and abcg2 expression in esophageal squamous cell carcinoma. Dis. Esophagus. 2012, 25, 638–644. [Google Scholar] [CrossRef]
- Tsunoda, S.; Okumura, T.; Ito, T.; Kondo, K.; Ortiz, C.; Tanaka, E.; Watanabe, G.; Itami, A.; Sakai, Y.; Shimada, Y. Abcg2 expression is an independent unfavorable prognostic factor in esophageal squamous cell carcinoma. Oncology 2006, 71, 251–258. [Google Scholar] [CrossRef]
- An, Y.; Ongkeko, W.M. Abcg2: The key to chemoresistance in cancer stem cells? Expert Opin. Drug Metab. Toxicol. 2009, 5, 1529–1542. [Google Scholar] [CrossRef]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (bcrp/abcg2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Bunting, K.D. Abc transporters as phenotypic markers and functional regulators of stem cells. Stem Cells 2002, 20, 11–20. [Google Scholar] [CrossRef]
- Ding, X.W.; Wu, J.H.; Jiang, C.P. Abcg2: A potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010, 86, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Hrycyna, C.A.; Yan, Q.W.; Medina-Perez, W.Y.; Robey, R.W.; van de Laar, A.; Litman, T.; Dean, M.; Bates, S.E. Acquired mutations in the mxr/bcrp/abcp gene alter substrate specificity in mxr/bcrp/abcp-overexpressing cells. Cancer Res. 2001, 61, 6635–6639. [Google Scholar] [PubMed]
- Pozza, A.; Perez-Victoria, J.M.; Sardo, A.; Ahmed-Belkacem, A.; Di Pietro, A. Purification of breast cancer resistance protein abcg2 and role of arginine-482. Cell Mol. Life Sci. 2006, 63, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Robey, R.W.; Belinsky, M.G.; Shchaveleva, I.; Ren, X.Q.; Sugimoto, Y.; Ross, D.D.; Bates, S.E.; Kruh, G.D. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-d-glucuronide) by abcg2: Effects of acquired mutations at r482 on methotrexate transport. Cancer Res. 2003, 63, 4048–4054. [Google Scholar] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (abcg2) in drug transport. AAPS J. 2005, 7, E118–E133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Visconte, V.; Anwer, F.; Kelly, K.R.; Nawrocki, S.T. Rational cotargeting of hdac6 and bet proteins yields synergistic antimyeloma activity. Blood Adv. 2019, 3, 1318–1329. [Google Scholar] [CrossRef]
- Mahalingam, D.; Carew, J.S.; Espitia, C.M.; Cool, R.H.; Giles, F.J.; de Jong, S.; Nawrocki, S.T. Heightened jnk activation and reduced xiap levels promote trail and sunitinib-mediated apoptosis in colon cancer models. Cancers (Basel) 2019, 11, 895. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wendlandt, E.; Tricot, G.; Zhan, F.; Carew, J.S.; Nawrocki, S.T. Junctional adhesion molecule-a is overexpressed in advanced multiple myeloma and determines response to oncolytic reovirus. Oncotarget 2015, 6, 41275–41289. [Google Scholar] [CrossRef]
- Gupta, P.; Gao, H.L.; Ashar, Y.V.; Karadkhelkar, N.M.; Yoganathan, S.; Chen, Z.S. Ciprofloxacin enhances the chemosensitivity of cancer cells to abcb1 substrates. Int. J. Mol. Sci. 2019, 20, 268. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Zhang, Y.K.; Zhang, X.Y.; Wang, Y.J.; Lu, K.W.; Hall, T.; Peng, R.; Yang, D.H.; Xie, N.; Chen, Z.S. Voruciclib, a potent cdk4/6 inhibitor, antagonizes abcb1 and abcg2-mediated multi-drug resistance in cancer cells. Cell Physiol. Biochem. 2018, 45, 1515–1528. [Google Scholar] [CrossRef]
- Zhang, Y.K.; Zhang, G.N.; Wang, Y.J.; Patel, B.A.; Talele, T.T.; Yang, D.H.; Chen, Z.S. Bafetinib (inno-406) reverses multidrug resistance by inhibiting the efflux function of abcb1 and abcg2 transporters. Sci. Rep. 2016, 6, 25694. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kathawala, R.J.; Espitia, C.M.; Jones, T.M.; Islam, S.; Gupta, P.; Zhang, Y.-K.; Chen, Z.-S.; Carew, J.S.; Nawrocki, S.T. ABCG2 Overexpression Contributes to Pevonedistat Resistance. Cancers 2020, 12, 429. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020429
Kathawala RJ, Espitia CM, Jones TM, Islam S, Gupta P, Zhang Y-K, Chen Z-S, Carew JS, Nawrocki ST. ABCG2 Overexpression Contributes to Pevonedistat Resistance. Cancers. 2020; 12(2):429. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020429
Chicago/Turabian StyleKathawala, Rishil J., Claudia M. Espitia, Trace M. Jones, Shariful Islam, Pranav Gupta, Yun-Kai Zhang, Zhe-Sheng Chen, Jennifer S. Carew, and Steffan T. Nawrocki. 2020. "ABCG2 Overexpression Contributes to Pevonedistat Resistance" Cancers 12, no. 2: 429. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020429