
Pharmacological or TRIB3-Mediated Suppression of ATF4 Transcriptional Activity Promotes Hepatoma Cell Resistance to Proteasome Inhibitor Bortezomib

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Pharmacological Perturbation of the eIF2α–ATF4 UPR Branch Affects HepG2 Cell Sensitivity to Bortezomib
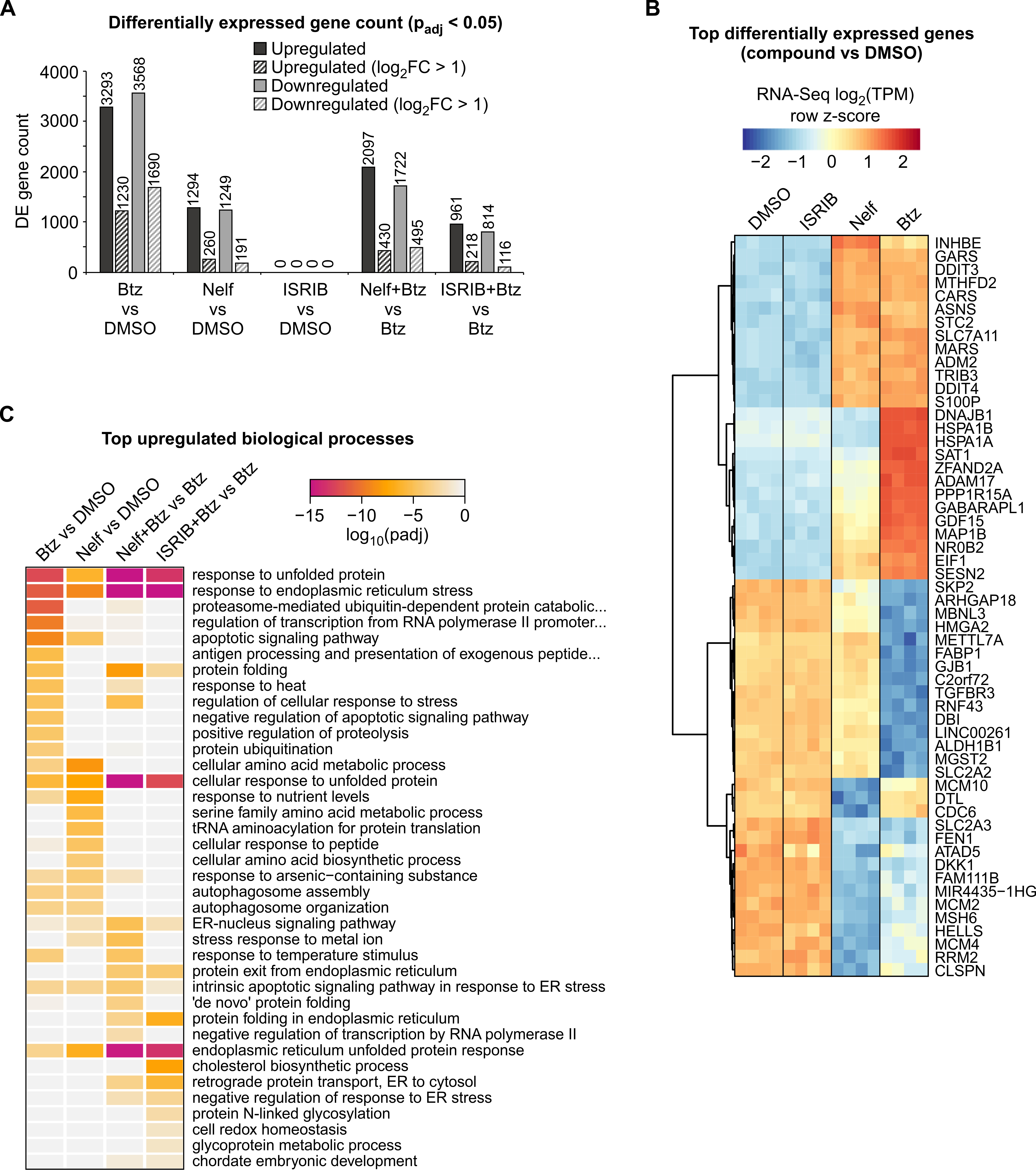
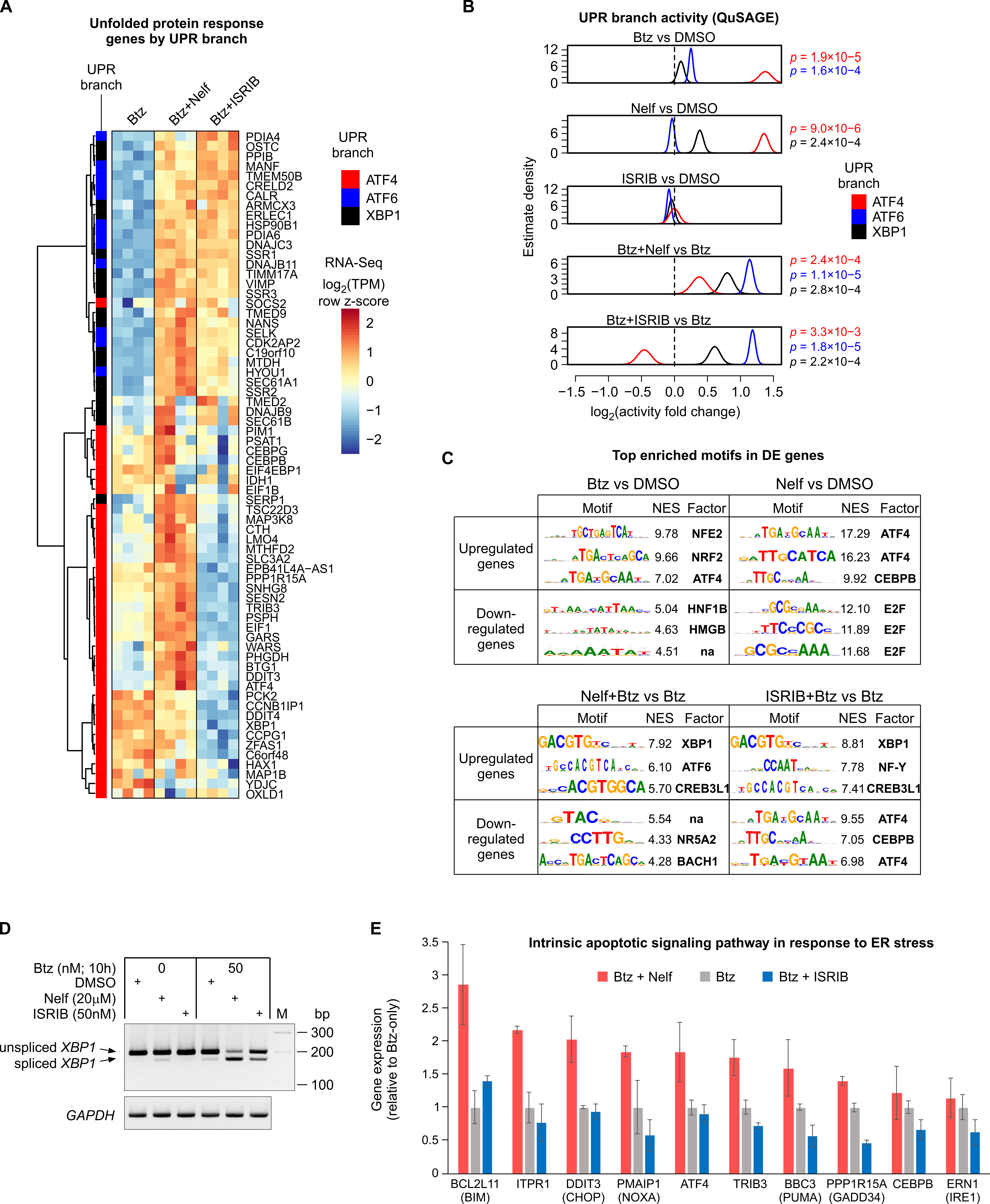
2.2. Transcriptional Profiling of Hepatoma Cells Treated with Bortezomib in Combination with Nelfinavir or ISRIB
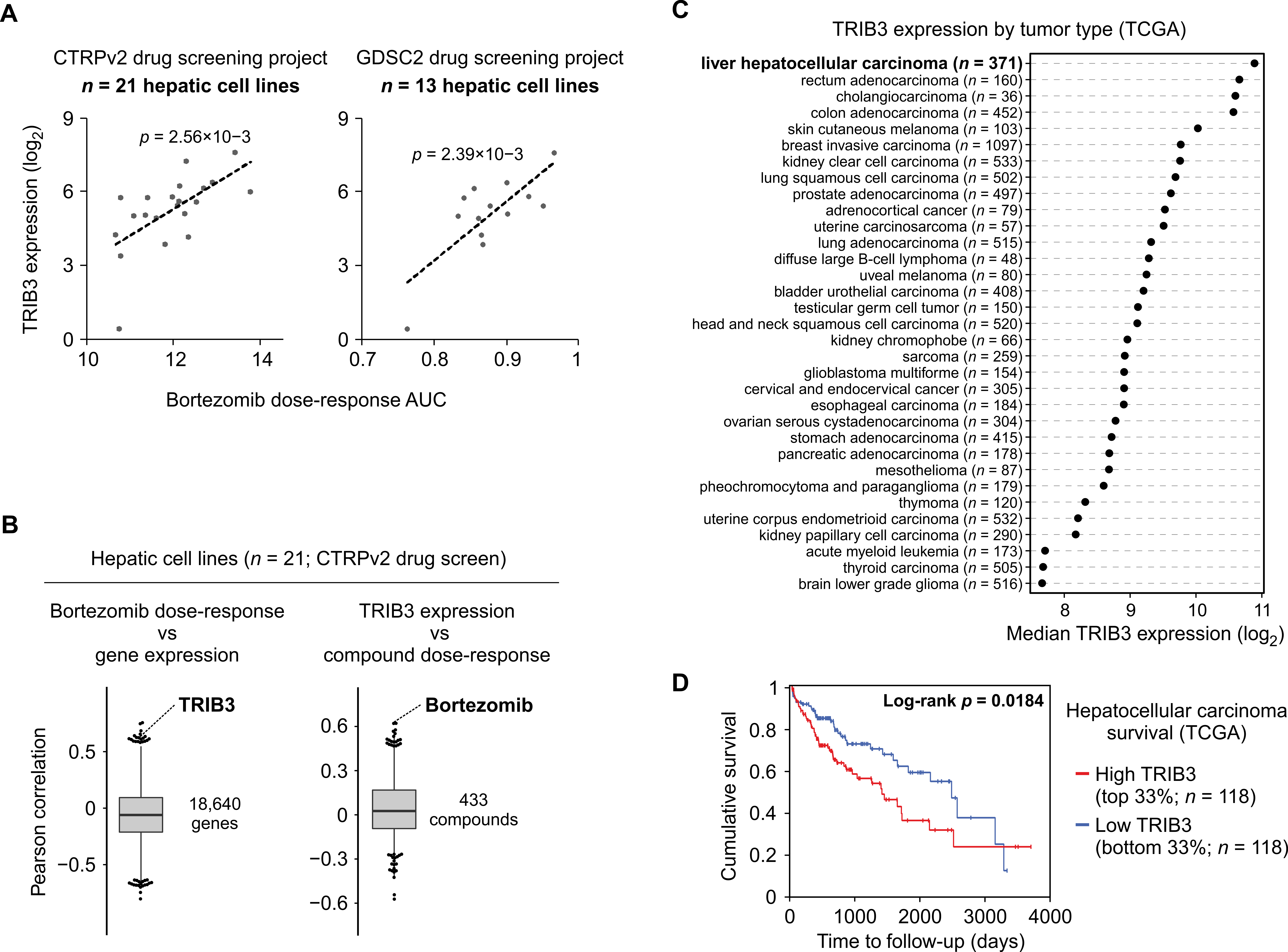
2.3. The ATF4 Regulatory Factor TRIB3 Is Highly Expressed in Hepatoma Cells and Correlates with Resistance to Bortezomib
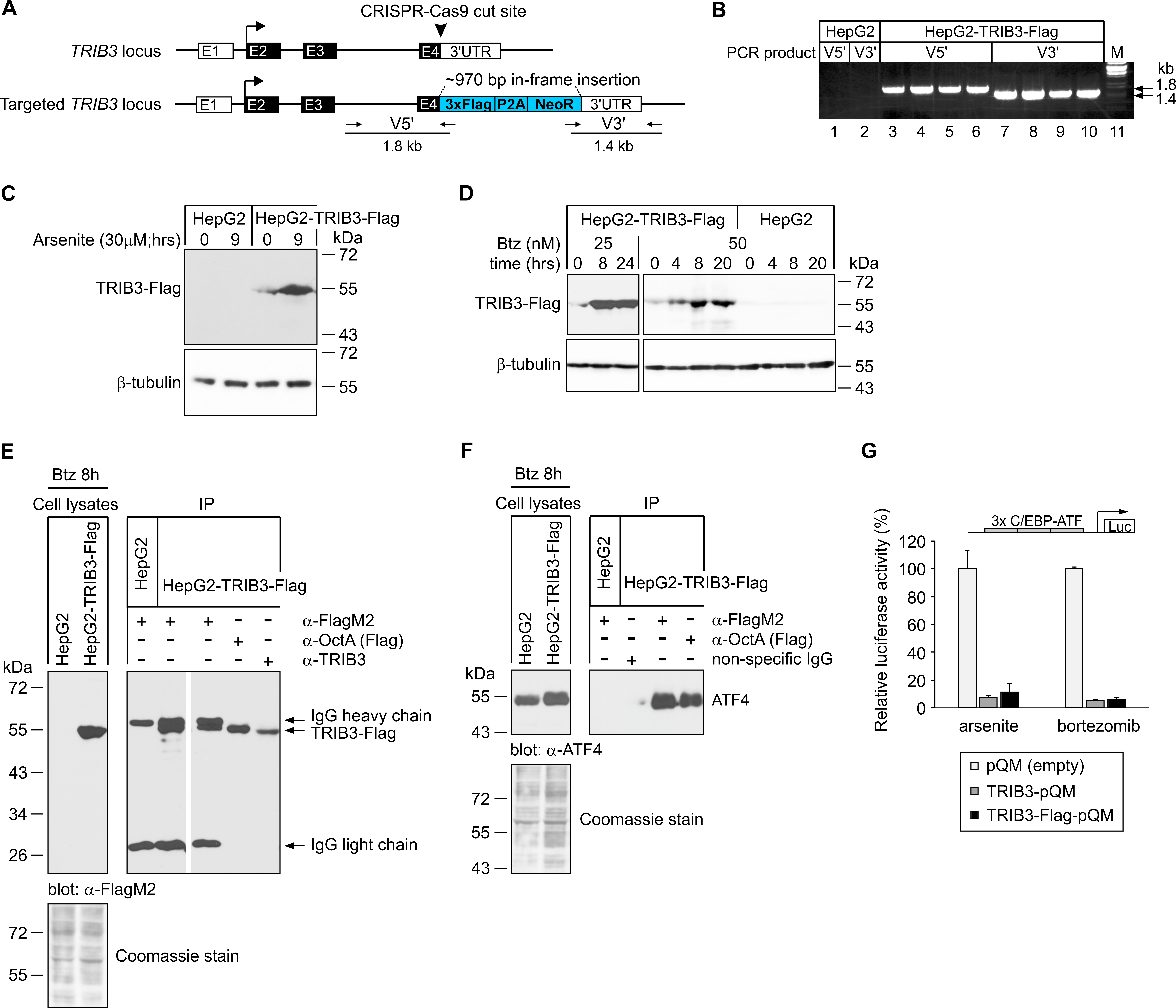
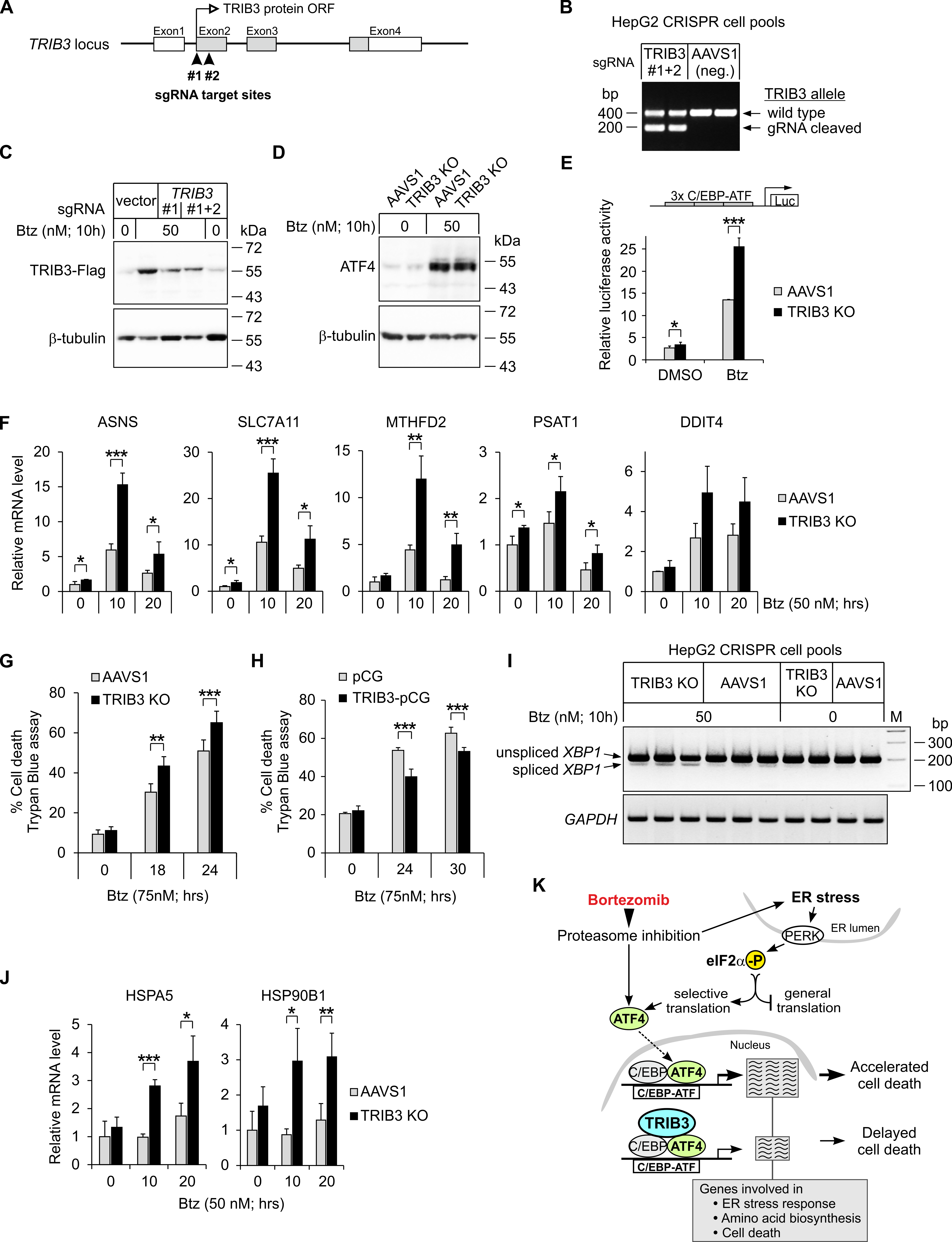
2.4. Generation and Characterization of HepG2 Cells Expressing Endogenous TRIB3 Fused with Flag-Tag (HepG2-TRIB3-Flag Cells)
2.5. TRIB3 Co-Localizes with ATF4 on Chromatin Genome-Wide
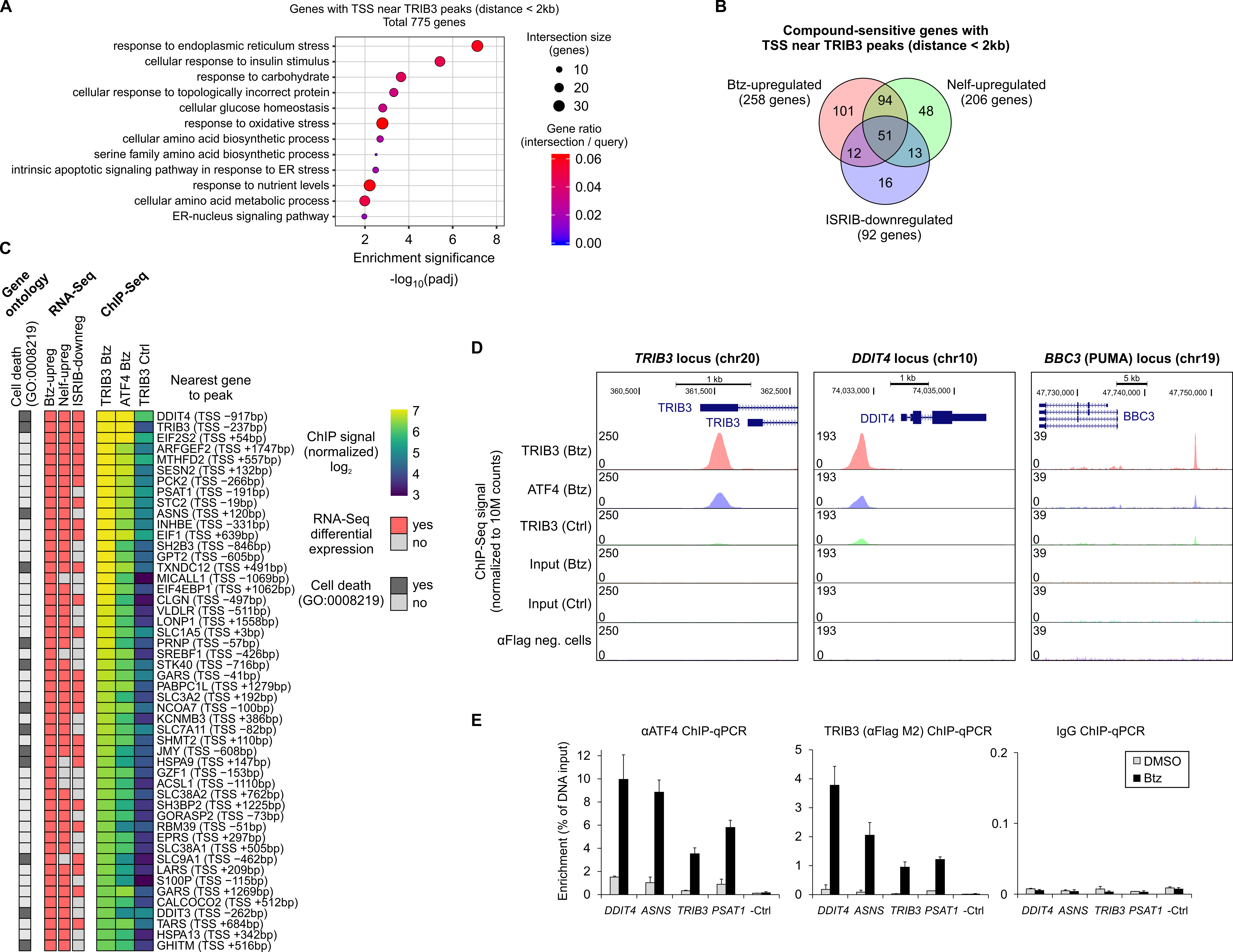
2.6. Enrichment of TRIB3 Binding Near Bortezomib-Upregulated Genes Involved in the ER Stress Response and Cell Death
2.7. TRIB3 Controls ATF4 to Maintain Balanced Activity of the UPR Branches and Cell Viability upon Exposure to Bortezomib
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Plasmid Construction
4.3. Epitope Tagging of Endogenous TRIB3 in Cell Culture
4.4. Chromatin Immunoprecipitation, ChIP-qPCR and Library Construction for ChIP-Seq
4.5. ChIP-Seq Data Analysis
4.6. CRISPR/Cas9-Mediated Gene Disruption in Cell Culture
4.7. RNA-Seq Library Preparation and Sequencing
4.8. RNA-Seq Data Analysis
4.9. Processing of Publicly Available Data Sets
4.10. Cell Viability Assays
4.11. RT-PCR and RT-qPCR
4.12. Dual-Luciferase Reporter Assay
4.13. Western Blotting
4.14. Protein Immunoprecipitation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutsadakis, I.A. Proteasome expression and activity in cancer and cancer stem cells. Tumour Biol. 2017, 39, 1010428317692248. [Google Scholar] [CrossRef] [Green Version]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Yerlikaya, A.; Kimball, S.R.; Stanley, B.A. Phosphorylation of eIF2alpha in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. Biochem. J. 2008, 412, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Tsai, J.C.; Kampmann, M.; Hearn, B.R.; Vedantham, P.; Jaishankar, P.; Sokabe, M.; Mendez, A.S.; Newton, B.W.; Tang, E.L.; et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 2015, 4, e07314. [Google Scholar] [CrossRef]
- de Gassart, A.; Bujisic, B.; Zaffalon, L.; Decosterd, L.A.; di Micco, A.; Frera, G.; Tallant, R.; Martinon, F. An inhibitor of HIV-1 protease modulates constitutive eIF2alpha dephosphorylation to trigger a specific integrated stress response. Proc. Natl. Acad. Sci. USA 2016, 113, E117–E126. [Google Scholar] [CrossRef] [Green Version]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Örd, D.; Örd, T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem. Biophys. Res. Commun. 2005, 330, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.J.; Mayekar, M.K.; Martin, N.; Saylor, K.L.; Gonit, M.; Jailwala, P.; Kasoji, M.; Haines, D.C.; Quinones, O.A.; Johnson, P.F. C/EBPgamma is a critical regulator of cellular stress response networks through heterodimerization with ATF4. Mol. Cell. Biol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Jousse, C.; Deval, C.; Maurin, A.C.; Parry, L.; Cherasse, Y.; Chaveroux, C.; Lefloch, R.; Lenormand, P.; Bruhat, A.; Fafournoux, P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J. Biol. Chem. 2007, 282, 15851–15861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Muerkoster, S.S.; Brosch, M.; Roder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.X.; Yang, J.H.; Saitsu, H. Bortezomib-resistance is associated with increased levels of proteasome subunits and apoptosis-avoidance. Oncotarget 2016, 7, 77622–77634. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Shashova, E.E.; Lyupina, Y.V.; Glushchenko, S.A.; Slonimskaya, E.M.; Savenkova, O.V.; Kulikov, A.M.; Gornostaev, N.G.; Kondakova, I.V.; Sharova, N.P. Proteasome functioning in breast cancer: Connection with clinical-pathological factors. PLoS ONE 2014, 9, e109933. [Google Scholar] [CrossRef]
- Niewerth, D.; Franke, N.E.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Degenhardt, J.; Anderl, J.; Schimmer, A.D.; Zweegman, S.; et al. Higher ratio immune versus constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica 2013, 98, 1896–1904. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nunez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef] [Green Version]
- Yaari, G.; Bolen, C.R.; Thakar, J.; Kleinstein, S.H. Quantitative set analysis for gene expression: A method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Res. 2013, 41, e170. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Goncalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Örd, D.; Örd, T. Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp. Cell Res. 2003, 286, 308–320. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Xu, L.G.; Zhai, Z.; Shu, H.B. SINK is a p65-interacting negative regulator of NF-kappaB-dependent transcription. J. Biol. Chem. 2003, 278, 27072–27079. [Google Scholar] [CrossRef] [Green Version]
- Bezy, O.; Vernochet, C.; Gesta, S.; Farmer, S.R.; Kahn, C.R. TRB3 blocks adipocyte differentiation through the inhibition of C/EBPbeta transcriptional activity. Mol. Cell. Biol. 2007, 27, 6818–6831. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Ohoka, N.; Hayashi, H.; Sato, R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J. Lipid Res. 2008, 49, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Savic, D.; Partridge, E.C.; Newberry, K.M.; Smith, S.B.; Meadows, S.K.; Roberts, B.S.; Mackiewicz, M.; Mendenhall, E.M.; Myers, R.M. CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Res. 2015, 25, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Örd, T.; Örd, D.; Kõivomägi, M.; Juhkam, K.; Örd, T. Human TRB3 is upregulated in stressed cells by the induction of translationally efficient mRNA containing a truncated 5′-UTR. Gene 2009, 444, 24–32. [Google Scholar] [CrossRef]
- Shan, J.; Zhang, F.; Sharkey, J.; Tang, T.A.; Örd, T.; Kilberg, M.S. The C/ebp-Atf response element (CARE) location reveals two distinct Atf4-dependent, elongation-mediated mechanisms for transcriptional induction of aminoacyl-tRNA synthetase genes in response to amino acid limitation. Nucleic Acids Res. 2016, 44, 9719–9732. [Google Scholar] [CrossRef] [Green Version]
- Palii, S.S.; Chen, H.; Kilberg, M.S. Transcriptional control of the human sodium-coupled neutral amino acid transporter system A gene by amino acid availability is mediated by an intronic element. J. Biol. Chem. 2004, 279, 3463–3471. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Chen, C.; Kilberg, M.S. Characterization of the nutrient-sensing response unit in the human asparagine synthetase promoter. Biochem. J. 2003, 372, 603–609. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Cherasse, Y.; Maurin, A.C.; Chaveroux, C.; Jousse, C.; Carraro, V.; Parry, L.; Deval, C.; Chambon, C.; Fafournoux, P.; Bruhat, A. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007, 35, 5954–5965. [Google Scholar] [CrossRef] [PubMed]
- Örd, T.; Örd, T. Mammalian pseudokinase TRIB3 in normal physiology and disease: Charting the progress in old and new avenues. Curr. Protein. Pept. Sci. 2017, 18, 819–842. [Google Scholar] [CrossRef] [PubMed]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Saeki, I.; Terai, S.; Fujisawa, K.; Takami, T.; Yamamoto, N.; Matsumoto, T.; Hirose, Y.; Murata, Y.; Yamasaki, T.; Sakaida, I. Bortezomib induces tumor-specific cell death and growth inhibition in hepatocellular carcinoma and improves liver fibrosis. J. Gastroenterol. 2013, 48, 738–750. [Google Scholar] [CrossRef]
- Huang, H.; Liu, N.; Yang, C.; Liao, S.; Guo, H.; Zhao, K.; Li, X.; Liu, S.; Guan, L.; Liu, C.; et al. HDAC inhibitor L-carnitine and proteasome inhibitor bortezomib synergistically exert anti-tumor activity in vitro and in vivo. PLoS ONE 2012, 7, e52576. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Coucke, C.; Laukens, D.; Devisscher, L.; Paridaens, A.; Bogaerts, E.; Vandierendonck, A.; Raevens, S.; Verhelst, X.; van Steenkiste, C.; et al. Next-generation proteasome inhibitor oprozomib synergizes with modulators of the unfolded protein response to suppress hepatocellular carcinoma. Oncotarget 2016, 7, 34988–35000. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.P.; Mahoney, M.R.; Szydlo, D.; Mok, T.S.; Marshke, R.; Holen, K.; Picus, J.; Boyer, M.; Pitot, H.C.; Rubin, J.; et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma, Invest. New Drugs 2012, 30, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Örd, D.; Örd, T.; Biene, T.; Örd, T. TRIB3 increases cell resistance to arsenite toxicity by limiting the expression of the glutathione-degrading enzyme CHAC1. Biochim. Biophys. Acta 2016, 1863, 2668–2680. [Google Scholar] [CrossRef]
- Örd, T.; Örd, D.; Örd, T. TRIB3 limits FGF21 induction during in vitro and in vivo nutrient deficiencies by inhibiting C/EBP-ATF response elements in the Fgf21 promoter. Biochim. Biophys. Acta 2018, 1861, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; Behan, F.M.; Kirca, M.; Smith, S.; Reynolds, J.V.; Long, A.; Kelleher, D. An integrative genomic approach in oesophageal cells identifies TRB3 as a bile acid responsive gene, downregulated in Barrett’s oesophagus, which regulates NF-kappaB activation and cytokine levels. Carcinogenesis 2010, 31, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasha, M.; Eid, A.H.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 3296294. [Google Scholar] [CrossRef]
- Luo, J.; Xia, Y.; Luo, J.; Li, J.; Zhang, C.; Zhang, H.; Ma, T.; Yang, L.; Kong, L. GRP78 inhibition enhances ATF4-induced cell death by the deubiquitination and stabilization of CHOP in human osteosarcoma. Cancer Lett. 2017, 410, 112–123. [Google Scholar] [CrossRef]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; Lopiccolo, J.; Wilson, W., 3rd; Hollander, M.C.; Dennis, P.A. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Canal, M.; Romani-Aumedes, J.; Martin-Flores, N.; Perez-Fernandez, V.; Malagelada, C. RTP801/REDD1: A stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front Cell Neurosci. 2014, 8, 313. [Google Scholar] [CrossRef] [Green Version]
- Britto, F.A.; Dumas, K.; Giorgetti-Peraldi, S.; Ollendorff, V.; Favier, F.B. Is REDD1 a metabolic double agent? Lessons from physiology and pathology. Am. J. Physiol. Cell Physiol. 2020, 319, C807–C824. [Google Scholar] [CrossRef]
- Hershko, T.; Chaussepied, M.; Oren, M.; Ginsberg, D. Novel link between E2F and p53: Proapoptotic cofactors of p53 are transcriptionally upregulated by E2F. Cell Death Differ. 2005, 12, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Ri, M.; Masaki, A.; Mori, F.; Ito, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; Iida, S. Lower expression of activating transcription factors 3 and 4 correlates with shorter progression-free survival in multiple myeloma patients receiving bortezomib plus dexamethasone therapy. Blood Cancer J. 2015, 5, e373. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imrichova, H.; Hulselmans, G.; Atak, Z.K.; Potier, D.; Aerts, S. i-cisTarget 2015 update: Generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 2015, 43, W57–W64. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. Peerj Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Örd, D.; Meerits, K.; Örd, T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp. Cell Res. 2007, 313, 3556–3567. [Google Scholar] [CrossRef]
- Örd, T.; Örd, D.; Kuuse, S.; Plaas, M.; Örd, T. Trib3 is regulated by IL-3 and affects bone marrow-derived mast cell survival and function. Cell. Immunol. 2012, 280, 68–75. [Google Scholar] [CrossRef]
- Örd, T.; Örd, D.; Adler, P.; Vilo, J.; Örd, T. TRIB3 enhances cell viability during glucose deprivation in HEK293-derived cells by upregulating IGFBP2, a novel nutrient deficiency survival factor. Biochim. Biophys. Acta 2015, 1853, 2492–2505. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Örd, T.; Örd, D.; Kaikkonen, M.U.; Örd, T. Pharmacological or TRIB3-Mediated Suppression of ATF4 Transcriptional Activity Promotes Hepatoma Cell Resistance to Proteasome Inhibitor Bortezomib. Cancers 2021, 13, 2341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13102341
Örd T, Örd D, Kaikkonen MU, Örd T. Pharmacological or TRIB3-Mediated Suppression of ATF4 Transcriptional Activity Promotes Hepatoma Cell Resistance to Proteasome Inhibitor Bortezomib. Cancers. 2021; 13(10):2341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13102341
Chicago/Turabian StyleÖrd, Tiit, Daima Örd, Minna U. Kaikkonen, and Tõnis Örd. 2021. "Pharmacological or TRIB3-Mediated Suppression of ATF4 Transcriptional Activity Promotes Hepatoma Cell Resistance to Proteasome Inhibitor Bortezomib" Cancers 13, no. 10: 2341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13102341