Hypoxia-Induced Centrosome Amplification Underlies Aggressive Disease Course in HPV-Negative Oropharyngeal Squamous Cell Carcinomas

 , ,
, ,
Abstract
:1. Introduction
2. Results
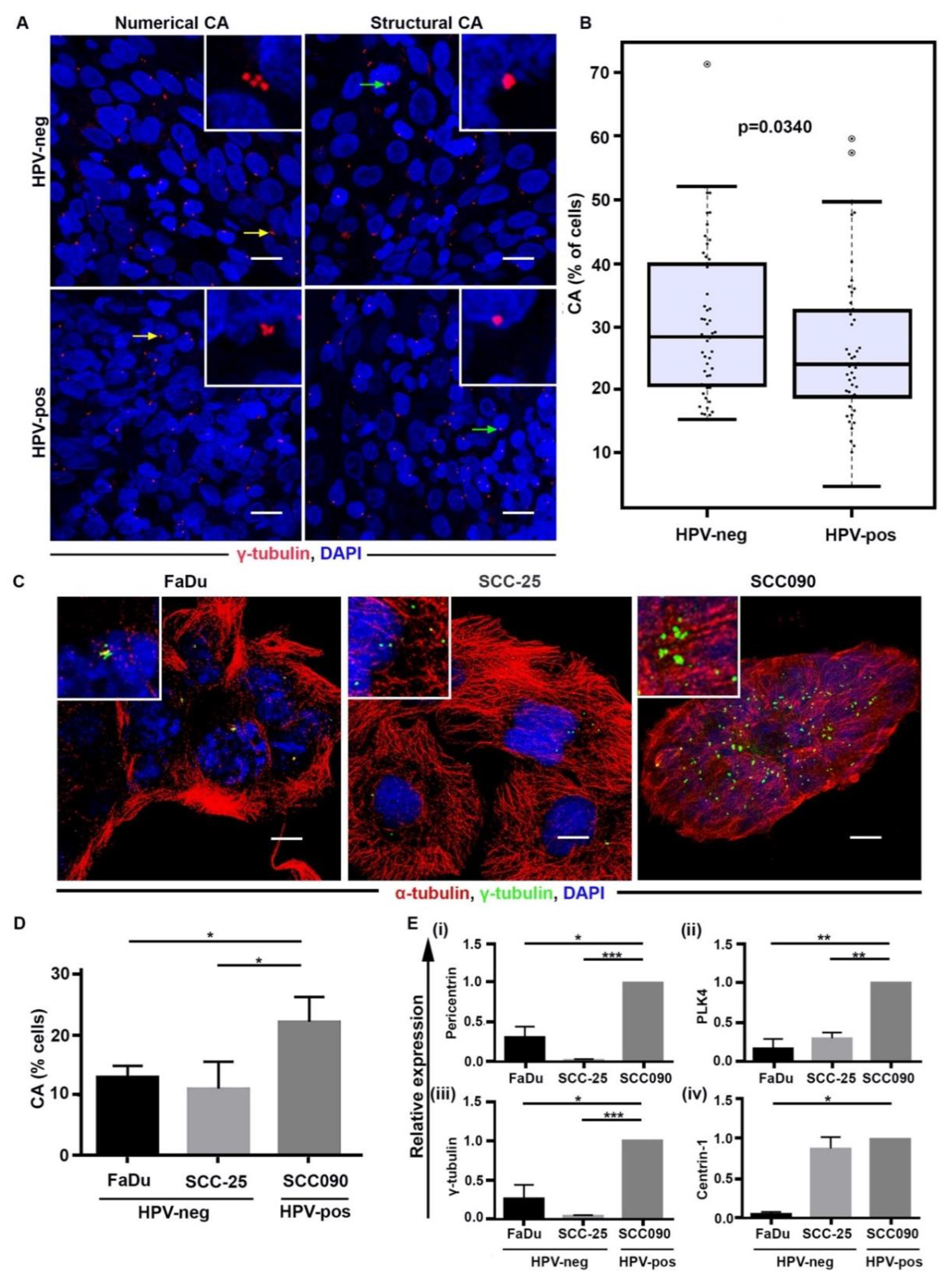
2.1. Discordant Relationship between CA in Cultured Cells versus Patient Samples for HPV-Pos and HPV-Neg OPSCC
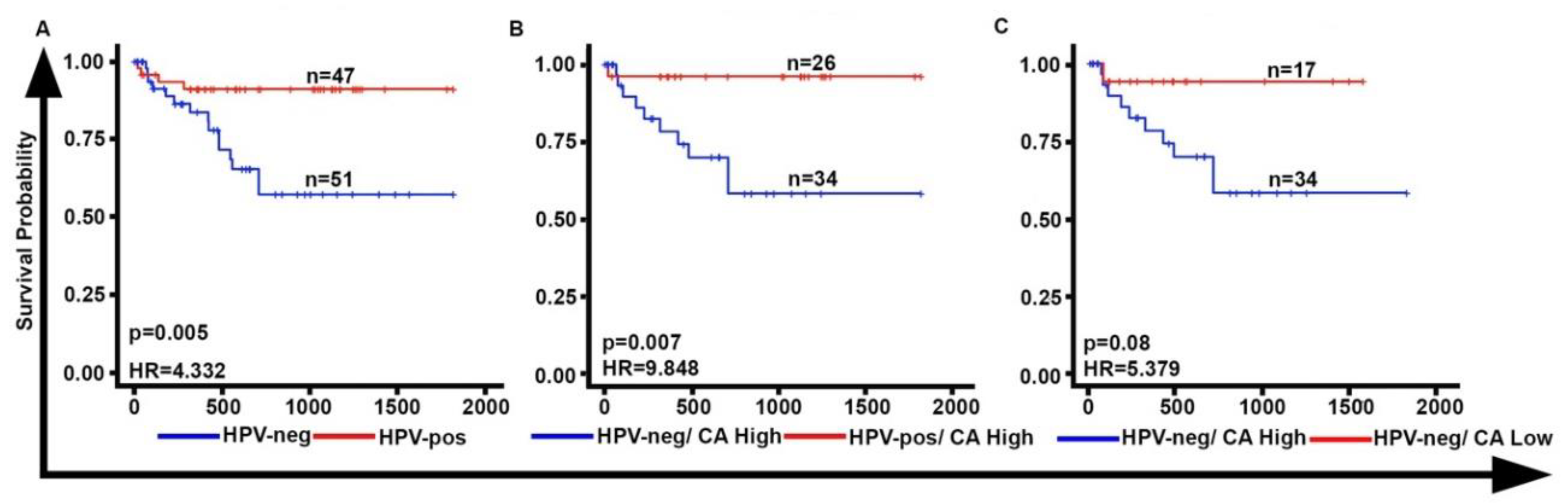
2.2. CA Is Associated with Poor Overall Survival in HPV-Neg OPSCCs
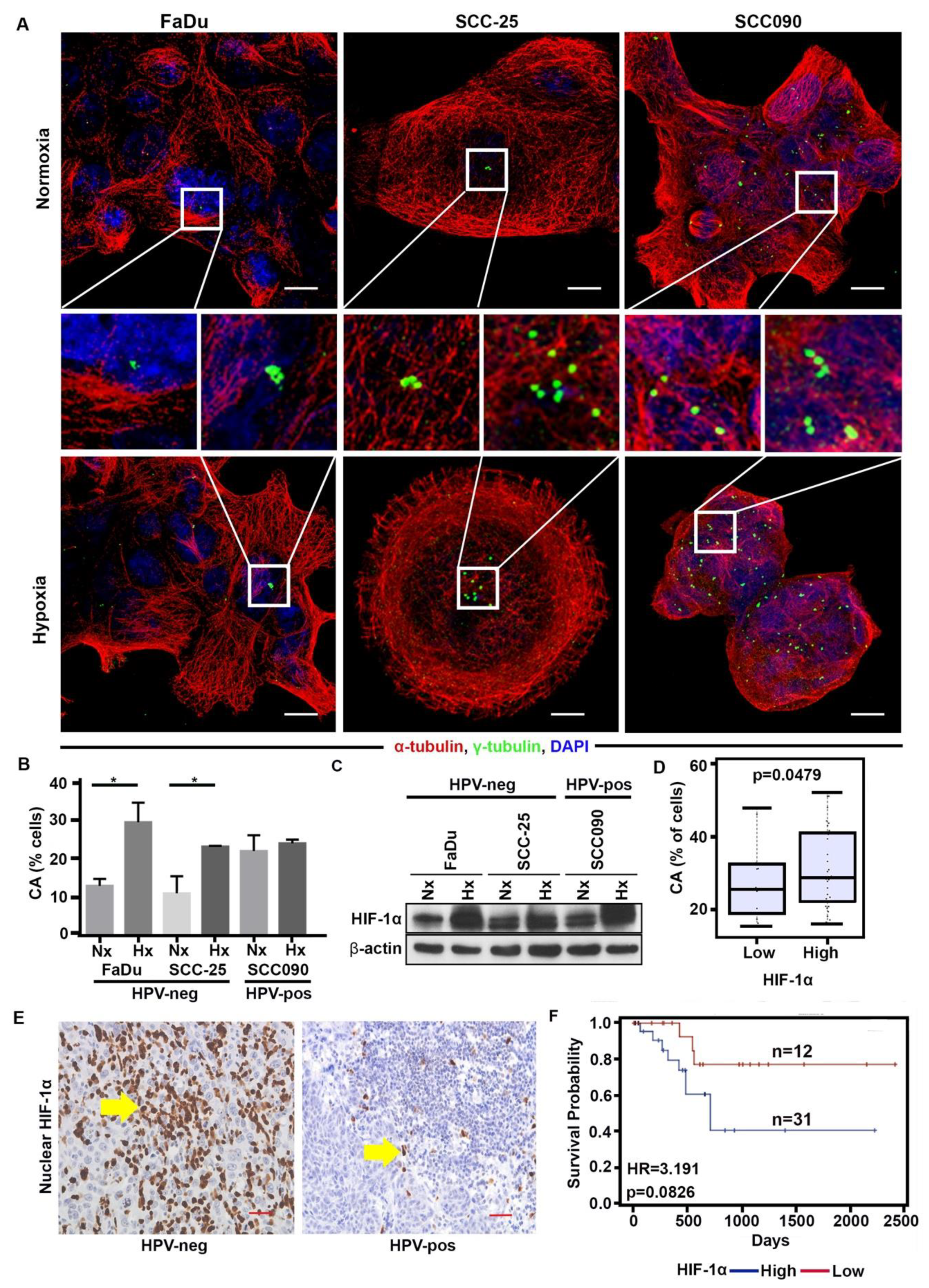
2.3. Hypoxia Enhances CA in HPV-Neg OPSCCs via HIF-1α
2.4. HIF-1α Upregulation Is Associated with Downregulation of miR-34a, Increase in CA and Expression of Cyclin D1
3. Discussion
4. Materials and Methods
4.1. Clinical Tissue Samples
4.2. HPV Analysis
4.3. Immunohistofluorescence Staining and Quantitation
4.4. Immunohistochemistry of Cyclin D1 and HIF-1α and Scoring
4.5. Cell Culture and Hypoxia Treatment
4.6. RNA Extraction and Quantitative Real Time PCR
4.7. miRNA Extraction and Quantitative Real Time PCR
4.8. Immunocytofluorescence Staining
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 123–141. [Google Scholar] [CrossRef] [PubMed]
- van Monsjou, H.S.; Balm, A.J.; van den Brekel, M.M.; Wreesmann, V.B. Oropharyngeal squamous cell carcinoma: A unique disease on the rise? Oral Oncol. 2010, 46, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus-Associated Cancers—United States, 2008–2012. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Lowy, D.R.; Munger, K. Prognostic implications of HPV in oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 82–84. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.H.; O’Sullivan, B. Overview of the 8th Edition TNM Classification for Head and Neck Cancer. Curr. Treat. Options Oncol. 2017, 18, 40. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Rothenberg, S.M.; Ellisen, L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Investig. 2012, 122, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Munger, K.; Scheffner, M.; Huibregtse, J.M.; Howley, P.M. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 1992, 12, 197–217. [Google Scholar]
- Duensing, S.; Munger, K. Human papillomaviruses and centrosome duplication errors: Modeling the origins of genomic instability. Oncogene 2002, 21, 6241–6248. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- D’Assoro, A.B.; Lingle, W.L.; Salisbury, J.L. Centrosome amplification and the development of cancer. Oncogene 2002, 21, 6146–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, A.; Rida, P.C.; Aneja, R. Heading off with the herd: How cancer cells might maneuver supernumerary centrosomes for directional migration. Cancer Metastasis Rev. 2013, 32, 269–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vugt, M.A.; Medema, R.H. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005, 24, 2844–2859. [Google Scholar] [CrossRef] [Green Version]
- Nurwidya, F.; Takahashi, F.; Minakata, K.; Murakami, A.; Takahashi, K. From tumor hypoxia to cancer progression: The implications of hypoxia-inducible factor-1 expression in cancers. Anat. Cell Biol. 2012, 45, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Hanns, E.; Job, S.; Coliat, P.; Wasylyk, C.; Ramolu, L.; Pencreach, E.; Suarez-Carmona, M.; Herfs, M.; Ledrappier, S.; Macabre, C.; et al. Human Papillomavirus-related tumours of the oropharynx display a lower tumour hypoxia signature. Oral Oncol. 2015, 51, 848–856. [Google Scholar] [CrossRef]
- Klein, A.; Flugel, D.; Kietzmann, T. Transcriptional regulation of serine/threonine kinase-15 (STK15) expression by hypoxia and HIF-1. Mol. Biol. Cell 2008, 19, 3667–3675. [Google Scholar] [CrossRef] [Green Version]
- Rosario, C.; Swallow, C.J. Abstract 4045: Hypoxia induces Plk4 expression and promotes immortalization of proliferating cells. Cancer Res. 2013, 4045. [Google Scholar] [CrossRef]
- Mittal, K.; Choi, D.H.; Ogden, A.; Donthamsetty, S.; Melton, B.D.; Gupta, M.V.; Pannu, V.; Cantuaria, G.; Varambally, S.; Reid, M.D.; et al. Amplified centrosomes and mitotic index display poor concordance between patient tumors and cultured cancer cells. Sci. Rep. UK 2017, 7, 43984. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Choi, D.H.; Klimov, S.; Pawar, S.; Kaur, R.; Mitra, A.K.; Gupta, M.V.; Sams, R.; Cantuaria, G.; Rida, P.C.G.; et al. A centrosome clustering protein, KIFC1, predicts aggressive disease course in serous ovarian adenocarcinomas. J. Ovarian Res. 2016, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, K.; Ogden, A.; Reid, M.D.; Rida, P.C.G.; Varambally, S.; Aneja, R. Amplified centrosomes may underlie aggressive disease course in pancreatic ductal adenocarcinoma. Cell Cycle 2015, 14, 2798–2809. [Google Scholar] [CrossRef] [PubMed]
- Pannu, V.; Mittal, K.; Cantuaria, G.; Reid, M.D.; Li, X.; Donthamsetty, S.; McBride, M.; Klimov, S.; Osan, R.; Gupta, M.V.; et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget 2015, 6, 10487–10497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, T.R. TCGA Releases Head and Neck Cancer Data. Cancer Discov. 2015, 5, 340–341. [Google Scholar]
- Eustace, A.; Mani, N.; Span, P.N.; Irlam, J.J.; Taylor, J.; Betts, G.N.; Denley, H.; Miller, C.J.; Homer, J.J.; Rojas, A.M. A 26-gene hypoxia signature predicts benefit from hypoxia-modifying therapy in laryngeal cancer but not bladder cancer. Clin. Cancer Res. 2013, 19, 4879–4888. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Khanna, S.; Roy, S.; Sen, C.K. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J. Biol. Chem. 2011, 286, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Shivdasani, R.A. MicroRNAs: Regulators of gene expression and cell differentiation. Blood 2006, 108, 3646–3653. [Google Scholar] [CrossRef]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Rokavec, M.; Jiang, L.; Horst, D.; Hermeking, H. Antagonistic Effects of p53 and HIF1A on microRNA-34a Regulation of PPP1R11 and STAT3 and Hypoxia-induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells. Gastroenterology 2017, 153, 505–520. [Google Scholar] [CrossRef]
- Nelsen, C.J.; Kuriyama, R.; Hirsch, B.; Negron, V.C.; Lingle, W.L.; Goggin, M.M.; Stanley, M.W.; Albrecht, J.H. Short term cyclin D1 overexpression induces centrosome amplification, mitotic spindle abnormalities, and aneuploidy. J. Biol. Chem. 2005, 280, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, M.; Rida, P.C.; Aneja, R. Turning the headlights on novel cancer biomarkers: Inspection of mechanics underlying intratumor heterogeneity. Mol. Asp. Med. 2015, 45, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, L.M.; Gleich, L.L.; Fukasawa, K.; Chadwell, J.; Miller, M.A.; Stambrook, P.J.; Gluckman, J.L. Centrosome hyperamplification in head and neck squamous cell carcinoma: A potential phenotypic marker of tumor aggressiveness. Laryngoscope 2000, 110, 1798–1801. [Google Scholar] [CrossRef]
- Syed, M.I.; Syed, S.; Minty, F.; Harrower, S.; Singh, J.; Chin, A.; McLellan, D.R.; Parkinson, E.K.; Clark, L.J. Gamma tubulin: A promising indicator of recurrence in squamous cell carcinoma of the larynx. Otolaryngol. Head Neck Surg. 2009, 140, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.; Gais, P.; Steuer-Vogt, M.K.; Boulesteix, A.L.; Deutschle, T.; Hampel, R.; Wagenpfeil, S.; Rauser, S.; Walch, A.; Bink, K.; et al. Centrosome abnormalities in head and neck squamous cell carcinoma (HNSCC). Acta Otolaryngol. 2009, 129, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bonilla-Velez, J.; Mroz, E.A.; Hammon, R.J.; Rocco, J.W. Impact of human papillomavirus on oropharyngeal cancer biology and response to therapy: Implications for treatment. Otolaryngol. Clin. N. Am. 2013, 46, 521–543. [Google Scholar] [CrossRef] [Green Version]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef]
- Knuth, J.; Sharma, S.J.; Wurdemann, N.; Holler, C.; Garvalov, B.K.; Acker, T.; Wittekindt, C.; Wagner, S.; Klussmann, J.P. Hypoxia-inducible factor-1alpha activation in HPV-positive head and neck squamous cell carcinoma cell lines. Oncotarget 2017, 8, 89681–89691. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Najy, A.J.; Huang, W.; Sethi, S.; Snyder, M.; Sakr, W.; Dyson, G.; Huttemann, M.; Lee, I.; Ali-Fehmi, R.; et al. HPV-associated differential regulation of tumor metabolism in oropharyngeal head and neck cancer. Oncotarget 2017, 8, 51530–51541. [Google Scholar] [CrossRef] [Green Version]
- Brockton, N.; Dort, J.; Lau, H.; Hao, D.; Brar, S.; Klimowicz, A.; Petrillo, S.; Diaz, R.; Doll, C.; Magliocco, A. High stromal carbonic anhydrase IX expression is associated with decreased survival in P16-negative head-and-neck tumors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 249–257. [Google Scholar] [CrossRef]
- Du, R.; Sun, W.; Xia, L.; Zhao, A.; Yu, Y.; Zhao, L.; Wang, H.; Huang, C.; Sun, S. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS ONE 2012, 7, e30771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, L.; Ley, J.; Wildes, T.M.; Schaffer, A.; Robinson, A.; Chun, S.E.; Lee, W.; Lewis, J., Jr.; Trinkaus, K.; Adkins, D. Phase I trial of palbociclib, a selective cyclin dependent kinase 4/6 inhibitor, in combination with cetuximab in patients with recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 58, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietbergen, M.M.; Leemans, C.R.; Bloemena, E.; Heideman, D.A.; Braakhuis, B.J.; Hesselink, A.T.; Witte, B.I.; Baatenburg de Jong, R.J.; Meijer, C.J.; Snijders, P.J.; et al. Increasing prevalence rates of HPV attributable oropharyngeal squamous cell carcinomas in the Netherlands as assessed by a validated test algorithm. Int. J. Cancer. 2013, 132, 1565–1567. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics | HPV-Neg | HPV-Pos | p Value |
|---|---|---|---|
| Gender, n (%) | |||
| Female | 11 (21.57) | 19 (40.43) | 0.0430 |
| Male | 40 (78.43) | 28 (59.57) | |
| Tumor Grade, n (%) | |||
| 1 | 5 (9.80) | 1 (2.13) | 0.2957 |
| 2 | 22 (43.14) | 27 (57.45) | |
| 3 | 18 (32.29) | 13 (27.66) | |
| N/A | 6 (11.76) | 6 (12.77) | |
| Tumor Stage, n (%) | |||
| II | 1 (1.96) | 0 (0.00) | 0.1933 |
| III | 15 (29.41) | 9 (19.15) | |
| IV | 28 (54.90) | 35 (74.47) | |
| N/A | 7 (13.73) | 3 (6.38) | |
| Smoking, n (%) | |||
| 0 | 6 (11.76) | 5 (10.64) | <0.0001 |
| 1 | 3 (5.58) | 29 (61.70) | |
| 2 | 4 (7.84) | 6 (12.77) | |
| 3 | 38 (74.51) | 7 (14.89) | |
| Alcohol, n (%) | |||
| No | 42 (82.35) | 46 (97.87) | 0.0393 |
| Yes | 8 (15.69) | 1 (2.13) | |
| N/A | 1 (1.96) | 0 (0.00) | |
| CA, n (%) | |||
| High | 34 (66.67) | 26 (55.32) | 0.2494 |
| Low | 17 (33.33) | 21 (44.58) | |
| Chemotherapy, n (%) | |||
| None | 32 (62.75) | 18 (38.30) | 0.0070 |
| Concomitant | 12 (23.53) | 26 (55.32) | |
| Neoadjuvant | 2 (3.92) | 0 (0.00) | |
| Adjunctive | 2 (3.92) | 0 (0.00) | |
| N/A | 3 (5.88) | 3 (6.39) | |
| Radiotherapy, n (%) | |||
| None | 5 (9.80) | 2 (4.26) | 0.1828 |
| Primary | 11 (21.57) | 14 (29.79) | |
| Adjuvant | 26 (50.98) | 29 (61.70) | |
| Palliative | 6 (11.76) | 1 (2.13) | |
| N/A | 3 (5.88) | 1 (2.13) | |
| Variable | Univariate Analysis | Multivariate Analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| p-Value | Hazard Ratio | 95% Hazard Ratio Confidence Limits | p-Value | Hazard Ratio | 95% Hazard Ratio Confidence Limits | ||||
| CA | High vs. low | 0.05 | 5.38 | 1.69 | 42.17 | 0.02 | 7.38 | 1.32 | 41.28 |
| Gender | Female | 0.35 | 0.37 | 0.05 | 2.90 | 0.61 | 1.64 | 0.23 | 11.49 |
| Age at diagnosis | High vs. low | 0.37 | 0.96 | 0.86 | 1.06 | 0.27 | 1.09 | 0.93 | 1.29 |
| Overall stage | IV vs. other | 0.77 | 0.84 | 0.27 | 2.64 | 0.49 | 1.87 | 0.32 | 11.04 |
| Alcohol abuse | Yes vs. no | 0.46 | 1.79 | 0.38 | 8.37 | 0.18 | 7.12 | 0.39 | 129.29 |
| Radiotherapy | Yes | 0.81 | 1.29 | 0.16 | 10.20 | 0.99 | 0.00 | 0.00 | - |
| Smoking | No vs. yes | 0.95 | 0.96 | 0.26 | 3.57 | 0.85 | 1.16 | 0.24 | 5.69 |
| Tumor size | >vs. ≤4 | 0.35 | 1.86 | 0.50 | 6.88 | 0.56 | 0.58 | 0.09 | 3.62 |
| Nodal metastasis | Present vs. absent | 0.94 | 0.92 | 0.11 | 7.85 | 0.17 | 0.29 | 0.05 | 1.71 |
| A | |||
| Variables | Multivariate Analysis | ||
| p-Value | Hazard Ratio | ||
| CA | High vs. low | 0.063 | 4.433 |
| Gender | Male | 0.939 | 1.070 |
| Age at diagnosis | High vs. low | 0.227 | 1.076 |
| Overall stage | IV | 0.852 | 1.163 |
| Alcohol abuse | Yes vs. no | 0.166 | 0.160 |
| Radiotherapy | Yes | 0.994 | 0.000 |
| HIF-1α | Yes | 0.994 | 0.991 |
| Cyclin D1 | Yes | 0.287 | 3.343 |
| B | |||
| Variable | Cyclin D1 | HIF-1α | CA |
| −2 Log L | 74.741 | 76.340 | 74.547 |
| C | |||
| Variable | −2 Log L | ||
| Null | 1912.9 | ||
| Weighted Index of CA7 | 1897.6 | ||
| Basic Sum CA7 | 1911.0 | ||
| Hyp26 Score | 1911.9 | ||
| CCNDI | 1907.1 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittal, K.; Choi, D.H.; Wei, G.; Kaur, J.; Klimov, S.; Arora, K.; Griffith, C.C.; Kumar, M.; Imhansi-Jacob, P.; Melton, B.D.; et al. Hypoxia-Induced Centrosome Amplification Underlies Aggressive Disease Course in HPV-Negative Oropharyngeal Squamous Cell Carcinomas. Cancers 2020, 12, 517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020517
Mittal K, Choi DH, Wei G, Kaur J, Klimov S, Arora K, Griffith CC, Kumar M, Imhansi-Jacob P, Melton BD, et al. Hypoxia-Induced Centrosome Amplification Underlies Aggressive Disease Course in HPV-Negative Oropharyngeal Squamous Cell Carcinomas. Cancers. 2020; 12(2):517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020517
Chicago/Turabian StyleMittal, Karuna, Da Hoon Choi, Guanhao Wei, Jaspreet Kaur, Sergey Klimov, Komal Arora, Christopher C. Griffith, Mukesh Kumar, Precious Imhansi-Jacob, Brian D. Melton, and et al. 2020. "Hypoxia-Induced Centrosome Amplification Underlies Aggressive Disease Course in HPV-Negative Oropharyngeal Squamous Cell Carcinomas" Cancers 12, no. 2: 517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12020517