
Tribbles Pseudokinase 3 Regulation and Contribution to Cancer


1
Inserm, UMR981, F-94805 Villejuif, France
2
Gustave Roussy, F-94805 Villejuif, France
3
Orsay, Université Paris Saclay, F-91400 Gif-sur-Yvette, France
4
Department of Medical Oncology, Gustave Roussy, F-94805 Villejuif, France
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(8), 1822; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081822
Submission received: 18 March 2021
/
Revised: 8 April 2021
/
Accepted: 8 April 2021
/
Published: 11 April 2021
(This article belongs to the Special Issue Pseudokinases, Tribbles Proteins and Cancer)
Abstract
:Simple Summary
Accumulating evidence supports a key function for Tribbles proteins in oncogenesis, both in leukemia and solid tumors. However, the exact role of these proteins is hard to define since in a context-dependent manner they can function as both oncogenes and tumor suppressors. Their complex role arises from the capacity to interact with a wide range of target molecules thereby acting as molecular scaffolds and signaling regulators of multiple pathways. This review focuses on one particular Tribbles family member, namely, TRIB3, addressing its gene and protein expression, as well as its role in cancer development and progression.
Abstract
The first Tribbles protein was identified as critical for the coordination of morphogenesis in Drosophila melanogaster. Three mammalian homologs were subsequently identified, with a structure similar to classic serine/threonine kinases, but lacking crucial amino acids for the catalytic activity. Thereby, the very weak ATP affinity classifies TRIB proteins as pseudokinases. In this review, we provide an overview of the regulation of TRIB3 gene expression at both transcriptional and post-translational levels. Despite the absence of kinase activity, TRIB3 interferes with a broad range of cellular processes through protein–protein interactions. In fact, TRIB3 acts as an adaptor/scaffold protein for many other proteins such as kinase-dependent proteins, transcription factors, ubiquitin ligases, or even components of the spliceosome machinery. We then state the contribution of TRIB3 to cancer development, progression, and metastasis. TRIB3 dysregulation can be associated with good or bad prognosis. Indeed, as TRIB3 interacts with and regulates the activity of many key signaling components, it can act as a tumor-suppressor or oncogene in a context-dependent manner.
Keywords:
pseudokinase; mTOR; Akt; rapamycin; ER stress; tumor suppressor; oncogene; RNA splicing; FK506 binding protein; signaling pathway1. Introduction
The Tribbles protein was firstly identified because of its critical role in coordinating morphogenesis in Drosophila melanogaster (fruit fly) [1,2,3]. Tribbles-deficient embryos enter mitosis early in the mesodermal cells that impairs morphogenetic movement of gastrulation, when on the contrary, Tribbles overexpression greatly reduces the number of cells per wing. In mammals, three Tribbles protein homologs exhibit a high degree of amino acid similarity: TRIB1 (alias C8fW or SKIP1), TRIB2 (C5fW or SKIP2), and TRIB3 (NIPK, SINK, or SKIP3). The kinase-like domain is highly conserved through evolution suggesting an important role in the function for the Tribbles family of proteins [4]. As suggested in Drosophila, a relationship with cell cycle or cell survival in mammalian cells was also reported. TRIBs exhibit either unique or overlapping functions (reviewed in [5]). Such similarities and discrepancies in a pathological context upgrade TRIBs as prognostic or predictive biomarkers of disease and putative valuable therapeutic targets.
In humans, TRIB3 expression is ubiquitous. Human tissue profiling indicates the thyroid gland, bone marrow, and peripheral blood leukocytes as sites of high TRIB3 mRNA expression [6]. The Human Protein Atlas project also assessed the expression level of TRIB3 mRNA and the abundance of TRIB3 protein in a variety of tissues (www.proteinatlas.org accessed on 6 January 2021). Clinical studies report an increased TRIB3 expression in tumor tissue as compared to normal tissue and a correlation with poor prognosis. In fact, colorectal cancer patients with high TRIB3 protein and mRNA expression levels are likely to experience a recurrence of the disease and displayed poorer overall survival [7]. In the same way, in renal cell carcinoma [8], gastric cancer [9], oral tongue squamous cell carcinoma [10], and non-small-cell lung carcinoma [11], the upregulated TRIB3 expression correlates with tumor stage, lymph node metastasis, disease recurrence, and thus with unfavorable prognosis. Nevertheless, despite the association of TRIB3 upregulation with bad prognosis, it can act as a tumor-suppressor or oncogene in a context-dependent manner.
2. TRIB3 Gene and Transcripts
The first approved name of the human TRIB3 gene was “Tribbles homolog 3 (Drosophila)”, recalling the Drosophila melanogaster homolog, but in 2016, the HUGO Gene Nomenclature Committee reconsidered it to “Tribbles pseudokinase 3”, putting in evidence its function. The human TRIB3 gene (gene ID: 57761) is located in chromosome 20 at the position 20p13 and spans a region of 16.9 kb. Thanks to alternative transcription start sites (TSS) and alternative splicing, it generates four transcripts (Figure 1).
The Ensembl genome browser annotates TRIB3-201 transcript as the principal transcript, and TRIB3-202 as the alternative transcript (Table 1). The protein encoded by TRIB3-202 transcript harbors 27 additional amino acids at the N-term in comparison to the one encoded by TRIB3-201. However, there is no experimental evidence indicating that this difference in sequence translates in functional diversity in the two proteins. Furthermore, the existence of protein isoforms related to TRIB3-203 and TRIB3-204 transcripts (Transcript Support Levels (TSL) of 5 and 3, respectively) has never been proven empirically, resulting in TRIB3-201 being the only transcript that encodes the TRIB3 protein.
Alternative transcription start sites add complexity to the human TRIB3 gene expression. In silico analysis of the 5′-Untranslated Transcribed Region (5′UTR) suggests three putative alternative promoters with assigned transcripts (Figure 2). One putative promoter could generate a very short isoform of TRIB3 (encoding a protein truncated to 60 aa) for which no matching information is available to our knowledge. In contrast, the other two promoters allow the transcription of up to ten putative transcripts that finally match the TRIB3-201 principal transcript and the TRIB3-202 alternative transcript. Since all of the transcriptional differences of TRIB3 isoforms condensed in the first exon that is not protein coding, all of the TRIB3 generated proteins are identical.
3. TRIB3 Protein Structure
The 3D structure for TRIB3 has not been established yet, but insights from the crystal structure of its homolog TRIB1 suggest that Tribbles proteins fold into the canonical conformation of protein kinases [13]. Indeed, the TRIB3 protein presents a structure similar to a classic serine-threonine protein kinase with N- and C-terminal domains flanking a kinase-like domain in the center of the protein (Figure 3).
The kinase-like domain is highly conserved through evolution suggesting an important role in the function for the Tribbles family of proteins [4]. However, even if the catalytic core contains the classic substrate-binding domain, it presents several substitutions of amino acids that are crucial for the catalytic activity of conventional kinases (Figure 3). The most evident differences are (1) the lack of the highly conserved Asp-Phe-Gly (DFG) motif which is required for the binding of Mg2+ and subsequent activation of canonical kinases; and (2) the lack of the glycine-rich loop (GXGXXG) usually involved in ATP-binding. In vitro experiments confirmed no phospho-transferase activity in presence of several different classical Ser/Thr kinase substrates [14]. However, even though it has been demonstrated that TRIB3 can very weakly auto-phosphorylate under particular in vitro conditions, independently of divalent cations, cellular substrates of TRIB3 have not been identified yet [15]. Based on all these characteristics, TRIB3 is classified as a pseudokinase.
The N-terminal domain contains a large amount of proline (P), glutamic acid (E), serine (S), and threonine (T) residues. Such PEST regions are involved in Ubiquitin-mediated degradation of a variety of substrates, thus controlling the half-life of target proteins [16,17]. A nuclear localization signal (NLS) sequence allows TRIB3 to localize to the nucleus where it interacts with various transcription factors such as ATF4, CHOP, C/EBPβ, PPARγ, p65/RelA, or SMAD3. The C-terminal domain of TRIB3 contains two conserved sequences: Mitogen Activated Protein Kinase Kinase (MEK1/MAPKK) binding motif and Constitutive Photomorphogenesis Protein 1 Homolog (COP1) binding motif recognized by this subclass of E3 ubiquitin ligase.
4. Regulation of TRIB3 Expression
The best-described modalities of TRIB3 regulation up to date are through several forms of cellular stress. Evidence in the literature suggest that endoplasmic reticulum (ER) stress [18,19], oxidative stress, hypoxia, essential amino acid deficiency [20], oversupply or lack of glucose [21,22], or free fatty acids excess [23] contribute to the activation of the TRIB3 promoter and lead to TRIB3 upregulation. In addition, post-translational modifications of TRIB3 contribute to the regulation of protein location, stability, confirmation, and activity.
4.1. Transcriptional Regulation
A recently published, in silico analysis of the TRIB3 promoter region indicates six unique putative promoters but three of them with no transcript assigned (Figure 2). Further investigations of the region spanning the promoters #2 and #3 detected binding motifs for a large amount of transcription factors (Figure 4).
Several of these transcriptional factors effectively trigger TRIB3 expression levels in different contexts. The best described activator is ATF4 (Activation Transcription Factor 4) that is recruited to TRIB3 promoter thanks to a basic leucine zipper domain (bZIP) c/EBP-ATF response element [24]. In conditions of severe or prolonged stress, ATF4 forms a heterodimer with CHOP (C/EBP homologous protein) that promotes cell death via the induction of several pro-apoptotic genes and the suppression of the anti-apoptotic Bcl-2 proteins [25,26]. The transcriptional coactivator PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator-1 alpha) also promotes TRIB3 expression which leads to induced insulin resistance in liver cells [27]. The cell-type specific transcription factor NFATc1 (Nuclear factor of activated T-cells, cytoplasmic 1) promotes TRIB3 expression in vascular smooth muscle cells upon phenamil treatment resulting in an attenuation of pulmonary artery hypertension in rats [28]. Another example is FoxO1 (Forkhead box protein O1), which according to the cellular context can either promote or repress TRIB3 expression. Hence, it controls the insulin sensitivity in hepatocytes by inhibiting the expression of TRIB3, whereas it promotes cell death in neurons under NGF deprivation by inducing the expression of TRIB3 [29,30].
Even though there is extensive literature describing the activation of TRIB3 in several stress conditions, little is known about its repression. In a recent publication from our group we demonstrated that the transcriptional co-repressor GCF2 (GC-Binding Factor 2) alias LRRFIP1 (Leucine Rich Repeat (In FLII) Interacting Protein 1) participates in TRIB3 downregulation in cancer cells incubated with rapamycin or its derivatives (rapalogs) [12]. The canonical target of rapalogs is mTOR (mechanistic target of rapamycin). They bind with high affinity to the intracellular receptor FKBP12, a member of the FK506-binding protein (FKBP) family [31], and the resulting complex binds to the FKBP12-Rapamycin Binding (FRB) domain of mTOR that leads to its allosteric inhibition. Interestingly, rapamycin can also bind FKBP25 with high affinity [32] that in turn regulates GCF2 recruitment to the promoter of TRIB3 [12]. Moreover, the consensus-binding motif for GCF2 overlaps the RNA polymerase II binding motif (TF2B) near the TSS in the TRIB3 promoter, probably interfering with the recruitment of RNA polymerase II, and the initiation of transcription.
4.2. Post-Translational Regulation
Several enzymatic modifications occurring at a post-translational stage may regulate TRIB3 function or location. Thus, high-throughput proteomic analyses followed by mass-spectrometry confirmed the presence of seven phosphorylation sites on TRIB3 protein (Figure 5). However, to date, there is no information about the associated kinases or the functional significance of such phosphorylation. The histone-Lysine N-Methyltransferase SMYD1 (SET and MYND Domain Containing 1) triggers TRIB3 methylation at K16 in the N-term region (Figure 5). Following this methylation, TRIB3 acts as a co-repressor to SMYD1 and inhibits its transcription [33]. The acetylation of TRIB3 by PCAF (P300/CBP-Associated Factor) results in increased expression of TRIB3 [33,34]. A yeast two-hybrid screen identified an interaction of TRIB3 with the E3 ubiquitin ligase Seven in absentia homolog 1 (SIAH1) in mammalian cells, and demonstrated that it promotes proteasome-dependent degradation [35].
Finally, upon apoptosis-inducing treatment (TNFα/CHX or anti-Fas antibody), TRIB3 is cleaved by endoproteases of the caspases family at a site located 20 aa from the C-term [36]. This cleavage leads to accelerated cell death by emphasizing pro-caspase-3 and -7 activation. In contrast, upon ER stress-inducing treatment, TRIB3 is no longer cleaved. The pro-caspase-3 undergoes nuclear translocation leading to its non-activation and prevention of apoptosis induction [36].
5. TRIB3 as A Scaffold for Diverse Signaling Proteins
TRIB3 interferes with a broad range of cellular processes through non-catalytic mechanisms. Instead, it takes part in pleiotropic signaling network thanks to diverse and varied protein–protein interactions. Known TRIB3 partners are kinase-dependent proteins, transcription factors, ubiquitin ligases, spliceosome machinery components, as well as many other proteins. Since TRIB3 lacks an active catalytic site, it contributes to several processes as a scaffold protein or an adaptor. In fact, TRIB3 generally regulates other proteins by altering their subcellular localization or impeding the interactions with their enzymatic partners. The outcome of these regulations is strictly context dependent since TRIB3 can act both as activator and as repressor.
5.1. TRIB3 Regulation of Kinase-Dependent Proteins
Thanks to its C-term MEK1-binding domain, TRIB3 is able to deactivate multiple mitogen-activated protein kinases (MAPKs). In fact, the best-described function of TRIB3 is the negative regulation of Akt (thymoma viral proto-oncogene/protein kinase B) activity (Figure 6). The PI3K/Akt signal transduction pathway is pivotal for cell response to various extracellular stimuli. Active Akt plays a pro-survival role thanks to its multifunctional downstream signaling nodes. However, its effective activation requires phosphorylation on two key residues, namely, Thr308 and Ser473. Firstly, PI3K (phosphoinositide-3-kinase) activates PDK1 (Pyruvate Dehydrogenase Kinase 1), which phosphorylates AKT at Thr308. Then, for optimal activation, mTORC2 phosphorylates Akt on Ser437 [37]. Several evidence suggest that TRIB3 disrupts the phosphorylation at both Thr308 and Ser473 residues through a direct interaction with Akt [38]. In addition, TRIB3 can inhibit the Akt phosphorylation at Ser473 by binding to the mTORC2 component, RICTOR [39]. The TRIB3-mediated Akt inhibition does not inhibit all Akt substrates in the same way, which is an additional testament for its context-dependent regulation. For instance, FoxO and BAD phosphorylation is impaired, whereas GSK3β and PRAS40 phosphorylation remains intact.
In parallel to the modulation of the PI3K/Akt signaling pathway, TRIB3 binds also directly to extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), and dose-dependently regulates their phosphorylation state, leading to the modulation of cell proliferation and migration of lung adenocarcinoma cells, for example [40]. It should be noted that TRIB3 does not affect the levels of p-P38 MAPK. Similar effects of TRIB3 onto MAPK-dependent pro-survival and pro-migratory effects were reported in renal cell carcinoma [8], or breast cancer, completed with interactions with TGFβ pathway [41].
5.2. TRIB3 Regulation of Transcription Factors
In addition to the interaction with several protein kinases, TRIB3 binds and regulates the expression level of different transcription factors. In this manner, it contributes to the modulation of multiple cell behaviors that have whole-body impact. Through a COP1 (constitutive photomorphogenic protein 1) conserved binding motif present at the C-terminal domain, TRIB3 can recruit the E3 ubiquitin ligase COP1 and favor ubiquitination and proteasomal degradation of transcription factors [42,43,44]. For example, TRIB3 interacts with PPARα (peroxisome proliferator-activated receptor alpha) and promotes its ubiquitin-dependent degradation [45]. This favors the survival of leukemic cells and meditates the progression of acute myeloid leukemia (AML). Furthermore, it participates to a tight control of adipogenesis by interacting with PPARγ (peroxisome proliferator-activated receptor gamma), leading to the downregulation of its transcriptional activities. This negative regulation of PPARγ inhibits adipocyte differentiation [42].
TRIB3 regulates inflammatory signaling through NF-kB (nuclear factor-kappa B). However, the relationship between TRIB3 and NF-kB is quite complex. In certain instances, TRIB3 increases NF-kB activity, and leads, for example, to the induction of apoptosis in pancreatic β cells [46]. In contrast, in esophageal cancer cells, TRIB3 inhibits NF-kB signaling by interacting with RelA, a member of NF-kB family [47]. Moreover, TRIB3 can indirectly inhibit NF-kB as the downstream effector of the ATF4-CHOP pathway [48,49].
ATF4 (activating transcription factor-4) binds to and activates the promoter of chaperone encoding genes that help to restore cellular homeostasis. Under sustained stress, ATF4 also binds to and activates the promoter of CHOP (CCAAT-enhancer-binding protein homologous protein), which in turn boosts endoplasmic reticulum (ER) stress-induced apoptotic cell death. Furthermore, CHOP can form heterodimers with ATF4, C/EBPβ (CCAAT/enhancer binding protein), and C/EBPγ, then binds to the promoter of TRIB3 and contributes to its positive regulation [19]. In turn, TRIB3 inhibits the transcriptional activity of ATF4 and CHOP by binding directly their transactivation domains without affecting their DNA binding ability [18,50]. In this manner, a negative feedback-loop is established where TRIB3 represses its own promoter and fine-tunes its own expression. In conclusion, we could imagine TRIB3 as a sensor of ER-stress induced cell death. For instance, when the stress is mild, ATF4/CHOP-containing complexes induce TRIB3 expression, but the TRIB3-dependent negative-feedback loop inhibits their transcriptional activity that finally promotes cell survival. However, when the stress is severe, sustained TRIB3 overexpression leads to cell death.
In colorectal cancer cells, TRIB3 interacts with TCF4 but also with β-catenin in a dose-dependent manner [51]. This transcriptional complex induces the expression of genes related to Wnt signaling pathway and cancer stemness. Moreover, activated β-catenin increased expression of TRIB3, indicating a positive-feedback loop. In liver cancer cells, an increased expression level of TRIB3 correlates with increased expression levels of PPARγ, ATF4, ATF5, eIF2α, CHOP, and several other transcription factors related to activation of Wnt/β-catenin signaling pathway [52].
5.3. TRIB3 Regulation of Pre-mRNA Splicing
In the context of cancer cell resistance to rapamycin, a non-targeted liquid chromatography tandem mass chromatography (LC-MS/MS) proteomics combined to functional annotation clusters was conducted to complete the TRIB3 protein interaction network [12]. Surprisingly, in addition to the well-known TRIB3 interactors described above, the majority of the identified partners were related to the pre-mRNA splicing process. More specifically, TRIB3 interacts with core spliceosomal proteins that form the PRP19 and U2 small nuclear ribonucleoproteins (U2 snRNP) complexes, and thus is involved in pre-mRNA splicing modulation. Rapamycin treatment leads to impairment of the pre-mRNA splicing efficacy by downregulating TRIB3 expression in an mTOR independent manner [12].
Interestingly, a recent study in Saccharomyces cerevisiae reported that rapamycin triggers the accumulation of unusual introns as stabilized linear non-coding RNAs, associated with components of the spliceosome [53]. This accumulation of introns interferes with key biological functions in yeast, like, for instance, cell survival [54], whereas intron retention (IR) in higher eukaryotic organisms is coupled to non-sense-mediated mRNA decay (NMD) and regulates gene expression. Evidence in the literature suggest that IR can also regulate RNA stability and translation, as well as the generation of distinct protein isoforms [55]. In mammalian cells, pharmacological mTOR inhibition can lead to intron retention in lipogenic genes. In this context, IR impacts the stability of the mRNA by triggering NMD and disrupts the lipid metabolism thereby affecting cellular growth [56].
6. TRIB3 and Cancer
Since TRIB3 is a negative regulator of key processes and signaling pathways, it has a critical role in several pathologies. For instance, the overexpression of TRIB3 is involved in several metabolic dysfunctions such as diabetes type II and cardiovascular diseases [57]. In addition, considerable number of studies suggest that TRIB3 dysregulation plays a role in cancer development, progression, and metastasis.
6.1. Genetic Alterations and Expression
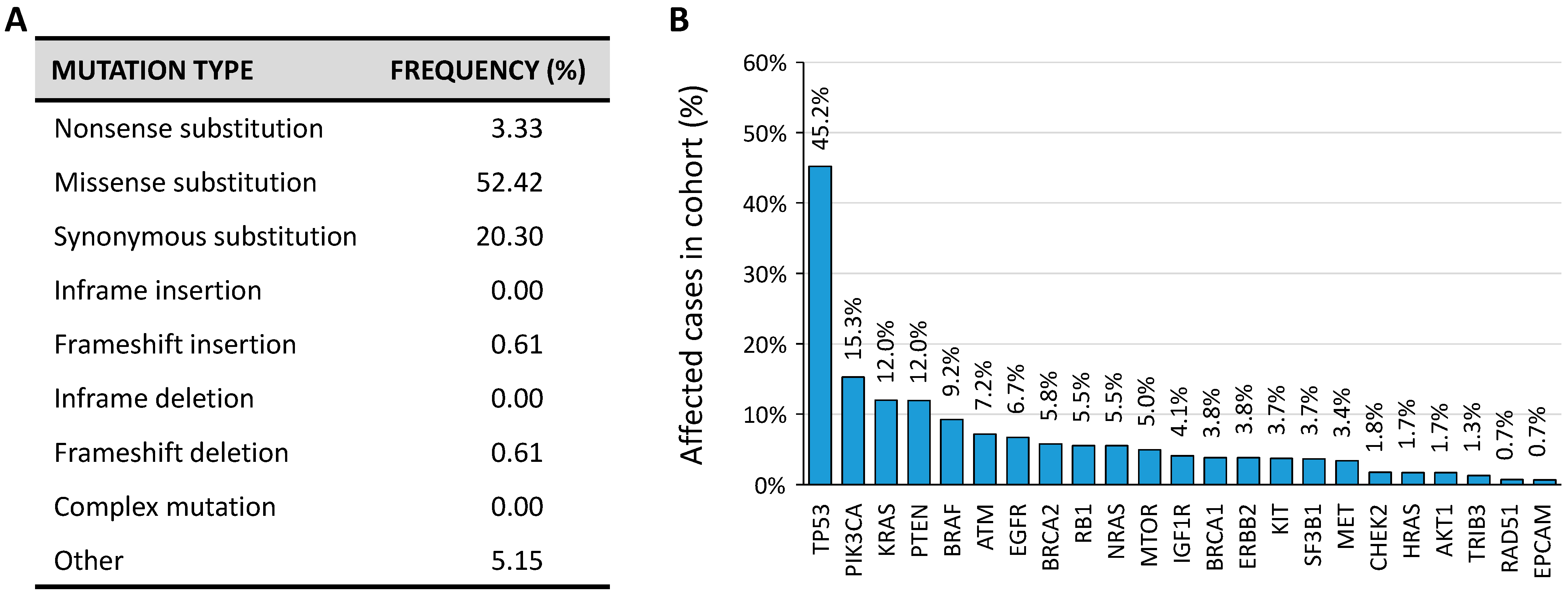
According to the Catalogue Of Somatic Mutations In Cancer (COSMIC) database, several somatic mutations are present in the TRIB3 gene (Figure 7A). However, in the current version of The Cancer Genome Atlas Research Network (TCGA) that comprises cancer samples for 33 different cancer types (last access January 2021) TRIB3 is mutated in 1.28% of cancer samples (Figure 7B).
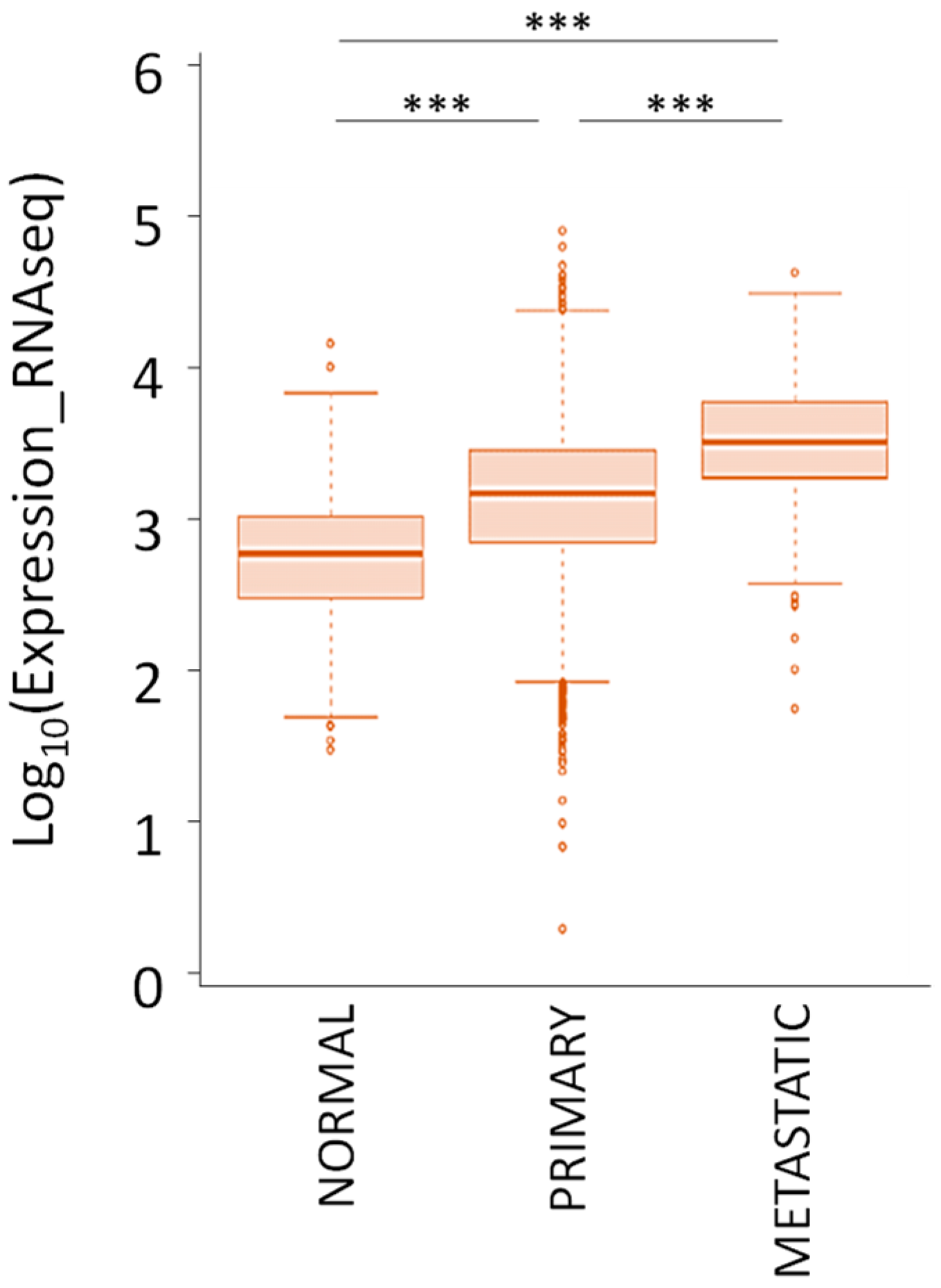
In the context of genetic alterations, two different studies reported a copy number gain for the TRIB3 locus in 16% and 40% of patients [58,59]. As a result, the colorectal, liver, and lung cancer exhibit higher TRIB3 mRNA level as compared with non-tumor sample. In accordance, data from TCGA confirm that TRIB3 mRNA expression is higher in the tumor tissues compared to the normal tissues (Figure 8). In addition, TRIB3 expression is even higher in metastatic lesions compared to primary tumors.
Finally, evidence in the literature for gastric, liver, colorectal, renal cell, and non-small-cell lung carcinoma suggest increased TRIB3 protein level in the tumor tissue compared to the adjacent non-tumor tissue [14]. Taken together, these data indicate that in cancer, TRIB3 is upregulated both at mRNA and protein level.
6.2. TRIB3 Expression Levels and Patient’s Prognosis
Clinical studies report an increased TRIB3 expression in tumor tissue as compared to normal tissue, and this correlates with poor prognosis. In fact, colorectal cancer patients with high TRIB3 protein and mRNA expression levels are likely to experience a recurrence of the disease and display poorer overall survival [7]. In the same way, in renal cell carcinoma [8], gastric cancer [9], oral tongue squamous cell carcinoma [10], and non-small-cell lung carcinoma [11], the upregulated TRIB3 expression correlates with tumor stage, lymph node metastasis, disease recurrence, and thus with unfavorable prognosis. Nevertheless, despite the association of upregulation with poor prognosis, TRIB3 can act as a tumor-suppressor or oncogene in a context-dependent manner. However, no evidence could currently predict in which scenario TRIB3 would promote or inhibit cancer progression.
6.3. Is TRIB3 A Tumor Suppressor?
In some cellular and animal models of cancer, TRIB3 repression or deletion leads to enhanced cell proliferation and tumor formation in xenograft models, suggesting a potential role as tumor suppressor for TRIB3. The principal mechanism by which TRIB3 inhibits tumor progression probably relies on the inhibition of the phosphorylation/activation of Akt resulting in reduced pro-survival action [60,61]. Another mechanism by which TRIB3 can contribute to tumor suppression is via the interaction with the anti-viral cytidine deaminase APOBEC3A (Apolipoprotein B mRNA Editing Enzyme Catalytic subunit 3A). In cancer cells, two members from this family APOBEC3A and APOBEC3B are prevalent, act as potent mutators, and are associated with the burden of signature C to T mutations [62]. TRIB3 has been reported to restrict APOBEC3A activity by promoting its degradation via a proteasome-independent manner [63]. Such a TRIB3-APOBEC3A complex proposes TRIB3 as an important guardian of the genome integrity.
6.4. Is TRIB3 An Oncogene?
TRIB3 can contribute to tumor progression via the attenuation of the major intracellular degradative process named autophagy that has a potential preventive role against early stage cancer [64]. In an elegant study, Hua et al. reported that TRIB3 interacts with the autophagosome cargo protein p62, and restricts its co-aggregation with selected substrates tagged for degradation [65]. Thereby, the p62/TRIB3 2 interaction induces the blocking of the autophagic flux that interestingly leads to the accumulation of several tumor-promoting factors. The interruption of this p62/TRIB3 interaction attenuates xenograft tumor growth and metastatic dissemination, confirming the role of TRIB3 in this context as an oncogene [65,66].
In addition, TRIB3 can contribute to tumor progression via the positive regulation of signaling pathways like Notch and TGF-β. TRIB3 was identified as a master regulator of JAG1 (Jagged canonical Notch ligand 1) gene expression, and Notch activation in breast cancer. It induces cell survival and tumor xenograft growth [41,67]. The same authors demonstrated that MAPK-ERK was the predominant pathway promoting TRIB3-dependent Notch activation, and TGFβ-SMAD4 signaling in the regulation of JAG1 expression [41,67]. Similarly, in aggressive lung cancer cell lines, TRIB3 depletion is correlated with Notch1 downregulation and inhibition of tumor growth and metastatic dissemination [11]. In hepatocellular carcinoma cell lines, TRIB3 overexpression can increase the activity of TGFβ-SMAD3 by reducing the degradation of SMAD3 and promoting its translocation to the nucleus, which in turn promotes epithelial–mesenchymal transition [68].
7. Conclusions
Tribbles pseudokinase 3 clearly has a role in tumor progression, either as tumor suppressor or as oncogene. Nevertheless, in the wake of the impaired catalytic activity of its kinase-like domain, TRIB3 acts as a collaborating effector and modulates the localization or activity of other intracellular molecules. TRIB3 acts like a “jack of all trades” through a wide interaction network including components of central signaling pathways, such as Akt or MAPK, for example. In parallel, TRIB3 also interferes with gene expression at the transcriptional level, through direct interaction with various transcription factors or through impairment of the spliceosome machinery and pre-mRNA maturation. These complex interactive functions of TRIB3 are encouraging to better understand the processes in which it is involved, but also to explore novel cancer therapeutic approaches that target TRIB3.
Funding
This work was supported by INSERM, Gustave Roussy (taxe d’apprentissage), Opération Parrains Chercheurs, Odyssea, Dassault Foundation, and Breast Cancer Research Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Großhans, J.; Wieschaus, E. A Genetic Link between Morphogenesis and Cell Division during Formation of the Ventral Furrow in Drosophila. Cell 2000, 101, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Seher, T.C.; Leptin, M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr. Biol. 2000, 10, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Mata, J.; Curado, S.; Ephrussi, A.; Rørth, P. Tribbles Coordinates Mitosis and Morphogenesis in Drosophila by Regulating String/CDC25 Proteolysis. Cell 2000, 101, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, Z.; Czibula, A.; Kiss-Toth, E. Tribbles: Novel regulators of cell function; evolutionary aspects. Cell. Mol. Life Sci. 2006, 63, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss-Toth, E.; Bagstaff, S.M.; Sung, H.Y.; Jozsa, V.; Dempsey, C.; Caunt, J.C.; Oxley, K.M.; Wyllie, D.H.; Polgar, T.; Harte, M.; et al. Human Tribbles, a Protein Family Controlling Mitogen-activated Protein Kinase Cascades. J. Biol. Chem. 2004, 279, 42703–42708. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Zhou, J.; Ma, K.; Zhang, J.; Xie, H.; Zhang, K.; Li, L.; Cai, L.; Zhang, N.; Zhang, Z.; et al. TRIB3 Promotes the Proliferation and Invasion of Renal Cell Carcinoma Cells via Activating MAPK Signaling Pathway. Int. J. Biol. Sci. 2019, 15, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.-J.; Lin, R.-J.; Wang, H.-C.; Yuan, T.-M.; Chuang, S.-M. TRIB3 downregulation enhances doxorubicin-induced cytotoxicity in gastric cancer cells. Arch. Biochem. Biophys. 2017, 622, 26–35. [Google Scholar] [CrossRef]
- Zhang, J.; Wen, H.-J.; Guo, Z.-M.; Zeng, M.-S.; Li, M.-Z.; Jiang, Y.-E.; He, X.-G.; Sun, C.-Z. TRB3 overexpression due to endoplasmic reticulum stress inhibits AKT kinase activation of tongue squamous cell carcinoma. Oral Oncol. 2011, 47, 934–939. [Google Scholar] [CrossRef]
- Zhou, H.; Luo, Y.; Chen, J.-H.; Hu, J.; Luo, Y.-Z.; Wang, W.; Zeng, Y.; Xiao, L. Knockdown of TRB3 induces apoptosis in human lung adenocarcinoma cells through regulation of Notch 1 expression. Mol. Med. Rep. 2013, 8, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Stefanovska, B.; Vicier, C.E.; Dayris, T.; Ogryzko, V.; Scott, V.; Bouakka, I.; Delaloge, S.; Rocca, A.; Le Saux, O.; Trédan, O.; et al. Rapalog-mediated repression of Tribbles pseudokinase 3 regulates pre-mRNA splicing. Cancer Res. 2020, 80, 2190–2203. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Nakatani, Y.; Jamieson, S.A.; Dai, W.; Lucet, I.S.; Mace, P.D. Molecular Mechanism of CCAAT-Enhancer Binding Protein Recruitment by the TRIB1 Pseudokinase. Structure 2015, 23, 2111–2121. [Google Scholar] [CrossRef] [Green Version]
- Bowers, A.J.; Scully, S.; Boylan, J.F. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 2003, 22, 2823–2835. [Google Scholar] [CrossRef] [Green Version]
- Bailey, F.P.; Byrne, D.P.; Oruganty, K.; Eyers, C.E.; Novotny, C.J.; Shokat, K.M.; Kannan, N.; Eyers, P.A. The Tribbles 2 (TRB2) pseudokinase binds to ATP and autophosphorylates in a metal-independent manner. Biochem. J. 2015, 467, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.; Wells, R.; Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: The PEST hypothesis. Science 1986, 234, 364–368. [Google Scholar] [CrossRef]
- Wennemers, M.; Bussink, J.; Beucken, T.V.D.; Sweep, F.C.G.J.; Span, P.N. Regulation of TRIB3 mRNA and Protein in Breast Cancer. PLoS ONE 2012, 7, e49439. [Google Scholar] [CrossRef] [PubMed]
- Örd, D.; Örd, T. Characterization of human NIPK (TRB3, SKIP3) gene activation in stressful conditions. Biochem. Biophys. Res. Commun. 2005, 330, 210–218. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Deval, C.; Maurin, A.-C.; Parry, L.; Chérasse, Y.; Chaveroux, C.; Lefloch, R.; Lenormand, P.; Bruhat, A.; Fafournoux, P. TRB3 Inhibits the Transcriptional Activation of Stress-regulated Genes by a Negative Feedback on the ATF4 Pathway. J. Biol. Chem. 2007, 282, 15851–15861. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, X.; Franklin, J.L.; Messina, J.L.; Hill, H.S.; Moellering, D.R.; Walton, R.G.; Martin, M.; Garvey, W.T. Mammalian Tribbles homolog 3 impairs insulin action in skeletal muscle: Role in glucose-induced insulin resistance. Am. J. Physiol. Metab. 2010, 298, E565–E576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, T.; Örd, D.; Adler, P.; Vilo, J.; Örd, T. TRIB3 enhances cell viability during glucose deprivation in HEK293-derived cells by upregulating IGFBP2, a novel nutrient deficiency survival factor. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2492–2505. [Google Scholar] [CrossRef] [Green Version]
- Geng, T.; Hu, W.; Broadwater, M.H.; Snider, J.M.; Bielawski, J.; Russo, S.B.; Schwacke, J.H.; Ross, J.; Cowart, L.A. Fatty acids differentially regulate insulin resistance through endoplasm reticulum stress-mediated induction of tribbles homologue 3: A potential link between dietary fat composition and the pathophysiological outcomes of obesity. Diabetologia 2013, 56, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Örd, T.; Örd, T. Mammalian Pseudokinase TRIB3 in Normal Physiology and Disease: Charting the Progress in Old and New Avenues. Curr. Protein Pept. Sci. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mkrtchian, S. Targeting unfolded protein response in cancer and diabetes. Endocrine-Related Cancer 2015, 22, C1–C4. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.-H.; Satoh, H.; Herzig, S.; Lee, C.-H.; Hedrick, S.; Kulkarni, R.N.; Evans, R.M.; Olefsky, J.M.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef]
- Chan, M.C.; Weisman, A.S.; Kang, H.; Nguyen, P.H.; Hickman, T.; Mecker, S.V.; Hill, N.S.; Lagna, G.; Hata, A. The Amiloride Derivative Phenamil Attenuates Pulmonary Vascular Remodeling by Activating NFAT and the Bone Morphogenetic Protein Signaling Pathway. Mol. Cell. Biol. 2010, 31, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Han, S.; Kitamura, T.; Accili, M. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Investig. 2006, 116, 2464–2472. [Google Scholar] [CrossRef] [Green Version]
- Zareen, N.; Biswas, S.C.; Greene, L. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013, 20, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Bjornsti, M.-A.; Houghton, P.J. Rapamycins: Mechanism of action and cellular resistance. Cancer Biol. Ther. 2003, 2, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galat, A.; Thai, R. Rapamycin-binding FKBP25 associates with diverse proteins that form large intracellular entities. Biochem. Biophys. Res. Commun. 2014, 450, 1255–1260. [Google Scholar] [CrossRef]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 Facilitates Heart Development by Antagonizing Oxidative and ER Stress Responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. New Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Li, L.; Liu, Q.; Xing, G.; Kuai, X.; Sun, J.; Yin, X.; Wang, J.; Zhang, L.; He, F. E3 ubiquitin ligase SIAH1 mediates ubiquitination and degradation of TRB3. Cell. Signal. 2008, 20, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Takahama, S.; Endo, Y.; Sawasaki, T. Stress-Inducible Caspase Substrate TRB3 Promotes Nuclear Translocation of Procaspase-3. PLoS ONE 2012, 7, e42721. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Du, K. TRB3: A tribbles Homolog That Inhibits Akt/PKB Activation by Insulin in Liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsting, E.; Patel, S.V.; Declèves, A.-E.; Lee, S.J.; Rahman, Q.M.; Akira, S.; Satriano, J.; Sharma, K.; Vallon, V.; Cunard, R. Tribbles Homolog 3 Attenuates Mammalian Target of Rapamycin Complex-2 Signaling and Inflammation in the Diabetic Kidney. J. Am. Soc. Nephrol. 2014, 25, 2067–2078. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Fang, X.; Malik, W.S.; He, Y.; Li, X.; Xie, M.; Sun, W.; Xu, Y.; Liu, X. TRB3 interacts with ERK and JNK and contributes to the proliferation, apoptosis, and migration of lung adenocarcinoma cells. J. Cell. Physiol. 2020, 235, 538–547. [Google Scholar] [CrossRef]
- Izrailit, J.; Berman, H.K.; Datti, A.; Wrana, J.L.; Reedijk, M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGF pathways as fundamental Notch regulators in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Ohoka, N.; Hayashi, H.; Sato, R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARγ transcriptional activity. J. Lipid Res. 2008, 49, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Dedhia, P.H.; Keeshan, K.; Uljon, S.; Xu, L.; Vega, M.E.; Shestova, O.; Zaks-Zilberman, M.; Romany, C.; Blacklow, S.C.; Pear, W.S. Differential ability of Tribbles family members to promote degradation of C/EBPα and induce acute myelogenous leukemia. Blood 2010, 116, 1321–1328. [Google Scholar] [CrossRef]
- Örd, D.; Meerits, K.; Örd, T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp. Cell Res. 2007, 313, 3556–3567. [Google Scholar] [CrossRef]
- Luo, X.; Zhong, L.; Yu, L.; Xiong, L.; Dan, W.; Li, J.; Ye, J.; Chu, X.; Liu, C.; Liu, B. TRIB3 destabilizes tumor suppressor PPARα expression through ubiquitin-mediated proteasome degradation in acute myeloid leukemia. Life Sci. 2020, 257, 118021. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Zhang, W.; Xu, S.; Lin, H.; Wang, Z.; Liu, H.; Fang, Q.; Li, C.; Peng, L.; Lou, J. TRIB3 alters endoplasmic reticulum stress-induced β-cell apoptosis via the NF-κB pathway. Metabolism 2014, 63, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.P.; Behan, F.M.; Kirca, M.; Smith, S.; Reynolds, J.V.; Long, A.; Kelleher, D. An integrative genomic approach in oesophageal cells identifies TRB3 as a bile acid responsive gene, downregulated in Barrett’s oesophagus, which regulates NF-κB activation and cytokine levels. Carcinogenesis 2010, 31, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, L.-G.; Zhai, Z.; Shu, H.-B. SINK Is a p65-interacting Negative Regulator of NF-κB-dependent Transcription. J. Biol. Chem. 2003, 278, 27072–27079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzymski, T.; Paantjens, A.; Bod, J.; Harris, A.L. Multiple pathways are involved in the anoxia response of SKIP3 including HuR-regulated RNA stability, NF-κB and ATF4. Oncogene 2008, 27, 4532–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Örd, D.; Örd, T. Mouse NIPK interacts with ATF4 and affects its transcriptional activity. Exp. Cell Res. 2003, 286, 308–320. [Google Scholar] [CrossRef]
- Hua, F.; Shang, S.; Zhang, X.-W.; Liu, S.-S.; Yu, J.-M.; Wang, F.; Zhang, C.; Huang, B.; Hu, Z.-W.; Yang, Y.-W.; et al. TRIB3 Interacts With β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Z.; Yang, L.; Yang, Y.; Yang, J.; Niu, Z.; Zhang, X.; Song, Q.; Lei, Y.; Wu, H.; Guo, J. Activation of Wnt/β-catenin pathway causes insulin resistance and increases lipogenesis in HepG2 cells via regulation of endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2020, 526, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Fink, G.R.; Bartel, D.P. Excised linear introns regulate growth in yeast. Nat. Cell Biol. 2019, 565, 606–611. [Google Scholar] [CrossRef]
- Edwards, S.R.; Johnson, T.L. Intron RNA sequences help yeast cells to survive starvation. Nat. Cell Biol. 2019, 565, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Qual. Life Res. 2017, 136, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudente, S.; Sesti, G.; Pandolfi, A.; Andreozzi, F.; Consoli, A.; Trischitta, V. The mammalian tribbles homolog TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr. Rev. 2012, 33, 526–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.Z.A.; Mokhtar, N.M.; Sin, T.K.; Rose, I.M.; Sagap, I.; Harun, R.; Jamal, R. Integrated Analysis of Copy Number Variation and Genome-Wide Expression Profiling in Colorectal Cancer Tissues. PLoS ONE 2014, 9, e92553. [Google Scholar] [CrossRef]
- Loo, L.W.M.; Tiirikainen, M.; Cheng, I.; Lum-Jones, A.; Seifried, A.; Church, J.M.; Gryfe, R.; Weisenberger, D.J.; Lindor, N.M.; Gallinger, S.; et al. Integrated analysis of genome-wide copy number alterations and gene expression in microsatellite stable, CpG island methylator phenotype-negative colon cancer. Genes, Chromosom. Cancer 2013, 52, 450–466. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Gomez, E.P.; Davila, D.; Zúñiga-García, P.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. TRIB3 suppresses tumorigenesis by controlling mTORC2/AKT/FOXO signaling. Mol. Cell. Oncol. 2015, 2, e980134. [Google Scholar]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Pérez-Gómez, E.; Dávila, D.; Zúñiga-García, P.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. Loss of Tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. 2014, 22, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Aynaud, M.-M.; Suspène, R.; Vidalain, P.-O.; Mussil, B.; Guétard, D.; Tangy, F.; Wain-Hobson, S.; Vartanian, J.-P. Human Tribbles 3 Protects Nuclear DNA from Cytidine Deamination by APOBEC3A. J. Biol. Chem. 2012, 287, 39182–39192. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Li, K.; Yu, J.-J.; Lv, X.; Yan, J.; Zhang, X.-W.; Sun, W.; Lin, H.; Shang, S.; Wang, F.; et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015, 6, 7951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, F.; Li, K.; Yu, J.-J.; Hu, Z.-W. The TRIB3-SQSTM1 interaction mediates metabolic stress-promoted tumorigenesis and progression via suppressing autophagic and proteasomal degradation. Autophagy 2015, 11, 1929–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izrailit, J.; Jaiswal, A.; Zheng, W.; Moran, M.F.; Reedijk, M. Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene 2017, 36, 1048–1057. [Google Scholar] [CrossRef]
- Hua, F.; Mu, R.; Liu, J.; Xue, J.; Wang, Z.; Lin, H.; Yang, H.; Chen, X.; Hu, Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J. Cell Sci. 2011, 124, 3235–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Transcript variant analysis of the TRIB3 gene (ENSG00000101255.11). (Adapted from the Ensembl platform; https://www.ensembl.org accessed on 6 January 2021).
Figure 1.
Transcript variant analysis of the TRIB3 gene (ENSG00000101255.11). (Adapted from the Ensembl platform; https://www.ensembl.org accessed on 6 January 2021).

Figure 2.
In silico analysis of the human TRIB3 promoter region. The three putative alternative promoters and the related TRIB3 isoforms are indicated (Adapted from [12]). aa: amino acid.
Figure 2.
In silico analysis of the human TRIB3 promoter region. The three putative alternative promoters and the related TRIB3 isoforms are indicated (Adapted from [12]). aa: amino acid.

Figure 3.
Functional domains of TRIB3 protein. NLS = nuclear localization signal.

Figure 4.
In silico analysis of the human TRIB3 promoter region using the MatInspector Software (Genomatix Software GmbH, Munich; version v3.13). The matching transcription factor binding sites with a 5′ to 3′ (above the line) or a 3′ to 5′ (below the line) orientation relative to the transcription start site (TSS) are indicated.
Figure 4.
In silico analysis of the human TRIB3 promoter region using the MatInspector Software (Genomatix Software GmbH, Munich; version v3.13). The matching transcription factor binding sites with a 5′ to 3′ (above the line) or a 3′ to 5′ (below the line) orientation relative to the transcription start site (TSS) are indicated.

Figure 5.
TRIB3 post-translational modifications. Phosphorylation (blue circle), ubiquitylation (orange circle), and methylation (green circle) sites reported from www.phosphosite.org accessed on 18 June 2020).
Figure 5.
TRIB3 post-translational modifications. Phosphorylation (blue circle), ubiquitylation (orange circle), and methylation (green circle) sites reported from www.phosphosite.org accessed on 18 June 2020).

Figure 6.
Putative mechanisms of TRIB3-mediated AKT signaling inhibition. Red circle = phosphorylation. (A) Activated signaling pathway in absence of TRIB3. (B) Partial inhibition of the signaling pathway in the presence of TRIB3.
Figure 6.
Putative mechanisms of TRIB3-mediated AKT signaling inhibition. Red circle = phosphorylation. (A) Activated signaling pathway in absence of TRIB3. (B) Partial inhibition of the signaling pathway in the presence of TRIB3.

Figure 7.
(A) Proportion of TRIB3 somatic mutations according to the COSMIC database (Tate et al., 2019). (B) Non-exhaustive distribution of most frequently mutated genes according to the TCGA Research Network (https://www.cancer.gov/tcga accessed on 6 January 2021).
Figure 7.
(A) Proportion of TRIB3 somatic mutations according to the COSMIC database (Tate et al., 2019). (B) Non-exhaustive distribution of most frequently mutated genes according to the TCGA Research Network (https://www.cancer.gov/tcga accessed on 6 January 2021).

Figure 8.
TRIB3 mRNA expression in normal and tumor tissues from 33 different cancer types according to the TCGA Research Network (https://www.cancer.gov/tcga accessed on 18 June 2020). *** p-value < 0.001.
Figure 8.
TRIB3 mRNA expression in normal and tumor tissues from 33 different cancer types according to the TCGA Research Network (https://www.cancer.gov/tcga accessed on 18 June 2020). *** p-value < 0.001.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
TRIB3 Transcripts from Ensembl genome browser 101 (www.ensembl.org accessed on 6 January 2021).
Table 1.
TRIB3 Transcripts from Ensembl genome browser 101 (www.ensembl.org accessed on 6 January 2021).
| Name | Transcript ID | bp | Protein | UniProt | Flags |
|---|---|---|---|---|---|
| TRIB3-201 | ENST00000217233.8 | 2095 | 358aa | Q96RU7 | TSL:1, APPRIS P3 |
| TRIB3-202 | ENST00000422053.3 | 1533 | 385aa | J3KR25 | TSL:2, APPRIS ALT2 |
| TRIB3-203 | ENST00000449710.5 | 1070 | 271aa | B0QYQ2 | CDS 3′ incomplete, TSL:5 |
| TRIB3-204 | ENST00000615226.4 | 877 | 130aa | A0A087WTX3 | CDS 3′ incomplete, TSL:3 |
TSL1: all splice junctions of the transcript are supported by at least one non-suspect mRNA; SL2: the best supporting mRNA is flagged as suspect or the support is from multiple ESTs; TSL3: the only support is from a single EST; TSL5: no single transcript supports the model structure. APPRIS is a system to annotate alternatively spliced transcripts based on a range of computational methods. APPRIS P3: no clear principal variant identified, and more than one of the variants have distinct CCDS identifiers, APPRIS selects the variant with lowest CCDS identifier as the principal variant. APPRIS ALT2: candidate transcript that appears to be conserved in fewer than three tested species.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stefanovska, B.; André, F.; Fromigué, O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers 2021, 13, 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081822
AMA Style
Stefanovska B, André F, Fromigué O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers. 2021; 13(8):1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081822
Chicago/Turabian StyleStefanovska, Bojana, Fabrice André, and Olivia Fromigué. 2021. "Tribbles Pseudokinase 3 Regulation and Contribution to Cancer" Cancers 13, no. 8: 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081822
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.