The c-Myc/AKT1/TBX3 Axis Is Important to Target in the Treatment of Embryonal Rhabdomyosarcoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TBX3 Is Overexpressed in ERMS
2.2. Establishment of ERMS Cell Culture Models in which TBX3 Was Either Stably Knocked Down or Overexpressed
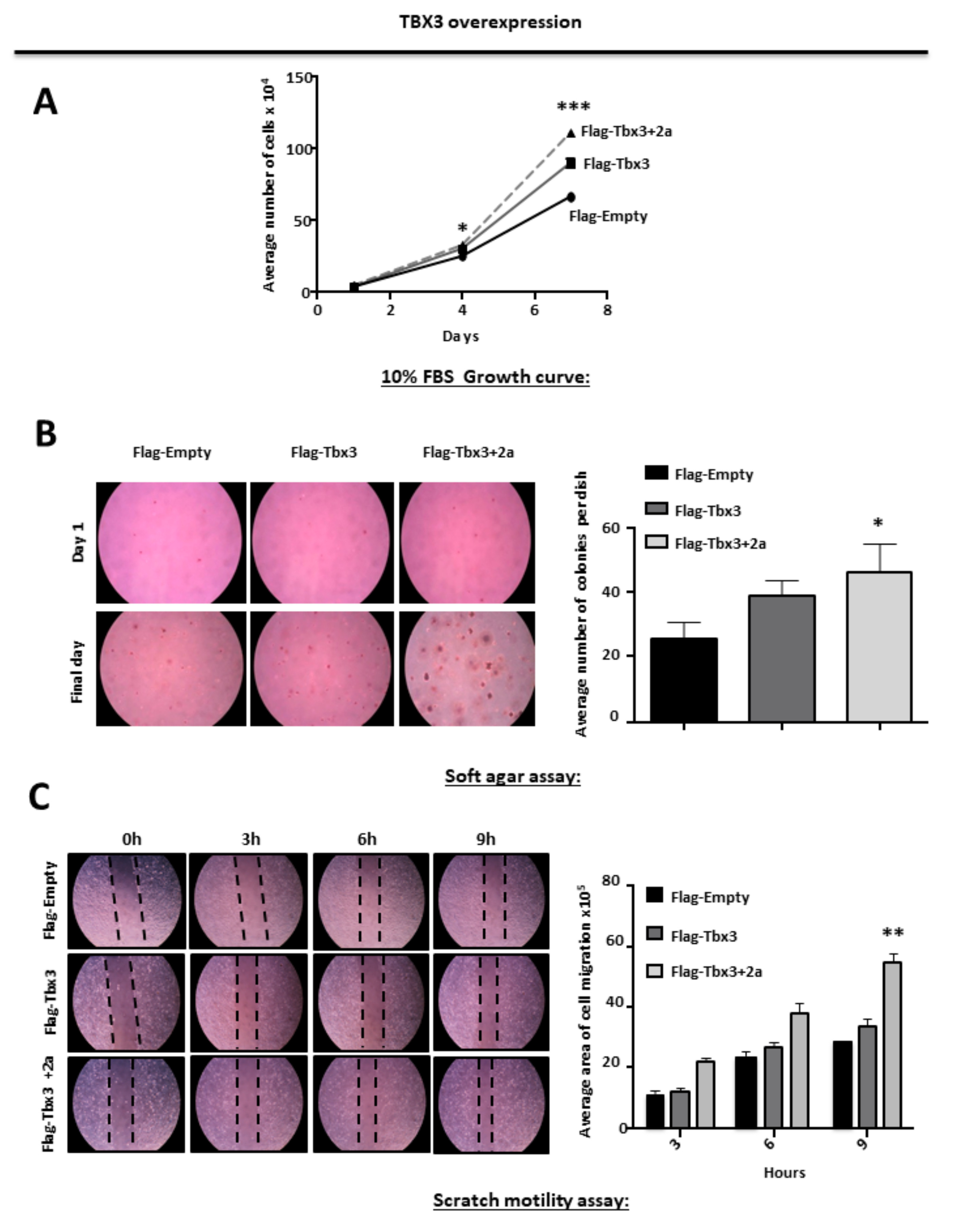
2.3. TBX3 Overexpression Promotes Proliferation, Anchorage Independent Growth and Migration of ERMS Cells
2.4. TBX3 Overexpression Promotes Tumour forming Ability and Invasiveness of ERMS Xenografts
2.5. TBX3 Knockdown Inhibits Cell Proliferation, Induces Apoptosis and Blocks Anchorage Independent Growth and Migration of ERMS Cells
2.6. c-Myc Transcriptionally Upregulates TBX3 in ERMS
2.7. AKT1 Is the Predominant AKT Isoform in ERMS and Post-Translationally Upregulates TBX3
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfections
4.2. Generation of Stable Cell Lines
4.3. Western Blot Analysis
4.4. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Proliferation Assay
4.6. Anchorage Independence Assay
4.7. In Vitro Cell Migration Assay
4.8. Xenograft Mouse Model
4.9. Immunohistochemistry
4.10. Fluorescence Assisted Cell Sorting
4.11. Treatments
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ries, L.; Smith, M.; Gurney, J.; Linet, M.; Tamra, T.; Young, J.; Bunin, G. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; National Cancer Institute: Bethesda, MD, USA, 1999. [Google Scholar]
- American Cancer Society. About Rhabdomyosarcoma; American Cancer Society: Atlanta, GA, USA, 2014. [Google Scholar]
- Ognjanovic, S.; Linabery, A.; Charbonneau, G.; Ross, J. Trends in childhood RMS incidence and survival in the US. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C. Childhood rhabdomyosarcoma: Recent advances and prospective views. J. Dent. Res. 2012, 91, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Stewart, E.; Shelat, A.A.; Qu, C.; Bahrami, A.; Hatley, M.; Wu, G.; Bradley, C.; McEvoy, J.; Pappo, A.; et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell 2013, 24, 710–724. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, D.S.; Gupta, A.A.; Rudzinski, E.R. What is new in the biology and treatment of pediatric rhabdomyosarcoma? Curr. Opin. Pediatr. 2014, 26, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.; Guttridge, D.C. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 2013, 280, 4323–4334. [Google Scholar] [CrossRef]
- Sun, X.; Guo, W.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Rhabdomyosarcoma: Advances in molecular and cellular biology. Sarcoma 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Vlahovic, G.; Meadows, K.L.; Hatch, A.J.; Jia, J.; Nixon, A.B.; Uronis, H.E.; Morse, M.A.; Selim, A.; Crawford, J.; Riedel, R.F.; et al. A phase I trial of the IGF-1R antibody ganitumab (AMG 479) in combination with everolimus (RAD001) and panitumumab in patients with advanced cancer. Oncologist 2018, 23, 782. [Google Scholar] [CrossRef] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H. Targeting the sonic hedgehog signaling pathway: Review of smoothened and GLI inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Bamshad, M.; Le, T.; Watkins, W.S.; Dixon, M.E.; Kramer, B.E.; Roeder, A.D.; Carey, J.C.; Root, S.; Schinzel, A.; Van Maldergem, L.; et al. The spectrum of mutations in TBX3: Genotype/phenotype relationship in ulnar-mammary syndrome. Am. J. Hum. Genet. 1999, 64, 1550–1562. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Huang, X.; Chen, C.; Gray, J.; Huang, T. TBX3 and its isoform TBX3 + 2a are functionally distinctive in inhibition of senescence and are overexpressed in a subset of breast cancer cell lines. Cancer Res. 2004, 64, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Hoogaars, W.; Barnett, P.; Rodriguez, M.; Clout, D.; Moorman, A.; Goding, C.R.; Christoffels, V. TBX3 and its splice variant TBX3 + Exon 2a are functionally similar. Pigment. Cell Melanoma Res. 2008, 21, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Krstic, M.; Kolendowski, B.; Cecchini, M.J.; Postenka, C.O.; Hassan, H.M.; Torchia, J.; Chambers, A.F.; Tuck, A.B. TBX3 promotes progression of pre-invasive breast cancer cells by inducing emt and directly up-regulating SLUG. J. Pathol. 2019, 248, 191–203. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, Y.; Chen, K. Tbx3 isoforms are involved in pluripotency maintaining through distinct regulation of nanog transcriptional activity. Biochem. Biophys. Res. Commun. 2014, 444, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.U.; Emechebe, U.; Thomas, K.R.; Moon, A.M. Mouse Tbx3 mutants suggest novel molecular mechanisms for ulnar-mammary syndrome. PLoS ONE 2013, 8, e67841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packham, E.A.; Brook, D.J. T-box genes in human disorders. Hum. Mol. Genet. 2003, 12, 37R–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.; Rimm, D.; Williams, K.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004, 64, 5270–5282. [Google Scholar] [CrossRef]
- Humtsoe, J.O.; Koya, E.; Pham, E.; Aramoto, T.; Zuo, J.; Ishikawa, T.; Kramer, R.H. Transcriptional profiling identifies upregulated genes following induction of epithelial-mesenchymal transition in squamous carcinoma cells. Exp. Cell Res. 2012, 318, 379–390. [Google Scholar] [CrossRef]
- Khan, S.F.; Damerell, V.; Omar, R.; Toit, M.D.; Khan, M.; Maranyane, H.; Mlaza, M.; Bleloch, J.; Bellis, C.; Bianca, D.B.; et al. The roles and regulation of TBX3 in development and disease. Gene 2019, 144223. [Google Scholar] [CrossRef]
- Liu, J.; Esmailpour, T.; Shang, X.; Gulsen, G.; Liu, A.; Huang, T. TBX3 over-expression causes mammary gland hyperplasia and increases mammary stem-like cells in an inducible transgenic mouse model. BMC Dev. Biol. 2011, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Aladowicz, E.; Lanfrancone, L.; Goding, C.R. Tbx3 represses E-Cadherin expression and enhances melanoma invasiveness. Cancer Res. 2008, 68, 7872–7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, C.A.; Labalette, C.; Armengol, C.; Cougot, D.; Wei, Y.; Cairo, S.; Pineau, P.; Neuveut, C.; De Reyniès, A.; Dejean, A.; et al. Tbx3 Is a downstream target of the Wnt/β-catenin pathway and a critical mediator of β-catenin survival functions in liver cancer. Cancer Res. 2007, 67, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, N.C.; Papaioannou, V.E. The T-box transcription factors TBX2 and TBX3 in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomnytska, M.; Dubrovska, A.; Hellman, U.; Volodko, N.; Souchelnytskyi, S. Increased expression of CSHMT, Tbx3 and utrophin in plasma of ovarian and breast cancer patients. Int. J. Cancer 2006, 118, 412–421. [Google Scholar] [CrossRef]
- Hansel, D.E.; Rahman, A.; House, M.; Ashfaq, R.; Berg, K.; Yeo, C.J.; Maitra, A. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin. Cancer Res. 2004, 10, 6152–6158. [Google Scholar] [CrossRef] [Green Version]
- Burgucu, D.; Guney, K.; Sahinturk, D.; Ozbudak, I.H.; Ozel, D.; Ozbilim, G.; Yavuzer, U. Tbx3 represses PTEN and is over-expressed in head and neck squamous cell carcinoma. BMC Cancer 2012, 12, 481. [Google Scholar] [CrossRef] [Green Version]
- Wansleben, S.; Peres, J.; Hare, S.; Goding, C.R.; Prince, S. T-box transcription factors in cancer biology. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 380–391. [Google Scholar] [CrossRef]
- Yarosh, W.; Barrientos, T.; Esmailpour, T.; Lin, L.; Carpenter, P.M.; Osann, K.; Anton-Culver, H.; Huang, T. TBX3 is overexpressed in breast cancer and represses P14ARF by interacting with histone deacetylases. Cancer Res. 2008, 68, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Willmer, T.; Hare, S.; Peres, J.; Prince, S. The T-box transcription factor TBX3 drives proliferation by direct repression of the P21WAF1 cyclin-dependent kinase inhibitor. Cell Div. 2016, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Peres, J.; Davis, E.; Mowla, S.; Bennett, D.C.; Li, J.A.; Wansleben, S.; Prince, S. The highly homologous T-box transcription factors, TBX2 and TBX3, have distinct roles in the oncogenic process. Genes Cancer 2010, 1, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Peres, J.; Prince, S. The T-box Transcription Factor, TBX3, Is Sufficient to Promote Melanoma Formation and Invasion. Mol. Cancer 2013, 12, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ballim, D.; Rodriguez, M.; Cui, R.; Goding, C.R.; Teng, H.; Prince, S. The anti-proliferative function of the Tgf-Β1 signaling pathway involves the repression of the oncogenic Tbx2 by its homologue Tbx3. J. Biol. Chem. 2014, 289, 35633–35643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkhofer, L.; Walter, K.; Costa, I.G.; Carrasco, M.C.R.; Eiseler, T.; Hafner, S.; Genze, F.; Zenke, M.; Bergmann, W.; Illing, A.; et al. Tbx3 fosters pancreatic cancer growth by increased angiogenesis and activin/nodal-dependent induction of stemness. Stem Cell Res. 2016, 17, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willmer, T.; Cooper, A.; Sims, D.; Govender, D.; Prince, S. The T-box transcription factor 3 is a promising biomarker and a key regulator of the oncogenic phenotype of a diverse range of sarcoma subtypes. Oncogenesis 2016, 5, e199. [Google Scholar] [CrossRef]
- Dang, C.V.; Resar, L.M.S.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the C-Myc oncogenic transcription factor. Exp. Cell Res. 1999, 253, 63–77. [Google Scholar] [CrossRef]
- Barrios, C.; Castresana, J.S.; Ruiz, J.; Kreicbergs, A. Amplification of the C-Myc proto-oncogene in soft tissue sarcomas. Oncology 1994, 51, 13–17. [Google Scholar] [CrossRef]
- Castresana, J.S.; Barrios, C.; Gómez, L.; Kreicbergs, A. Amplification of the C-Myc proto-oncogene in human chondrosarcoma. Diagn. Mol. Pathol. 1992, 1, 235–238. [Google Scholar] [CrossRef]
- Demir, D.; Yaman, B.; Anacak, Y.; Keçeci, B.; Kandiloǧlu, G.; Akalin, T. Prognostic significance of Bcl-2, C-Myc, survivin and tumor grade in synovial sarcoma. Turk. Patoloji Derg. Turk. J. Pathol. 2014, 30, 55–65. [Google Scholar] [CrossRef]
- Sollazzo, M.R.; Benassi, M.S.; Magagnoli, G.; Gamberi, G.; Molendini, L.; Ragazzini, P.; Merli, M.; Ferrari, C.; Balladelli, A.; Picci, P. Increased C-Myc oncogene expression in ewing’s sarcoma: Correlation with Ki67 proliferation index. Tumori 1999, 85, 167–173. [Google Scholar] [CrossRef]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol. Cancer 2006, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2015, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotzer, M.A.; Hogarty, M.D.; Janss, A.J.; Liu, X.; Zhao, H.; Eggert, A.; Sutton, L.N.; Rorke, L.B.; Brodeur, G.M.; Phillips, P.C. MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clin. Cancer Res. 2001, 7, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, E.M.J.J.; Klijn, J.G.M.; van Staveren, I.L.; Portengen, H.; Foekens, J.A. C-Myc amplification is a better prognostic factor than HER2/Neu amplification in primary breast cancer. Cancer Res. 1992, 52, 1107–1113. [Google Scholar] [PubMed]
- Schuijers, J.; Manteiga, J.C.; Weintraub, A.S.; Day, D.S.; Zamudio, A.V.; Hnisz, D.; Lee, T.I.; Young, R.A. Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 2018, 23, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanità, P.; Monache, S.D.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. C-Myc sustains transformed phenotype and promotes radioresistance of embryonal rhabdomyosarcoma cell lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef]
- Zhang, J.; Song, N.; Zang, D.; Yu, J.; Li, J.; Di, W.; Guo, R.; Zhao, W.; Wang, H. C-Myc promotes tumor proliferation and anti-apoptosis by repressing P21 in rhabdomyosarcomas. Mol. Med. Rep. 2017, 16, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Willmer, T.; Peres, J.; Mowla, S.; Abrahams, A.; Prince, S. The T-box factor TBX3 Is important in S-Phase and is regulated by c-MYC and cyclin A-CDK2. Cell Cycle 2015, 14, 3173–3183. [Google Scholar] [CrossRef] [Green Version]
- Myers, A.P.; Cantley, L.C. Targeting a common collaborator in cancer development. Sci. Transl. Med. 2010, 2, 48ps45. [Google Scholar] [CrossRef] [Green Version]
- Sarris, E.G.; Saif, M.W.; Syrigos, K.N. The biological role of PI3K pathway in lung cancer. Pharmaceuticals 2012, 5, 1236–1264. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T.M.; Koreckij, T.D.; Corey, E. Targeted therapy for advanced prostate cancer: Inhibition of the PI3K/Akt/MTOR pathway. Curr. Cancer Drug Targets 2010, 9, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, L.; Zhang, X.; Cui, Z. PI3K/AKT/MTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Onco. Targets Ther. 2017, 10, 2865–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, J.R.; Bellacosa, A. AKT plays a central role in tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10983–10985. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.-S.; Rosenblatt, K.; Huang, K.; Lahat, G. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2011, 30, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Morooka, T.; Hoshida, Y.; Zhang, B.; Qiu, Y.; Nakamichi, I.; Hamada, K.I.; Ueda, T.; Naka, N.; Kudawara, I.; et al. Prognostic significance of activated AKT expression in soft-tissue sarcoma. Clin. Cancer Res. 2006, 12, 3070–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grille, S.J.; Bellacosa, A.; Upson, J.; Klein-Szanto, A.J.; Van Roy, F.; Lee-Kwon, W.; Donowitz, M.; Tsichlis, P.N.; Larue, L. The protein kinase AKT induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003, 63, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.S.; Ren, W.; Korchin, B.; Lahat, G.; Dicker, A.; Lu, Y.; Mills, G.; Pollock, R.E.; Lev, D. Soft tissue sarcoma cells are highly sensitive to AKT blockade: A Role for P53-independent up-regulation of GADD45α. Cancer Res. 2008, 68, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Cen, L.; Hsieh, F.C.; Lin, H.J.; Chen, C.S.; Qualman, S.J.; Lin, J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br. J. Cancer 2007, 97, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Petricoin, E.F.; Espina, V.; Araujo, R.P.; Midura, B.; Yeung, C.; Wan, X.; Eichler, G.S.; Johann, D.J.; Qualman, S.; Tsokos, M.; et al. Phosphoprotein pathway mapping: AKT/Mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007, 67, 3431–3440. [Google Scholar] [CrossRef] [Green Version]
- Jahangiri, A.; Weiss, W.A. It takes two to tango: Dual inhibition of PI3K & MAPK in rhabdomyosarcoma. Clin. Cancer Res. 2013, 9, 5811–5813. [Google Scholar] [CrossRef]
- Marampon, F.; Bossi, G.; Ciccarelli, C.; Rocco, A.D.; Sacchi, A.; Pestell, R.G.; Zani, B.M. MEK/ERK inhibitor U0126 affects In Vitro and In Vivo growth of embryonal rhabdomyosarcoma. Mol. Cancer Ther. 2009, 8, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncol. 2015, 20, 227–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of RAS mutations in cancer. Cancer Res. 2012, 72, 2457–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesada, J.; Amato, R. The molecular biology of soft-tissue sarcomas and current trends in therapy. Sarcoma 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleveld, T.F.; Schild, L.; Koster, J.; Zwijnenburg, D.A.; Alles, L.K.; Ebus, M.E.; Volckmann, R.; Tijtgat, G.A.; van Sluis, P.; Versteeg, R.; et al. RAS-MAPK pathway-driven tumor progression is associated with loss of cic and other genomic aberrations in neuroblastoma. Cancer Res. 2018, 78, 6297–6307. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Helman, L.J. Levels of PTEN protein modulate AKT phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene 2003, 22, 8205–8211. [Google Scholar] [CrossRef] [Green Version]
- Toker, A. Achieving specificity in AKT signaling in cancer. Adv. Biol. Regul. 2013, 52, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yu, W.; Chen, X.; Peng, X.; Jeon, S.; Morris, J.; Guzman, G.; Hay, N. Spontaneous hepatocellular carcinoma after the combined deletion of AKT isoforms. Cancer Cell 2017, 29, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Peres, J.; Mowla, S.; Prince, S. The T-box transcription factor, TBX3, is a key substrate of AKT3 in melanomagenesis. Oncotarget 2015, 6, 1821–1833. [Google Scholar] [CrossRef] [Green Version]
- Omar, R.; Cooper, A.; Maranyane, H.M.; Zerbini, L.; Prince, S. COL1A2 Is a TBX3 target that mediates its impact on fibrosarcoma and chondrosarcoma cell migration. Cancer Lett. 2019, 459, 227–239. [Google Scholar] [CrossRef]
- Zacharias, M.; Brcic, L.; Eidenhammer, S.; Popper, H. Bulk tumour cell migration in lung carcinomas might be more common than epithelial-mesenchymal transition and be differently regulated. BMC Cancer 2018, 18, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouraklis, G.; Triche, T.J.; Wesley, R.; Tsokos, M. Myc oncogene expression and nude mouse tumorigenicity and metastasis formation are higher in alveolar than embryonal rhabdomyosarcoma cell lines. Pediatr. Res. 1999, 45, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Khan, J. OncoGenomics Database. Available online: https://pob.abcc.ncifcrf.gov/cgi-bin/JK. (accessed on 12 December 2019).
- Barretina, J.; Caponigro, G.; Stransky, N.; Kavitha, V.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tato, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis 1. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
- Villanueva, T.M. Long path to MYC inhibition approaches clinical trials. Nat. Rev. Drug Discov. 2019, 18, 337. [Google Scholar]
- Beaulieu, M.; Jauset, T.; Massó-vallés, D.; Martínez-martín, S.; Rahl, P.; Maltais, L.; Zacarias-fluck, M.F.; Casacuberta-serra, S.; Pozo, S.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11, 1–27. [Google Scholar] [CrossRef]
- Yu, K.; Discafani, C.; Zhang, W.; Skotnicki, J.; Frost, P. MTOR, a novel target in breast cancer: The effect of CCI-779, an MTOR inhibitor, in preclinical models of breast cancer. Endocr. Relat. Cancer 2001, 8, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. MTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 2005, 16, 525–537. [Google Scholar] [CrossRef]
- Frost, P.; Moatamed, F.; Hoang, B.; Shi, Y.; Gera, J.; Yan, H.; Frost, P.; Gibbons, J.; Lichtenstein, A. In Vivo antitumor effects of the MTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood 2004, 104, 4181–4188. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Li, Y.; Yue, P.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R. The Combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer In Vitro and In Vivo. PLoS ONE 2011, 6, e20899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Reckamp, K.; Yu, H.; Figlin, R.A. AKT inhibitors in clinical development for the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 1355–1366. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. article clonal evolution enhances leukemia-propagating cell frequency in t cell acute lymphoblastic leukemia through AKT/MTORC1 pathway activation. Cancer Cell 2014, 25, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target C-MYC. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Iii, R.J.S. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Crew, A.P.; Coleman, K.; Crews, C.M.; Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; et al. Hijacking the E3 ubiquitin ligase cereblon to article hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Politz, O.; Siegel, F.; Barfacker, L.; Ulf, B.; Hagerbarth, A.; Scott, W.J.; Michels, M.; Ince, S.; Liu, N.; Ince, S.; et al. BAY 1125976, a selective allosteric AKT1/2 inhibitor, exhibits high efficacy on akt signaling-dependent tumor growth in mouse models. Int. J. Cancer 2017, 140, 449–459. [Google Scholar] [CrossRef]
- Prince, S.; Wiggins, T.; Hulley, P.A.; Kidson, S.H. Stimulation of melanogenesis by tetradecanoylphorbol 13-acetate (TPA) in mouse melanocytes and neural crest cells. Pigment. Cell Res. 2003, 16, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.; Teng, H.; Bilican, B.; Parker, M.I.; Liu, B.; Carriera, S.; Goding, C.R.; Prince, S. Ectopic Tbx2 expression results in polyploidy and cisplatin resistance. Oncogene 2008, 27, 976–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sims, D.; Maranyane, H.M.; Damerell, V.; Govender, D.; Isaacs, A.W.; Peres, J.; Prince, S. The c-Myc/AKT1/TBX3 Axis Is Important to Target in the Treatment of Embryonal Rhabdomyosarcoma. Cancers 2020, 12, 501. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020501
Sims D, Maranyane HM, Damerell V, Govender D, Isaacs AW, Peres J, Prince S. The c-Myc/AKT1/TBX3 Axis Is Important to Target in the Treatment of Embryonal Rhabdomyosarcoma. Cancers. 2020; 12(2):501. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020501
Chicago/Turabian StyleSims, Danica, Hapiloe Mabaruti Maranyane, Victoria Damerell, Dhirendra Govender, Ashwin W. Isaacs, Jade Peres, and Sharon Prince. 2020. "The c-Myc/AKT1/TBX3 Axis Is Important to Target in the Treatment of Embryonal Rhabdomyosarcoma" Cancers 12, no. 2: 501. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020501