
Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer
by
 , , , and
, , , and
Chia-Jung Li
1,2 ,
,
Yen-Dun Tony Tzeng
3,4,*, 
Yi-Han Chiu
5,
Hung-Yu Lin
6,
Ming-Feng Hou
7 and
Pei-Yi Chu
8,9,10,11,* 1
Department of Obstetrics and Gynecology, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan
2
Institute of BioPharmaceutical Sciences, National Sun Yat-sen University, Kaohsiung 804, Taiwan
3
Department of Surgery, Kaohsiung Veterans General Hospital, Kaohsiung 813, Taiwan
4
Institute of Biomedical Sciences, National Sun Yat-sen University, Kaohsiung 804, Taiwan
5
Department of Microbiology, Soochow University, Taipei 111, Taiwan
6
Research Assistant Center, Show Chwan Memorial Hospital, Changhua 500, Taiwan
7
Center for Cancer Research, Division of Breast Surgery, Department of Surgery, Kaohsiung Medical University Chung-Ho Memorial Hospital, Kaohsiung 807, Taiwan
8
School of Medicine, College of Medicine, Fu Jen Catholic University, New Taipei City 242, Taiwan
9
Department of Pathology, Show Chwan Memorial Hospital, Changhua 500, Taiwan
10
Department of Health Food, Chung Chou University of Science and Technology, Changhua 510, Taiwan
11
National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan
*
Authors to whom correspondence should be addressed.
Cancers 2021, 13(12), 2978; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122978
Submission received: 15 May 2021
/
Revised: 10 June 2021
/
Accepted: 12 June 2021
/
Published: 14 June 2021
(This article belongs to the Special Issue Molecular Biomarkers for Risk and Prognosis of Cancer)
Abstract
:Simple Summary
Breast cancer emergencies have become a rapidly evolving field in medicine during the last ten years. Carcinogenesis is a multiparametric process that involves diverse factors such as genetic, environmental, or aging. Recent research that elucidates the tumor biology and molecular pathways that mediate cancer progression and drug resistance has led to the development of various molecular targeted therapies involving monoclonal antibodies, small molecule receptor tyrosine kinase inhibitors, and agents that block downstream signaling pathways in breast cancer.
Abstract
Triple negative breast cancer (TNBC) is a heterogeneous tumor characterized by early recurrence, high invasion, and poor prognosis. Currently, its treatment includes chemotherapy, which shows a suboptimal efficacy. However, with the increasing studies on TNBC subtypes and tumor molecular biology, great progress has been made in targeted therapy for TNBC. The new developments in the treatment of breast cancer include targeted therapy, which has the advantages of accurate positioning, high efficiency, and low toxicity, as compared to surgery, radiotherapy, and chemotherapy. Given its importance as cancer treatment, we review the latest research on the subtypes of TNBC and relevant targeted therapies.
1. Introduction
Breast cancer is the most common malignancy among women with an incidence rate of 10,492 in 100,000 cases in 2015, according to reports of the Ministry of Health and Welfare. The high morbidity and mortality rates cause a financial burden with regards to health insurance [1]. Several risk factors for breast cancer were investigated, including alcohol intake (relative risk: 1.10; 95% confidence interval: 1.06–1.14) [2], obesity (hazard ratio: 1.58, 95% confidence interval: 1.40–1.79) [3], age (hazard ratio: 10.1, 95% confidence interval: 8.49–11.94) [4], early menarche, and late menopause. Despite several years of bench to bedside investigations, breast cancer remains a challenge for all physicians, with the detailed mechanism of its tumorigenesis still unclear. Its molecular and biological attributes were investigated in detail to better understand the development and pathogenesis of breast cancer, including oncogenes, the tumor suppression genes, and downstream pathways that promote tumor proliferation, differentiation, and distant metastasis. The treatment of breast cancer requires a multidisciplinary approach involving surgery, radiation, and medical oncology to control tumor cell proliferation and metastasis. The new treatments for breast cancer are also discussed in this article, including the use of PARP, MAPK, CHK1, CDK 4/6 inhibitor, anticytotoxic T-lymphocyte-associated protein 4 (anti-CTLA4) antibodies, and anti-programmed cell death protein 1 (anti-PD1)/ligand 1 (anti-PD-L1) antibodies. We believe that elucidating the mechanism of breast cancer will provide an important foundation for therapeutic intervention. This article provides an update on research and clinical results using the molecular biology of tumorigenesis and summarizes current treatments to provide a new strategy for physicians.
2. Clinical Features of Breast Cancer
The common clinical presentation of breast cancer is usually a palpable breast mass observed upon on self-breast examination. Other associated symptoms included local pain or nipple discharge. In metastasis, fever, weight loss, night sweats, or bone pain may be present as a result of tumor progression. Imaging studies play an important role in early detection and timely intervention. Three image detection modalities for breast cancer were investigated, including breast ultrasound, mammography, and magnetic resonance imaging (MRI). A breast ultrasound is effective for characterizing malignant solid masses as an initial diagnostic evaluation with a sensitivity rate of 98.4% and negative predictive value of 99.5% [5,6]. The mammogram was used for elderly women to characterize soft tissue density, which may present with an irregular border, spiculated pattern, and local calcifications. According to mammographic findings, the radiologist will utilize the Breast Imaging Reporting and Data System (BI-RADS) for further diagnostic assessment (Table 1). The breast MRI is an important tool in preoperative survey and postoperative follow-up, especially to evaluate scar tissue. The sensitivity is reported to be high at more than 90%, but the specificity is moderate at about 72%. In high-risk breast cancer screening, breast MRI has an important role in preventing missed diagnoses [7]. Several prognostic and predictive factors for breast cancer were reported, including the tumor node metastasis (TNM) system, tumor morphology, and tumor markers. The TNM system categorizes breast cancer by location and invasion anatomy, including tumor size, lymph node involvement, and distant metastasis. The eighth edition of the TNM system is shown in Table 2, reported from the Union for International Cancer Control (UICC) in 2017.
The anatomic stage provides surgeons with important information needed for making decisions about surgical intervention and preventing complications. After surgical intervention, the target therapy and chemotherapy play a critical role in controlling tumor proliferation, local recurrence, and distant metastasis. The tumor biology was investigated and is important in prognosis and response to treatment. Biomarkers have been investigated and taken into account in modified staging systems to accurately predict clinical outcomes. The expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) are generally used in clinical management. The expression of hormone receptors, including ER and PR, are reflective of the response to adjuvant endocrine therapy and are associated with better outcomes. Currently, the expression of ER and PR levels is significantly related to the overall survival, disease-free survival, and time to treatment failure [8]. The status of ER expression is associated with the distant metastatic site. Bone and soft tissue are common distant metastatic sites of ER-positive breast cancer, compared to ER-negative breast cancer, which usually occurs in the brain and liver. The expression of the PR is independent of ER expression and also significantly related to clinical outcome. PR-negative breast cancers have a more aggressive prognosis. HER2 overexpression is a predictor of poor clinical outcomes if patients do not receive chemotherapy and target therapy. Other biomarkers, including Ki-67, luminal A, luminal B, and basal, are involved in clinical prognosis. In high-grade breast cancer with rapid progression, the mitotic count and expression of Ki-67 are elevated, reflecting a high proliferation rate, and this type of cancer may better respond to anthracycline treatment, which inhibits DNA and RNA synthesis. The expression of luminal A and luminal B genes is associated with ER-positive breast cancers. The luminal subtype tumors typically express luminal cytokeratin 8 and 18. Luminal A tumors are usually associated with high expression of ER and lower expression of HER-2 genes. Luminal B tumors are associated with lower expression of the ER gene and have poor prognosis, compared to the luminal A subtype. With the emergence of genomics and transcriptomics techniques, several thousands of genes have been identified and investigated to predict clinical prognosis. The detailed mechanisms of tumorigenesis and the interaction with the tumor microenvironment are discussed below.
3. Molecular Types of Triple Negative Breast Cancer
Breast tumor heterogeneity is one of the main reasons for the significant differences in the therapeutic effects of breast cancer patients. In 2011, Lehmann’s team examined the genetic sequence of TNBC patients through mRNA expression profiling clustering and found that TNBC can be classified into six subtypes: basal cell-like type 1 (BL1), basal cell Like type 2 (BL2), immunomodulatory subtype (IM), mesenchymal subtype (M), mesenchymal stem cell-like subtype (MSL), and luminal androgen receptor subtype (LAR) [9]. These six subtypes have different gene expression profiles and are involved in different signal transduction pathways (Figure 1). The BL1 and BL2 subtypes account for about 50% of TNBC. The BL1 subtype has high expression of cell proliferation-related genes, DNA damage repair genes, and Ki-67 protein. The BL2 subtype has high expression of multiple growth factor signal pathway genes and metabolic signal transduction genes. IM subtypes have high expression of multiple immune-related signal pathways and cytokine-related signal pathways. M and MSL subtypes have high expression of epithelial–mesenchymal transition (EMT) genes and cell differentiation-related genes, and are sensitive to PI3K/mTOR inhibitors [10]. The difference between M-type and MSL-type TNBC is the low expression of cell proliferation protein and tight junction protein in the MSL subtype, and the low mitotic index. The M subtype is related to cell motility, cell differentiation, and extracellular matrix receptors. LAR subtypes are rich in hormone-regulated signaling pathways, including steroid synthesis and androgen receptor signaling pathways.
Further classification of TNBC can bring more accurate diagnosis and personalized treatment to TNBC patients. In 2015, Burstein et al. performed a matrix analysis of gene expression in 198 TNBC patient microarrays at Baylor College of Medicine, USA, and grouped 80 core genes to obtain four more stable TNBC subtypes, namely basal like/immune suppressed (BLIS), basal like/immune activated (BLIA), mesenchymal (MES), and LAR [11]. Liu et al. in their 2016 study stated that by typing analysis of 145 TNBC cases, the superior typing method was obtained by classifying TNBC into four subtypes: BLIS, IM, MES, and LAR [12]. Lehmann’s team again refined the TNBC molecular subtypes from six (TNBCtype) to four (TNBCtype-4) tumor-specific subtypes (BL1, BL2, M, and LAR) in 2016 and demonstrated differences in diagnostic age, grading, local and distant disease progression, and histopathology [13]. In addition, in 2019, Kim et al. first classified TNBC into neutrophil-enriched subtype (NES) and macrophage-enriched subtype (MES), and found that immune checkpoint blockage (ICB) therapy has different efficacy on these two subtypes [14], providing a new idea for molecular typing of TNBC to guide clinical treatment. The subtypes and classification of different TNBC tumors should provide important value for future clinical decision making and for TNBC patients with conventional chemotherapy versus targeted and immunotherapy therapies currently in clinical trials.
Although the TNBC molecular classification defines four major subtypes, BL1 and BL2, M, and LAR, with distinct ontologies and different responses to therapy, the LAR definition seems to be one of the most reliable definitions among the different gene ontology-based classification systems. There are different classification systems based on DNA and RNA analysis that identify four molecularly defined TNBC subtypes: LAR, mesenchymal (MES), basal-like immunosuppression (BLIS), and basal-like immune activation (BLIA), characterized by different prognosis and potential therapeutic targets [11,15]. Interestingly, DNA analysis revealed specific gene amplification and targetable molecular expression. LAR was also found to be characterized by AR and MUC1 markers, suggesting the strategic importance of anti-AR therapies and the potential role of MUC1 vaccines as an effective treatment for this subtype [16]. Compared to other profiles, the LAR subtypes identified by this new classification system share the same genetic and biological features as those identified by Lehmann [9] and Pietenpol et al. [9,11].
4. Prognostic Biomarkers of TNBC
4.1. BRCA 1/2
BRCA1 and BRCA2 are human tumor suppressor genes located on the long arm of chromosomes 17 and 13, respectively. The structures of BRCA1 and BRCA2 are different, but their roles are related and important in the DNA repair process [17]. BRCA2 binds to a DNA single strand and directly affects the regulation of recombinase, whereas RAD51 localizes to DNA double-strand cleavage, which requires the formation of the BRCA1–PALB2–BRCA2 complex. Therefore, BRCA1/BRCA2 encodes a protein that maintains human genome stability and prevents mutant genes. The rearrangement is significant [18,19]. BRCA1/BRCA2 plays an important role in DNA double-strand break repair. BRCA1/BRCA2 is involved in the repair of damaged DNA. If the repair of DNA is inhibited, it will induce apoptosis to clear the cells that may be mutated; however, once BRCA1/BRCA2 is mutated, DNA repair cannot be completed, and the progression of tumor development is promoted [20]. Mutations in BRCA1/BRCA2 are recognized as risk factors for breast cancer induction, but the role in breast cancer prognosis is still controversial. Existing studies seem to favor BRCA1/BRCA2 as having little effect on the prognosis of breast cancer. Previous studies have indicated that there is no difference in overall survival between BRCA-associated breast cancer and sporadic breast cancer [21]. Similarly, there is a meta-analysis showing that the evidence from 66 studies does not support the poor prognosis of BRCA1/BRCA2 mutation carriers. The study analyzed the relationship between BRCA1 and BRCA2 single mutation carrying and double mutation carrying with recurrence-free survival, metastasis-free survival, and overall survival in breast cancer patients, which were not associated with clinical factors after adjustment [22]. Therefore, the role of BRCA mutations in breast cancer progression and BRCA mutation-targeted treatment effects remain to be confirmed by large-scale, multicenter, prospective studies.
4.2. ALDH1A1
The protein encoded by ALDH1A1 (aldehyde dehydrogenase 1 family member A1) belongs to the aldehyde dehydrogenase family [18]. Aldehyde dehydrogenase is a polymorphic enzyme responsible for the oxidation of aldehydes to carboxylic acids, which leave the liver and are metabolized by the body’s muscle and heart [23]. ALDH1A1 is also an important component of corneal lens protein, which helps maintain the transparency of the cornea. In the face of cancer, ALDH1A1 is regarded as a marker of breast cancer stem cells, and cancer stem cells have strong carcinogenicity and self-renewal ability, and play an important role in tumorigenesis, development, and prognosis [24]. Tan et al. showed that ALDH1A1 is highly expressed in breast cancer tissues, but it is not associated with prognosis. ALDH1 is expressed in 25% of tumors, and the ALDH1-positive rate in young patients is almost 14 times higher than that in elderly patients. ALDH1A1 is considered to be an independent ER-negative factor, but it is not associated with breast cancer recurrence and associated death [25]. Numerous studies suggest that the expression of ALDH1A1 suggests a poor prognosis for breast cancer [24,26]. Japanese studies have found that 26% of patients with invasive ductal carcinoma have ALDH1 expression, which is associated with larger tumors, higher histological grades, later TNM stage, HER2 overexpression, and hormone receptor negative status. The recurrence-free survival time and overall survival time of the ALDH1A1 expression group were shorter than those of the non-expression group [27]. The results of a meta-analysis of a total of 3274 breast cancer patients enrolled in 15 studies were consistent with those of the Japanese study. In addition, cases with ALDH1A1 expression are more likely to show development of lymph node metastasis, confirming that ALDH1A1 is a marker of poor tumor progression and prognosis [24].
A previous study pointed out that Ma et al. [28] showed shorter recurrence-free survival (RFS) and overall survival (OS) in survival analysis of ALDH1-expressing patients in 158 cases of TNBC. Ying et al. [24] reported that ALDH1 expression is a biomarker for predicting poor survival in breast cancer patients. However, Morimoto et al. [29] found no statistical difference in disease-free survival (DFS) and OS between ALDH1+ and ALDH1- cases in TNBC patients. Meanwhile, Resetkova et al. [30] showed that stromal expression of ALDH1 rather than tumor expression was associated with TNBC survival. Although there are differences and controversies between these studies, this may be due to different case characteristics, and assay sensitivity and assessment criteria, and sample size. The small number of relevant studies may also explain the lack of consistency in results. Regardless of the reasons for the discrepancies, further studies involving larger cohorts and using standardized and well-matched controls are needed to validate these results.
4.3. CXCR4
CXCR4 (C-X-C chemokine receptor type 4) is expressed in most tissues and parenchymal organs in the body and is a G protein-coupled receptor (GPCR) consisting of 352 amino acids. The chemokine receptor CXCR4 is a specific receptor for chemokine (C-X-C) ligand 12 (CXCL12, also known as chemokine stromal cell-derived factor-1) [31]. CXCL12 has a strong chemotactic effect on lymphocytes, and CXCR4 plays an important role in various physiological functions, such as the dynamic balance of immune cells, including T cells. In tissue regeneration, the CXCL12/CXCR4 system plays an important role in promoting stem cell homing [32].
CXCR4 has a clear suggestive effect on breast cancer metastasis and prognosis, and is highly expressed in a variety of tumor cells and involved in chemotaxis, invasion, angiogenesis, and cell proliferation. It has been shown that breast cancer patients with high CXCR4 expression have poor prognosis [33,34]. In breast cancer patients, CXCR4 overexpression is associated with lymph node status and poor prognosis [33]. In addition, almost 75% of TNBC patients exhibit high CXCR4 expression [34]. Furthermore, there is a strong association between CXCR4 overexpression and histological cancer grade in menopausal and TNBC patients. The five-year DFS and five-year OS rates were 57.7% and 58.3% for the low CXCR4 group and 42.1% and 44.7% for the high CXCR4 group, respectively [35]. Patients with high CXCR4 overexpression (≥6-fold) in TNBC had a significantly higher incidence of cancer recurrence and cancer-related death than the low CXCR4 group (<6-fold) [34]. This result indicates that overexpression of CXCR4 in cancer specimens predicts a poor prognosis for TNBC and may be a predictor of poor prognosis.
A meta-analysis of the results of 15 studies (3104 patients in total) found that the expression of CXCR4 in breast cancer tissues was significantly higher than that in adjacent tissues, and high expression of CXCR4 in whole cells or the cytoplasm often indicated poor prognosis of breast cancer [36]. Another systematic review confirms these findings and shows that CXCR4 overexpression is significantly associated with lymph node metastasis and distant metastasis, with lower overall survival and disease-free survival. There is no correlation with clinical features of breast cancer, tumor stage, ER, and PR [32]. However, because of the inevitable omission of the meta-analysis literature search process and the differences among different research populations, understanding the specific mechanism of action of CXCR4 and whether it can be changed by radiotherapy and chemotherapy still needs further research.
4.4. p16INK4a
Inactivation of multiple tumor suppressor genes plays a role in the transformation of normal breast tissue into cancer. According to the multiangle analysis of tumor pathological type, grade of malignancy, efficacy, and prognosis, p16INK4a also plays an important role. There is a high deletion rate of p16INK4a in breast cancer cell lines. The main inactivation modes are deletion, point mutation, abnormal expression, and CpG island methylation in the promoter region [37,38]. Tumor suppressor genes often exhibit high frequency deletions and mutations in tumor cells. It was previously thought that abnormal changes in the p16INK4a gene were dominated by gene deletion, and point mutations were not the main cause of genetic changes. The overexpression rate of p16INK4a is more prominent in tumors with higher malignancy, and it is more overexpressed in breast cancer [39]. Conversely, there have been reports of p16INK4a downregulation in breast cancer [40].
The p16INK4a gene often has low expression in normal human tissue cells, and p16INK4a protein is generally not detected. The abnormal expression of p16INK4a may be related to the occurrence and development of breast cancer. However, whether it can become an independent prognostic factor or a high specificity marker for breast cancer remains to be confirmed by further large-scale clinical analysis and prospective studies. Recent studies have indicated that the methylation of CpG islands in the p16INK4a promoter region is more frequent than that of point mutations and homozygous deletions, and may be more valuable for the mechanism of p16 methylation induction and reversal. The methylation rate of the p16INK4a in breast cancer was found to be 30%, which is consistent with the results of two different research teams. It was also suggested that the methylation rate of breast cancer tissue was significantly higher than that of normal tissues and correlated with lymph node metastasis. The difference was statistically significant, but it was not significantly associated with the degree of differentiation of breast cancer [41,42]. These studies have shown that the higher methylation rate of the p16INK4a has important significance for breast cancer. The detection of the methylation rate of the p16INK4a may have high clinical value for early diagnosis of breast cancer. Moreover, the inhibition of tumor growth by the p16INK4a may play a role in anti-angiogenesis and induction of tumor cell apoptosis, in addition to mediating the arrest function of the cell proliferation cycle [43].
In addition, high p16INK4a expression has been shown to be associated with aggressive behavior and poor outcome in TNBC in studies of TNBC; thus, p16INK4a has potential prognostic utility and predictive value. High expression of p16INK4a predicts progression from in situ ductal carcinoma to basal-like invasive breast cancer [44]. It was also reported that p16INK4a overexpression was positively correlated with Ki67 expression in p53-negative TNBC but not in p53-positive tumors, suggesting that patients with p53-negative TNBC with high p16INK4a expression have a poorer prognosis [45]. Moreover, diffuse p16INK4a immunostaining in most cases of basal-like breast cancer and TNBC and a positive correlation between Rb negativity and diffuse p16INK4a expression was found [44]. Previous studies have found a better prognostic correlation for low p16INK4a expression in tumor tissues of TNBC cancer patients [46]. However, there was no prognostic significance between p16INK4a and overall survival and recurrence-free survival, in 60 TNBC patients [47]. As evidenced by these findings, there are conflicting data regarding the correlation between p16INK4a and prognosis, and more studies are needed to fully define the role of p16INK4a in TNBC clinical outcomes and survival.
4.5. ATM
ATM is a gene that is mutated in ataxia telangiectasia (A-T) and has been associated with cancer since its discovery. Swift et al. found that patients heterozygous for ATM genes are more likely to develop breast cancer; ATM genes are being explored widely for their role in gene function [48]. Although many studies in homozygous patients have failed to find links between ATM genes and tumors, this association has been confirmed in ATM heterozygotes, particularly the link between ATM heterozygotes and breast cancer. Western women have a 10% to 12% risk of breast cancer in their lifetime. The family history of breast cancer plays a key role in the pathogenesis. In addition, 5–10% of breast cancer patients have high penetrance germline gene mutations. The most important of these are the breast cancer susceptibility genes BRCA1 and BRCA2, and ATM and CHK2 are also important risk factors [49]. The role of the ATM gene in familial breast cancer is based on the exclusion of the susceptibility genes BRCA1, BRCA2, and CHK2. The detection rate of mutations in the ATM gene was 12/443 in the case group and 2/521 in the control group. This result suggests that mutations in the ATM gene may increase the incidence of breast cancer [50]. In addition to studies of ATM polymorphisms and gene mutations, recent studies have suggested that ATM mRNA levels are associated with lower metastasis-free survival (MFS), and low-level expression of ATM protein results in a shorter MFS. miR-203 is a dominant indicator of downregulation of the ATM gene in breast cancer. ATM gene mRNA and ATM protein levels are independent prognostic factors for sporadic breast cancer and play a good guiding role in treatment [51].
The ATM gene encodes a phosphatidylinositol 3-kinase (PI3K) that regulates and participates in the repair of DNA damage and a key substrate for cell cycle control. Previous studies analyzing the ATM gene in European women (42,671 cases and 42,164 controls) found an association between ATM mutations (c.7271 T > G) and overall breast cancer risk; however, a specific association with TNBC could not be determined because the tumors were not stratified by subtype [52]. In addition, 158 women with TNBC were recruited for analysis among Polish women, one of whom carried an ATM mutation, while no ATM mutation was detected in 44 women with non-TNBC hereditary breast cancer [53]. Other studies have observed an enrichment of ATM mutations in patients with ER-positive tumors [54,55], with a five-fold increase in ATM mutations in non-TNBC patients compared to TNBC tumors [56]. At present, the discussion regarding the ATM gene is of great value in the diagnosis, treatment, and prognosis of breast cancer. Determining whether it is a biomarker of breast cancer susceptibility like BRCA1 and BACA2 is worth investigating.
4.6. PTEN
The PTEN gene is an important tumor suppressor gene and plays a key regulatory role in breast cancer. In animals, it was found that 49% of female mice developed breast cancer six months after PTEN gene knockout (PTEN+/−) [57]. The effect of stable transfection of exogenous PTEN on the proliferation of endogenous PTEN-deficient human breast cancer cells confirmed that PTEN significantly inhibited the proliferation of breast cancer cells [58]. Therefore, determining whether the PTEN gene can inhibit the proliferation of tumor cells when artificially introduced into the cells of breast cancer patients could provide a new avenue for treatment. Through high-resolution fluorescence microsatellite analysis, it was found that AKT activation was positively correlated with PTEN gene deletion and HER-2 overexpression in breast cancer patients and negatively correlated with PR. Simultaneous deletion of PTEN and HER-2 overexpression results in enhanced activation of AKT, leading to the possibility of PR-negative expression [59]. The total positive expression rate of the PTEN gene in breast cancer was 69%, and the expression level in invasive breast cancer was significantly lower than that in breast carcinoma in situ and benign proliferative lesions. The positive expression rate of PTEN gradually decreases as the tumor progresses [60]. Breast cancer with downregulated PTEN protein has a strong invasiveness and is prone to infiltration and metastasis [61]. The expression of the PTEN gene can be used as a potential prognostic indicator.
PTEN mutations are common in TNBC, with gene-mediated loss of function occurring in 15% of cases [62] PTEN has recently been shown to protect the genome from instability by maintaining chromosomal integrity. Although women with Cowden syndrome carrying PTEN germline mutations have a 50% lifetime risk of developing breast cancer [63], there is no consistent phenotype of breast cancer associated with PTEN mutations. Most PTEN-associated tumors are more likely to be tubulointerstitial than TNBC. Observations suggest that (1) there is no significant difference in the incidence of PTEN-causing mutations between women with TNBC (n = 692) and women with non-TNBC tumors (n = 2696) [56]; and (2) the observation of only one deleterious mutation in 267 women [64] supports the view that PTEN mutations are not associated with an increased risk of TNBC. Preclinical AKT inhibition has higher activity in PTEN-deficient models of cancer [65], but there are significant challenges in determining which PTEN mutations confer sufficient loss of function to be targetable and the best way to identify such loss of function.
5. Therapeutic Strategy for TNBC
5.1. HER2 Inhibitor
Since the first discovery of HER-2 in breast cancer, its importance in the occurrence and development of breast cancer has gradually been recognized. Current targeted therapy research focuses on blocking the HER-2 signaling pathway. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, is a novel drug for the treatment of metastatic breast cancer. Trastuzumab has achieved good results in both HER-2-positive early and late-stage breast cancer treatments. Trastuzumab is used alone or in combination with chemotherapy for patients with advanced breast cancer. Trastuzumab treatment can prolong patient survival with low side effects. In a further in-depth study, the results of a combination chemotherapy regimen using trastuzumab with docetaxel or taxane resulted in overall survival of patients that was higher than that obtained using chemotherapy alone [66]. Therefore, trastuzumab combined with paclitaxel is the first-line treatment for HER-2 positive breast cancer patients. The investigators conducted a four-year follow-up study showing that patients who received trastuzumab in the observation group were more likely to have a higher survival rate (including disease-free survival) than those who did not receive treatment [67]. Since the patient’s treatment depends on his or her personal will, it is impossible to carry out rigorous statistical analysis, but patients receiving treatment have increases in disease-free survival and overall survival.
HER-2-targeted therapy has not benefited patients with low HER-2 expression in TNBC treatment strategies; however, combination therapy may be efficacious. The primary analysis of a phase IIb trial investigating the HER2-derived vaccine nelipepimut-S (NPS) did not benefit the intention-to-treat population (NPS; n = 55 vs. placebo; n = 44), but a subgroup analysis showed benefit in patients with TNBC [68]. Another study enrolled 136 people receiving NPS/GM-CSF and 139 people receiving placebo/GM-CSF. The results of the study confirmed that concomitant administration of trastuzumab and NPS with GM-CSF was safe and had no additional overall toxicity. The combination of HER-2-targeted NPS and trastuzumab was safe. In HER-2 low-expressing breast cancer, no significant differences in DFS were observed in the intention-to-treat analysis [69]; however, significant clinical benefits were observed in TNBC patients. These findings warrant further investigation in a phase III randomized trial.
Targeting RTK Signaling
Lapatinib is an RTKI that reversibly blocks EGFR and HER-2 [70], inhibits downstream MAPK/Erk1/2 and PI3K/AKT pathways [71], and enhances trastuzumab-dependent cell-mediated cytotoxicity [72]. In March 2007, the FDA approved lapatinib in combination with capecitabine for HER-2-positive breast cancer previously treated with anthracyclines, paclitaxel, and trastuzumab [73] In February 2010, the FDA approved lapatinib in combination with letrozole for the first-line treatment of hormone receptor (HR)-positive and HER-2 overexpressing postmenopausal metastatic breast cancer [74].
Lenatinib is a TKI that irreversibly inhibits HER-1, HER-2, and HER-4. Early clinical studies have shown that lenatinib exerts more effective inhibition than lapatinib in possible resistance pathways in HER-2-positive patients previously treated with trastuzumab or anti-HER-2. In July 2017, the FDA approved lenatinib for extended postoperative treatment with trastuzumab adjuvant therapy in patients with early-stage HER-2-positive breast cancer [75].
Tucatinib is a new oral selective TKI that is more selective for HER-2 than EGFR [76]. Recently, the FDA announced the granting of a New Drug Priority Approval Application (NDA) for tucatinib in combination with trastuzumab and capecitabine for the treatment of locally advanced unresectable or metastatic HER-2-positive breast cancer, including patients with brain metastases.
Pazopanib is a novel multitarget TKI that targets VEGFR-1, VEGFR-2, VEGFR-3, PDGFR/β, and c-Kit, and has been shown to inhibit tumor growth and angiogenesis in in vitro studies [77].
5.2. VEGF Inhibitor
TNBC is a highly proliferating tumor that requires the involvement of angiogenesis in its development, invasion, and metastasis processes. The expression level of vascular endothelial growth factor (VEGF) is an independent prognostic factor in early breast cancer. High expression of VEGF suggests high tumor malignancy, easy recurrence and metastasis, short disease-free survival, and low overall survival. VEGF-A is highly expressed in TNBC cells, so TNBC is generally considered to be sensitive to VEGF-targeting drugs [78]. Platelet-derived growth factor (PDGF) is one of the angiogenic factors. Abnormal activation of PDGF or PDGFR induces tumor angiogenesis and promotes migration and invasion of tumor cells. Overexpression of VEGF in tumor tissues can promote the migration and proliferation of vascular endothelial cells, promote angiogenesis, increase tumor growth, and significantly increase vascular permeability, allowing tumor cells to enter the blood vessels for tumor invasion and metastasis, providing more favorable conditions [79]. Currently, antiangiogenic drugs include bevacizumab and ramucirumab, and tyrosine kinase inhibitors such as sunitinib and sorafenib. These antiangiogenic drugs are likely to have higher antitumor activity in TNBC. In a phase II study of paclitaxel, bevacizumab, and gemcitabine in the treatment of HER2-negative breast cancer, the clinical benefit rate in the TNBC subgroup was 84.6%, and approximately 82.5% of patients achieved a total survival of 18 months [80]. In another phase III trial, RIBBON-2 in the TNBC subgroup, the bevacizumab combination group, and the chemotherapy group showed a reduction in the risk of disease progression by 51%. It also increased progression-free survival by 3.3 months and overall survival by 5.3 months [81]. These results demonstrate the efficacy of antiangiogenic drugs in inhibiting tumors in TNBC.
5.3. PARP Inhibitor
PARP is a key enzyme for repairing broken DNA single strands, and in wild-type cells containing BRCA1/2, broken DNA double strands can be repaired by homologous recombination. However, in BRCA1/2-deficient cells, the homologous recombination function fails and therefore PARP is relied upon to repair the broken DNA single strand. Therefore, PARP inhibitors have the ability to prevent self-repair in BRCA1/2 mutated breast cancer cells and accelerate apoptosis of tumor cells, thereby enhancing the efficacy of chemotherapy as well as radiotherapy [82].
As a novel drug, PARP inhibitors can cause double-strand breaks in BRCA-mutant breast cancer cells, leading to cell death due to synthetic lethality [83]. Olaparib is an effective PARP-1 and PARP-2 inhibitor [84] and was the first to be used by the FDA and the European Medicines Agency (EMEA) (Table 3). A PARP inhibitor was clinically approved for treatment of relapsed high-grade serous ovarian cancer. Olapani was first used in the clinical study of BRCA-related breast cancer, ClinicalTrials.gov Identifier: NCT00494234. Patients received olaparib (100 mg/time, 2 times/d (n = 27) and 400 mg/time, twice/d (n = 27)), and the results indicated a dose-dependent objective response rate (ORR) of 22% (100 mg group) and 41% (400 mg group). The 400 mg group had a median progression-free survival of 5.7 months (95% CI: 4.6–7.4), and that of the 100 mg group was 3.8 months (95% CI: 1.9–5.5) [85]. After a preliminary determination of the therapeutically effective dose, the OlympiAD study became the latest basis for FDA-approved olrapani, which included BRCA-mutant HER2-negative advanced breast cancer patients. The study group (n = 205) received olaparib (300 mg/second, 2 times/d monotherapy) while the control group (n = 97) only received capecitabine, eribulin, or vinorelbine in three cycle regimen single-agent chemotherapy. The study showed that the study group and the control group were 7.0 and 4.2 months (HR = 0.58, 95% CI: 0.33 to 0.80, p < 0.001). Therefore, the study indicated that olaparib has significant advantages in terms of efficacy and safety compared to other chemotherapeutic drugs [86].
Intractable TNBC requires more effective treatment strategies, so new drugs are being developed to sensitize sporadic TNBC to PARP inhibition. A clinical trial (ClinicalTrials.gov Identifier: NCT01623349) is evaluating olaparib and BKM120 (buparlisib) or BYL719 (PI3 kinase inhibitor) in advanced sporadic TNBC and high-grade plasma ovarian cancer [87]. Another clinical trial (ClinicalTrials.gov Identifier: NCT01434316) combined veliparib and dinaciclib (CDK inhibitor), in which CDK inhibition was assumed to sensitize TNBC to PARP inhibition [88]. After the phase I dose ascertainment portion, the study will enroll patients with BRCA-mutated and nonmutated advanced breast cancer in a dose expansion cohort. Another PARP inhibitor, talazoparib (BMN 673), effectively modulates PARP transcription. In addition, veriparib (veliparib), a novel potent PARP-1 and PARP-2 inhibitor, had a 51% partial remission rate in the treatment group in a clinical phase II trial of breast cancer treatment, which was significantly higher than the standard chemotherapy group (26%) [89]. This study suggests that veriparib is more effective than standard therapy for TNBC with a high risk of recurrence. In addition, multicenter clinical trials are evaluating the efficacy and safety of PARP inhibitors in combination with chemotherapy, immunosuppressants, and inhibitors of other targets (e.g., VEGFR, HSP90, P13K/AKT/mTOR, etc.) for the treatment of TNBC [90]. Regimens such as olaparib in combination with VEGFR or P13K inhibitors can effectively inhibit the rapid proliferation and growth of TNBC cells by affecting the blood oxygen supply to tumor cells and blocking molecules required for cell growth [91].
5.4. CHK1 Inhibitor
Checkpoint kinase 1 (CHK1) belongs to the serine/threonine protein kinase family and plays an important role in the regulation of cell cycle arrest by checkpoints caused by DNA damage or the presence of un-replicated DNA. The CHK1 inhibitor UNC01 abolished the production of G2 checkpoints dependent on DNA damage in cisplatin treatment and increased the sensitivity of cisplatin by nearly 60-fold. In addition, the TCGA database analysis found that the p53 mutation rate was very high in TNBC. The CHK1 inhibitor (UCN01 or AZD7762) increases the sensitivity of TNBC xenografts with p53 mutations to chemotherapy and achieves good preclinical effects [92]. These all indicate that CHK1 is a very promising target in patients with p53 mutations in TNBC. Based on the recognition that CHK1 is a drug target, other CHK1 inhibitors, AZD7762, PF-477736, SCH900776, and LY2606368, are undergoing phase I and II clinical trials.
5.5. EGFR Inhibitor
EGFR is a transmembrane tyrosine kinase growth factor receptor involved in cell proliferation and differentiation in normal tissues. It also participates in the adhesion and movement of normal tissue cells and initiates many downstream cell signal transduction pathways [110,111]. EGFR is a member of the ErbB family of membrane tyrosine kinase receptors. Studies have shown that most TNBCs have overexpressed EGFR, and early TNBC patients with EGFR overexpression usually have worse overall survival and disease-free survival than normal expression patients. Therefore, EGFR overexpression is often associated with poor prognosis in TNBC [112,113]. Currently, the targeted therapeutic drugs that block the EGFR signaling pathway in breast cancer research are mainly anti-EGFR monoclonal antibodies (cetuximab). A phase II open randomized clinical trial evaluated the efficacy of platinum citrate cetuximab in patients with TNBC. The combination group had a 10% increase in complete release rate compared with the single-agent group, progression-free survival was extended by 2.2 months, and overall survival was extended by 3.5 months, but cetuximab may cause adverse reactions [95]. These cytological and clinical trials have demonstrated the enormous potential of EGFR inhibitors in the targeted therapy of TNBC.
5.6. Targeting Based on Cell Proliferation and Survival-Dependent Pathways in TNBC
5.6.1. Targeting of the PI3K/AKT/mTOR Signaling Pathway
Distortion of the PI3K/AKT/mTOR signaling pathway is common in TNBC and has become a potential pathway for TNBC resistance to chemotherapy; phosphorylation of mTOR often suggests a poor prognosis in early TNBC [114,115]. Everolimus is an oral mTOR inhibitor that inhibits the downstream signaling of mTOR molecules in cells and arrests the cell cycle in the G1 or S phase, thereby inhibiting PI3K/AKT/mTOR pathway activity. A phase II clinical trial was performed in patients with TNBC (n = 50) treated with paclitaxel fluorouracil + epirubicin + cyclophosphamide (T-FEC), followed by randomization into the everolimus group (n = 23) and blank control group (n = 27). The results showed that the recurrence rate in the two groups was 47.8% and 29.6%, respectively, and the pCR rate was 30.4% and 25.9%, respectively [96]. Therefore, the study concluded that the pCR rate in patients with TNBC after combined treatment with everolimus did not improve significantly. In addition to monotherapy studies, a phase II neoadjuvant clinical study published in 2017 randomized TNBC patients into two groups; the study group (n = 96) was administered everolimus combined with cisplatin + paclitaxel, while the control group (n = 49) was administered placebo treatment. The results showed that the combination of everolimus with TNBC neoadjuvant therapy did not improve pCR or clinical response rates, but increased the incidence of adverse events [116]. Thus, inhibition of RTK upstream of mTOR leads to rebound activation of AKT [117]; conversely, inhibition of AKT activity initiates FOXO-dependent transcription and RTK activation [118]. Although blocking PI3K activity reduces AKT activation, it also leads to enhanced MAPK signaling [119]. In conclusion, this evidence is a good theoretical basis for studying dual inhibitors of the PI3K/AKT/mTOR pathway, which can control the activation of the target pathway and respond to its feedback loop, thereby preventing or delaying resistance. For example, combined mTOR and AKT inhibitors have shown synergistic efficacy in allograft models of basal-like patient origin [120]. A number of PI3K/mTOR inhibitors continue to be developed clinically, including gedatolisib (PF-05212384), which has therapeutic activity in breast cancer and acceptable safety and tumorigenic activity [121].
5.6.2. Targeting of MAPK Signaling Pathway
Excessive activation of MAPK is involved in abnormal proliferation and apoptosis of cancer cells, which in turn contributes to TNBC malignancy. Ras and Raf mutations are infrequent in TNBC; instead, activation of MAPK signaling pathways is often thought to be caused by multiple mechanisms of upstream receptor tyrosine kinase activation or by activation or mutation of upstream proteins, such as PI3K/AKT/mTOR and related proteins [122]. Previous studies have shown that flutamide and C1-1040 have synergistic effects in the trastuzumab model, causing a decrease in ERK phosphorylation levels during combination therapy with trastuzumab [123]. One study found that ERK inhibitor in combination with Forskoli increased the sensitivity of TNBC cells to adriamycin [124]. In addition, PAD1 is used to treat metastatic breast cancer by regulating MEK1–ERKl/2–MMP2 signaling in TNBC [125]. A growing body of preclinical evidence supports targeting the Ras/MAPK cell signaling pathway in TNBC subtypes, although large genomic surveys (TCGA public database) suggest that typical mutations in this pathway are rare [126,127]. Because of the early spread of TNBC, targeted treatment in the neoadjuvant setting may offer the effective therapeutic punch needed to eliminate micrometastatic disease and reduce mortality.
5.6.3. Targeting the JAK2/STAT3 Signaling Pathway
In patients with TNBC, JAK1, and JAK2 are usually overexpressed. The JAK/STAT pathway is involved in important biological processes such as cell proliferation, differentiation, apoptosis, and immune regulation. Phosphorylated STAT3 is found in more than 50% of breast tumors and is associated with invasive phenotypes and poor prognosis [128]. Previous studies have found that abnormalities in the IL-6/JAK2/STAT3 pathway play a crucial role in TNBC [129]. This suggests JAK2/STAT3 to be a potential therapeutic target. The JAK1/2 inhibitor ruxolitinib is now approved for the treatment of myelofibrosis and is expected to improve the treatment of TNBC as a single-targeted drug [130].
5.6.4. Targeting of the Src Signaling Pathway
Src tyrosine kinase is overexpressed in TNBC and is involved in disease progression. Preclinical studies have shown that the combination of Dasatinib and cisplatin, a combination of Src, c-kit, and PDGFRβ, can inhibit the growth of basal-like tumor cells or TNBC tumor cells [131,132,133]. In a phase II clinical trial using Dasatinib, anthracyclines, and paclitaxel in combination for treatment of metastatic TNBC, disease control rates were found to be 9.3%, partial response rates were 4.3%, and progression-free survival was around 8.3 weeks [100,101]. (Figure 2)
6. Immune Checkpoint Blockade
The tumor microenvironment is important for TNBC to achieve its biological behavior. It is not only the battlefield of immune surveillance and immune escape between tumor cells and the organism, but also the site where many prognosis-related effector cells, cytokines, and inflammatory mediators function, and the site of host immune killing and tumor immune tolerance [134,135]. Immune checkpoint inhibitors (ICBs) can inhibit the activity of immune checkpoint molecules to release the activity of T cells and restore the killing effect on tumor cells for antitumor purpose. Currently, there are two main types of ICBs: CTLA-4 receptor inhibitors and PD-1/PD-L1 inhibitors. Expression of PD-L1 on tumor cells and on immune cells surrounding the tumor indicates that an antitumor immune response has originally occurred [136]. High expression of immunosuppressive molecules such as PD-L1, CTLA4, TIM-3, and IDO by cells in the tumor cell microenvironment is one of the mechanisms of immune escape from tumor cells [137,138]. PD-L1 and CTLA-4 are immune checkpoints expressed on the surface of antigen-presenting cells in the initiation and effector phases of T-cell activation, respectively. Previous studies have shown that approximately 50% of PD-L1 transmembrane proteins in breast cancer with androgen receptor (AR)-negative prevalence and TNBC cells express PD-L1, whose receptor is PD-1, and also bind PD-L2 as other ligands, and express CTLA-4. Both may coexpress PIK3A and PTEN, which may allow dual blockade of these pathways for the chance of a more effective tumor response [137,138,139]. Showing that PD-L1 upregulation is more common in basal breast cancer and is associated with a higher T-cell cytotoxic immune response. PD-L1 upregulation is associated with better survival and chemotherapy response. Reactivation of inactive TIL by PD-L1 inhibitors suggests that PD-L1 upregulation may be a promising strategy for breast cancer [140].
7. Conclusions and Future Perspective
In this review, we describe some of the main strategies for addressing the challenges in TNBC treatment. TNBC is a high-risk type of breast cancer with a high invasiveness, malignancy, recurrence rate, and heterogeneity. TNBC has unique molecular biological characteristics, and multiple signaling pathways are involved in the development of the disease. Research on TNBC has been extensive, and both in vitro investigations and clinical trials have yielded good results. However, no targeted drugs have been used in clinical treatment. The use of clinical medications requires more innovation, such as selecting drugs based on medical, genetic, and clinical factors specific to the patient, and making clinical medications more targeted, safe, and effective. There are still many obstacles left to overcome in the area of TNBC-targeted therapy in order to achieve personalized drug treatments for patients with TNBC.
Author Contributions
Conceptualization, Y.-H.C., Y.-D.T.T., H.-Y.L., and P.-Y.C.; writing—original draft preparation, C.-J.L. and Y.-D.T.T.; writing—review and editing, C.-J.L., M.-F.H., and P.-Y.C.; visualization, H.-Y.L. and M.-F.H.; supervision, P.-Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Ministry of Science and Technology (MOST 109-2314-B-442-001 and MOST 109-2314-B-075B-002), National Health Research Institutes (NHRI-109BCCO-MF-202015-01), Show Chwan Memorial Hospital, Taiwan (SRD-109023), and Kaohsiung Veterans General Hospital (VGHKS109-106 and KSVGH110-142).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Toriola, A.T.; Colditz, G.A. Trends in breast cancer incidence and mortality in the United States: Implications for prevention. Breast Cancer Res. Treat. 2013, 138, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Ellison, R.C.; Zhang, Y.; McLennan, C.E.; Rothman, K.J. Exploring the relation of alcohol consumption to risk of breast cancer. Am. J. Epidemiol. 2001, 154, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, K.; Mao, K.; Su, F.; Liu, Q.; Jacobs, L.K. The prognostic value of age for invasive lobular breast cancer depending on estrogen receptor and progesterone receptor-defined subtypes: A NCDB analysis. Oncotarget 2016, 7, 6063–6073. [Google Scholar] [CrossRef] [Green Version]
- Stavros, A.T.; Thickman, D.; Rapp, C.L.; Dennis, M.A.; Parker, S.H.; Sisney, G.A. Solid breast nodules: Use of sonography to distinguish between benign and malignant lesions. Radiology 1995, 196, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Flobbe, K.; Bosch, A.; Kessels, A.H. The additional diagnostic value of ultrasonography in the diagnosis of breast cancer. Arch. Intern. Med. 2003, 163, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishna, S.; Agarwal, S.; Parikh, P.M.; Kaur, K.; Panwar, S.; Sharma, S.; Dey, A.; Saxena, K.K.; Chandra, M.; Sud, S. Role of magnetic resonance imaging in breast cancer management. South Asian J. Cancer 2018, 7, 69–71. [Google Scholar] [CrossRef]
- Bartlett, J.M.; Brookes, C.L.; Robson, T.; van de Velde, C.J.; Billingham, L.J.; Campbell, F.M.; Grant, M.; Hasenburg, A.; Hille, E.T.; Kay, C.; et al. Estrogen receptor and progesterone receptor as predictive biomarkers of response to endocrine therapy: A prospectively powered pathology study in the Tamoxifen and Exemestane Adjuvant Multinational trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1531–1538. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Chu, P.Y.; Yiang, G.T.; Wu, M.Y. The Molecular Mechanism of Epithelial-Mesenchymal Transition for Breast Carcinogenesis. Biomolecules 2019, 9, 476. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef]
- Trotta, L.; Norberg, A.; Taskinen, M.; Beziat, V.; Degerman, S.; Wartiovaara-Kautto, U.; Valimaa, H.; Jahnukainen, K.; Casanova, J.L.; Seppanen, M.; et al. Diagnostics of rare disorders: Whole-exome sequencing deciphering locus heterogeneity in telomere biology disorders. Orphanet J. Rare Dis. 2018, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- Gerratana, L.; Basile, D.; Buono, G.; de Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; de Santo, I.; del Mastro, L.; et al. Androgen receptor in triple negative breast cancer: A potential target for the targetless subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1 and BRCA2—Update and implications on the genetics of breast cancer: A clinical perspective. Clin. Genet. 2014, 85, 1–4. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Wang, Z.; Liu, M.; Qi, Z.; Meng, J.; Sun, J.; Yang, G. Aurora-A: A potential DNA repair modulator. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 2831–2836. [Google Scholar] [CrossRef]
- Park, J.Y.; Zhang, F.; Andreassen, P.R. PALB2: The hub of a network of tumor suppressors involved in DNA damage responses. Biochim. Biophys. Acta 2014, 1846, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, L.; Panchal, S.; Goodwin, P. Prognosis of BRCA-associated breast cancer: A summary of evidence. Breast Cancer Res. Treat. 2010, 119, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, A.J.; Schmidt, M.K.; van’t Veer, L.J.; Tollenaar, R.A.; van Leeuwen, F.E. Worse breast cancer prognosis of BRCA1/BRCA2 mutation carriers: What’s the evidence? A systematic review with meta-analysis. PLoS ONE 2015, 10, e0120189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabb, D.W.; Matsumoto, M.; Chang, D.; You, M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 2004, 63, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, D.L.; Duan, J.J.; Xu, S.L.; Zhang, J.F.; Yang, X.J.; Zhang, X.; Cui, Y.H.; Bian, X.W.; Yu, S.C. ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 2014, 14, 444. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.Y.; Thike, A.A.; Tan, P.H. ALDH1 expression is enriched in breast cancers arising in young women but does not predict outcome. Br. J. Cancer 2013, 109, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Bi, L.R.; Xu, N.; Yang, H.M.; Zhang, H.T.; Ding, Y.; Shi, A.P.; Fan, Z.M. The expression of aldehyde dehydrogenase 1 in invasive primary breast tumors and axillary lymph node metastases is associated with poor clinical prognosis. Pathol. Res. Pract. 2013, 209, 555–561. [Google Scholar] [CrossRef]
- Yoshioka, T.; Umekita, Y.; Ohi, Y.; Souda, M.; Sagara, Y.; Rai, Y.; Tanimoto, A. Aldehyde dehydrogenase 1 expression is a predictor of poor prognosis in node-positive breast cancers: A long-term follow-up study. Histopathology 2011, 58, 608–616. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, e6561. [Google Scholar] [CrossRef]
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009, 100, 1062–1068. [Google Scholar] [CrossRef]
- Resetkova, E.; Reis-Filho, J.S.; Jain, R.K.; Mehta, R.; Thorat, M.A.; Nakshatri, H.; Badve, S. Prognostic impact of ALDH1 in breast cancer: A story of stem cells and tumor microenvironment. Breast Cancer Res. Treat. 2010, 123, 97–108. [Google Scholar] [CrossRef]
- Jacobson, O.; Weiss, I.D. CXCR4 chemokine receptor overview: Biology, pathology and applications in imaging and therapy. Theranostics 2013, 3, 1–2. [Google Scholar] [CrossRef]
- Xu, T.P.; Shen, H.; Liu, L.X.; Shu, Y.Q. The impact of chemokine receptor CXCR4 on breast cancer prognosis: A meta-analysis. Cancer Epidemiol. 2013, 37, 725–731. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: A systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.D.; Panu, L.; Holm, N.T.; Li, B.D.; Johnson, L.W.; Zhang, S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J. Surg. Res. 2010, 159, 689–695. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Liu, G.; Zhu, X.; Chen, Y. High level of CXCR4 in triple-negative breast cancer specimens associated with a poor clinical outcome. Acta Med. Okayama 2013, 67, 369–375. [Google Scholar]
- Parker, C.C.; Kim, R.H.; Li, B.D.; Chu, Q.D. The chemokine receptor CXCR4 as a novel independent prognostic marker for node-positive breast cancer patients. J. Surg. Oncol. 2012, 106, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Jung, W.H.; Koo, J.S. Expression of p16 and pRB in invasive breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8209–8217. [Google Scholar]
- Wang, L.; Tang, L.; Xie, R.; Nie, W.; Chen, L.; Guan, X. p16 promoter hypermethylation is associated with increased breast cancer risk. Mol. Med. Rep. 2012, 6, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.W.; Sohn, J.H.; Kim, D.H.; Choi, Y.J.; Park, Y.L.; Kim, K.; Cho, Y.H.; Pyo, J.S.; Kim, J.H. Overexpressions of Cyclin B1, cdc2, p16 and p53 in human breast cancer: The clinicopathologic correlations and prognostic implications. Yonsei Med. J. 2011, 52, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Vallian, S.; Sedaghat, M.; Nassiri, I.; Frazmand, A. Methylation status of p16 INK4A tumor suppressor gene in Iranian patients with sporadic breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 991–996. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Lee, J.J.; Ko, E.; Cho, J.; Park, H.Y.; Lee, J.E.; Nam, S.J.; Kim, D.H.; Cho, E.Y. Methylation and Immunoexpression of p16(INK4a) Tumor Suppressor Gene in Primary Breast Cancer Tissue and Their Quantitative p16(INK4a) Hypermethylation in Plasma by Real-Time PCR. Korean J. Pathol. 2012, 46, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, X.; Zhang, J. Inhibition of Breast Tumor Cell Growth by Ectopic Expression of p16/INK4A via Combined Effects of Cell Cycle Arrest, Senescence and Apoptotic Induction, and Angiogenesis Inhibition. J. Cancer 2012, 3, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subhawong, A.P.; Subhawong, T.; Nassar, H.; Kouprina, N.; Begum, S.; Vang, R.; Westra, W.H.; Argani, P. Most basal-like breast carcinomas demonstrate the same Rb−/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am. J. Surg. Pathol. 2009, 33, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sugianto, J.; Sarode, V.; Peng, Y. Ki-67 expression is increased in p16-expressing triple-negative breast carcinoma and correlates with p16 only in p53-negative tumors. Hum. Pathol. 2014, 45, 802–809. [Google Scholar] [CrossRef]
- Karray-Chouayekh, S.; Baccouche, S.; Khabir, A.; Sellami-Boudawara, T.; Daoud, J.; Frikha, M.; Jlidi, R.; Gargouri, A.; Mokdad-Gargouri, R. Prognostic significance of p16INK4a/p53 in Tunisian patients with breast carcinoma. Acta Histochem. 2011, 113, 508–513. [Google Scholar] [CrossRef]
- Arima, Y.; Hayashi, N.; Hayashi, H.; Sasaki, M.; Kai, K.; Sugihara, E.; Abe, E.; Yoshida, A.; Mikami, S.; Nakamura, S.; et al. Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Int. J. Cancer 2012, 130, 2568–2579. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Reitnauer, P.J.; Morrell, D.; Chase, C.L. Breast and other cancers in families with ataxia-telangiectasia. N. Engl. J. Med. 1987, 316, 1289–1294. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Wooster, R.; Weber, B.L. Breast cancer genetics: What we know and what we need. Nat. Med. 2001, 7, 552–556. [Google Scholar] [CrossRef]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef]
- Rondeau, S.; Vacher, S.; De Koning, L.; Briaux, A.; Schnitzler, A.; Chemlali, W.; Callens, C.; Lidereau, R.; Bieche, I. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br. J. Cancer 2015, 112, 1059–1066. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkas, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Domagala, P.; Jakubowska, A.; Jaworska-Bieniek, K.; Kaczmarek, K.; Durda, K.; Kurlapska, A.; Cybulski, C.; Lubinski, J. Prevalence of Germline Mutations in Genes Engaged in DNA Damage Repair by Homologous Recombination in Patients with Triple-Negative and Hereditary Non-Triple-Negative Breast Cancers. PLoS ONE 2015, 10, e0130393. [Google Scholar] [CrossRef]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Kuo, W.H.; Huang, A.C.; Lu, Y.S.; Lin, C.H.; Kuo, S.H.; Wang, M.Y.; Liu, C.Y.; Cheng, F.T.; Yeh, M.H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [Green Version]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; Tsao, M.S.; Macpherson, D.; Suzuki, A.; Chapman, W.B.; Mak, T.W. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 2000, 60, 3605–3611. [Google Scholar]
- Li, S.; Shen, Y.; Wang, M.; Yang, J.; Lv, M.; Li, P.; Chen, Z. Loss of PTEN expression in breast cancer: Association with clinicopathological characteristics and prognosis. Oncotarget 2017, 8, 32043–32054. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, E.; Oki, E.; Kimura, Y.; Yamanaka, T.; Egashira, A.; Nishida, K.; Koga, T.; Morita, M.; Kakeji, Y.; Maehara, Y. Coexistence of the loss of heterozygosity at the PTEN locus and HER2 overexpression enhances the Akt activity thus leading to a negative progesterone receptor expression in breast carcinoma. Breast Cancer Res. Treat. 2007, 101, 249–257. [Google Scholar] [CrossRef]
- Yndestad, S.; Austreid, E.; Knappskog, S.; Chrisanthar, R.; Lilleng, P.K.; Lonning, P.E.; Eikesdal, H.P. High PTEN gene expression is a negative prognostic marker in human primary breast cancers with preserved p53 function. Breast Cancer Res. Treat. 2017, 163, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Vargas, A.C.; Reis-Filho, J.S.; Lakhani, S.R. Phenotype-genotype correlation in familial breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 27–40. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.; Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Anton, A.; Lluch, A.; et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. J. Clin. Oncol. 2005, 23, 4265–4274. [Google Scholar] [CrossRef]
- Chick, R.C.; Clifton, G.T.; Hale, D.F.; Vreeland, T.J.; Hickerson, A.T.; Kemp Bohan, P.M.; McCarthy, P.M.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; et al. Subgroup analysis of nelipepimut-S plus GM-CSF combined with trastuzumab versus trastuzumab alone to prevent recurrences in patients with high-risk, HER2 low-expressing breast cancer. Clin. Immunol. 2021, 225, 108679. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab versus Trastuzumab to Prevent Recurrences in Patients with High-Risk HER2 Low-Expressing Breast Cancer. Clin. Cancer Res. 2020, 26, 2515–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigtlaender, M.; Schneider-Merck, T.; Trepel, M. Lapatinib. Recent Results Cancer Res. 2018, 211, 19–44. [Google Scholar] [PubMed]
- Loibl, S.; Majewski, I.; Guarneri, V.; Nekljudova, V.; Holmes, E.; Bria, E.; Denkert, C.; Schem, C.; Sotiriou, C.; Loi, S.; et al. PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: Pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab. Ann. Oncol. 2016, 27, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Toh, U.; Tanaka, M.; Saimura, M.; Okumura, Y.; Saito, T.; Tanaka, T.; Teraoka, M.; Shimada, K.; Katayama, K.; et al. Role of HER2-Related Biomarkers (HER2, p95HER2, HER3, PTEN, and PIK3CA) in the Efficacy of Lapatinib plus Capecitabine in HER2-Positive Advanced Breast Cancer Refractory to Trastuzumab. Oncology 2017, 93, 51–61. [Google Scholar] [CrossRef]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Copeland, A.C.; Anders, C.K. Dual HER2-Targeting in the Adjuvant Setting: Where We Have Been and Where We Are Going. Oncology 2018, 32, 483–487. [Google Scholar]
- Deeks, E.D. Neratinib: First Global Approval. Drugs 2017, 77, 1695–1704. [Google Scholar] [CrossRef]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer 2020, 19, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Chellappan, D.K.; Chellian, J.; Ng, Z.Y.; Sim, Y.J.; Theng, C.W.; Ling, J.; Wong, M.; Foo, J.H.; Yang, G.J.; Hang, L.Y.; et al. The role of pazopanib on tumour angiogenesis and in the management of cancers: A review. Biomed. Pharm. 2017, 96, 768–781. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Ruggieri, S.; Tamma, R.; Simone, G.; Mangia, A. Angiogenesis and Antiangiogenesis in Triple-Negative Breast cancer. Transl. Oncol. 2016, 9, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Labanca, V.; Bertolini, F. A Combinatorial Investigation of the Response to Anti-angiogenic Therapy in Breast Cancer: New Strategies for Patient Selection and Opportunities for Reconsidering Anti-VEGF, Anti-PI3K and Checkpoint Inhibition. EBioMedicine 2016, 10, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Lobo, C.; Lopes, G.; Baez, O.; Castrellon, A.; Ferrell, A.; Higgins, C.; Hurley, E.; Hurley, J.; Reis, I.; Richman, S.; et al. Final results of a phase II study of nab-paclitaxel, bevacizumab, and gemcitabine as first-line therapy for patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2010, 123, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.; Valero, V.; Tiangco, B.; Dakhil, S.; Brize, A.; Rugo, H.S.; Rivera, R.; Duenne, A.; Bousfoul, N.; Yardley, D.A. Second-line bevacizumab-containing therapy in patients with triple-negative breast cancer: Subgroup analysis of the RIBBON-2 trial. Breast Cancer Res. Treat. 2012, 133, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Liposits, G.; Loh, K.P.; Soto-Perez-de-Celis, E.; Dumas, L.; Battisti, N.M.L.; Kadambi, S.; Baldini, C.; Banerjee, S.; Lichtman, S.M. PARP inhibitors in older patients with ovarian and breast cancer: Young International Society of Geriatric Oncology review paper. J. Geriatr. Oncol. 2019, 10, 337–345. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef]
- Johnson, N.; Shapiro, G.I. Cyclin-dependent kinases (cdks) and the DNA damage response: Rationale for cdk inhibitor-chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin. Targets 2010, 14, 1199–1212. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Han, H.S.; Dieras, V.; Robson, M.; Palacova, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Seong, R.K.; Choi, Y.K.; Shin, O.S. MDA7/IL-24 is an anti-viral factor that inhibits influenza virus replication. J. Microbiol. 2016, 54, 695–700. [Google Scholar] [CrossRef]
- Dent, R.A.; Lindeman, G.J.; Clemons, M.; Wildiers, H.; Chan, A.; McCarthy, N.J.; Singer, C.F.; Lowe, E.S.; Watkins, C.L.; Carmichael, J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013, 15, R88. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.M.; Ansari, J.A.K.; El-Aziz, N.M.A.; Abozeed, W.N.; Warith, A.M.A.; Alsaleh, K.; Nabholtz, J.M. Triple Negative Breast Cancer: A Tale of Two Decades. Anticancer Agents Med. Chem. 2017, 17, 491–499. [Google Scholar] [CrossRef]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Huober, J.; Fasching, P.A.; Hanusch, C.; Rezai, M.; Eidtmann, H.; Kittel, K.; Hilfrich, J.; Schwedler, K.; Blohmer, J.U.; Tesch, H.; et al. Neoadjuvant chemotherapy with paclitaxel and everolimus in breast cancer patients with non-responsive tumours to epirubicin/cyclophosphamide (EC) ± bevacizumab—Results of the randomised GeparQuinto study (GBG 44). Eur. J. Cancer 2013, 49, 2284–2293. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Akcakanat, A.; Liu, S.; Green, M.C.; Murray, J.L.; Chen, H.; Palla, S.L.; Koenig, K.B.; Brewster, A.M.; Valero, V.; et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann. Oncol. 2014, 25, 1122–1127. [Google Scholar] [CrossRef]
- Rodon, J.; Perez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharm. 2018, 82, 285–298. [Google Scholar] [CrossRef]
- Patani, N.; Douglas-Jones, A.; Mansel, R.; Jiang, W.; Mokbel, K. Tumour suppressor function of MDA-7/IL-24 in human breast cancer. Cancer Cell Int. 2010, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.C.; Gadi, V. Dasatinib in breast cancer: Src-ing for response in all the wrong kinases. Ann. Transl. Med. 2018, 6 (Suppl. S1), S60. [Google Scholar] [CrossRef]
- Morris, P.G.; Rota, S.; Cadoo, K.; Zamora, S.; Patil, S.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; et al. Phase II Study of Paclitaxel and Dasatinib in Metastatic Breast Cancer. Clin. Breast Cancer 2018, 18, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef]
- Moulder, S.L.; Borges, V.F.; Baetz, T.; McSpadden, T.; Fernetich, G.; Murthy, R.K.; Chavira, R.; Guthrie, K.; Barrett, E.; Chia, S.K. Phase I Study of ONT-380, a HER2 Inhibitor, in Patients with HER2+—Advanced Solid Tumors, with an Expansion Cohort in HER2+ Metastatic Breast Cancer (MBC). Clin. Cancer Res. 2017, 23, 3529–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escriva-de-Romani, S.; Arumi, M.; Bellet, M.; Saura, C. HER2-positive breast cancer: Current and new therapeutic strategies. Breast 2018, 39, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Lee, K.W.; Oh, D.Y.; Lee, J.S.; Im, S.A.; Kim, D.W.; Han, S.W.; Kim, Y.J.; Kim, T.Y.; Kim, J.H.; et al. Phase 1 Studies of Poziotinib, an Irreversible Pan-HER Tyrosine Kinase Inhibitor in Patients with Advanced Solid Tumors. Cancer Res. Treat. 2018, 50, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsdorf, M.; Ingvar, C.; Jorgensen, L.; Tuxen, M.K.; Jakobsen, E.H.; Saetersdal, A.; Kimper-Karl, M.L.; Kroman, N.; Balslev, E.; Ejlertsen, B. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res. Treat. 2011, 126, 463–470. [Google Scholar] [CrossRef]
- Schuler, M.; Awada, A.; Harter, P.; Canon, J.L.; Possinger, K.; Schmidt, M.; de Greve, J.; Neven, P.; Dirix, L.; Jonat, W.; et al. A phase II trial to assess efficacy and safety of afatinib in extensively pretreated patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Layman, R.M.; Ruppert, A.S.; Lynn, M.; Mrozek, E.; Ramaswamy, B.; Lustberg, M.B.; Wesolowski, R.; Ottman, S.; Carothers, S.; Bingman, A.; et al. Severe and prolonged lymphopenia observed in patients treated with bendamustine and erlotinib for metastatic triple negative breast cancer. Cancer Chemother. Pharm. 2013, 71, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Meattini, I.; Livi, L.; Saieva, C.; Franceschini, D.; Scotti, V.; Mangoni, M.; Loi, M.; di Brina, L.; Zei, G.; Bonomo, P.; et al. Prognostic role of human epidermal growth factor receptor 2 status in premenopausal early breast cancer treated with adjuvant tamoxifen. Clin. Breast Cancer 2013, 13, 247–253. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Shah, A.N.; Santa-Maria, C.A.; Cruz, M.R.; Mahalingam, D.; Carneiro, B.A.; Chae, Y.K.; Cristofanilli, M.; Gradishar, W.J.; Giles, F.J. Targeting Epidermal Growth Factor Receptor in triple negative breast cancer: New discoveries and practical insights for drug development. Cancer Treat. Rev. 2017, 53, 111–119. [Google Scholar] [CrossRef]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Basho, R.K.; Gilcrease, M.; Murthy, R.K.; Helgason, T.; Karp, D.D.; Meric-Bernstam, F.; Hess, K.R.; Herbrich, S.M.; Valero, V.; Albarracin, C.; et al. Targeting the PI3K/AKT/mTOR Pathway for the Treatment of Mesenchymal Triple-Negative Breast Cancer: Evidence from a Phase 1 Trial of mTOR Inhibition in Combination with Liposomal Doxorubicin and Bevacizumab. JAMA Oncol. 2017, 3, 509–515. [Google Scholar] [CrossRef]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel with or without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Guo, Z.; Luo, J.; Ellis, M.J.; Ma, C.X. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol. Cancer 2013, 12, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Chia, K.M.; Liu, J. Synergy between inhibitors of androgen receptor and MEK has therapeutic implications in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011, 13, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illiano, M.; Sapio, L.; Salzillo, A.; Capasso, L.; Caiafa, I.; Chiosi, E.; Spina, A.; Naviglio, S. Forskolin improves sensitivity to doxorubicin of triple negative breast cancer cells via Protein Kinase A-mediated ERK1/2 inhibition. Biochem. Pharm. 2018, 152, 104–113. [Google Scholar] [CrossRef]
- Qin, H.; Liu, X.; Li, F.; Miao, L.; Li, T.; Xu, B.; An, X.; Muth, A.; Thompson, P.R.; Coonrod, S.A.; et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. 2017, 409, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Luo, Q.; Xiao, Y.; Li, H.; Kong, L.; Ren, G. The implication from RAS/RAF/ERK signaling pathway increased activation in epirubicin treated triple negative breast cancer. Oncotarget 2017, 8, 108249–108260. [Google Scholar] [CrossRef] [Green Version]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Barni, S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 223–232. [Google Scholar] [CrossRef]
- Montero, J.C.; Esparis-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocana, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Haga, Y.; Higashisaka, K.; Yang, L.; Sekine, N.; Lin, Y.; Tsujino, H.; Nagano, K.; Tsutsumi, Y. Inhibition of Akt/mTOR pathway overcomes intrinsic resistance to dasatinib in triple-negative breast cancer. Biochem. Biophys. Res. Commun. 2020, 533, 672–678. [Google Scholar] [CrossRef]
- Kim, E.M.; Mueller, K.; Gartner, E.; Boerner, J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J. Surg. Res. 2013, 185, 231–239. [Google Scholar] [CrossRef]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef]
- Li, C.J.; Chen, H.M.; Lai, J.C. Diagnostic, Prognostic, and Predictive Biomarkers in Breast Cancer. J. Oncol. 2020, 2020, 1835691. [Google Scholar] [CrossRef]
- Marinelli, D.; Mazzotta, M.; Pizzuti, L.; Krasniqi, E.; Gamucci, T.; Natoli, C.; Grassadonia, A.; Tinari, N.; Tomao, S.; Sperduti, I.; et al. Neoadjuvant Immune-Checkpoint Blockade in Triple-Negative Breast Cancer: Current Evidence and Literature-Based Meta-Analysis of Randomized Trials. Cancers 2020, 12, 2497. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Lin, L.T.; Hou, M.F.; Chu, P.Y. PDL1/PD1 blockade in breast cancer: The immunotherapy era (Review). Oncol. Rep. 2021, 45, 5–12. [Google Scholar] [CrossRef]
- Botti, G.; Collina, F.; Scognamiglio, G.; Rao, F.; Peluso, V.; De Cecio, R.; Piezzo, M.; Landi, G.; De Laurentiis, M.; Cantile, M.; et al. Programmed Death Ligand 1 (PD-L1) Tumor Expression Is Associated with a Better Prognosis and Diabetic Disease in Triple Negative Breast Cancer Patients. Int. J. Mol. Sci. 2017, 18, 459. [Google Scholar] [CrossRef]
- Mori, H.; Kubo, M.; Yamaguchi, R.; Nishimura, R.; Osako, T.; Arima, N.; Okumura, Y.; Okido, M.; Yamada, M.; Kai, M.; et al. The combination of PD-L1 expression and decreased tumor-infiltrating lymphocytes is associated with a poor prognosis in triple-negative breast cancer. Oncotarget 2017, 8, 15584–15592. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Adelaide, J.; Chaffanet, M.; Ali, H.R.; Viens, P.; Caldas, C.; Birnbaum, D.; Bertucci, F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015, 6, 5449–5464. [Google Scholar] [CrossRef] [Green Version]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.I.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2018, 25, 34–42. [Google Scholar] [CrossRef]
Figure 1.
Association of different TNBC subtypes. TNBC gene expression subtypes are related to unique molecular characteristics and different TNBCs. Subtypes of TNBC: BL1, basal-like 1; BL2, basal-like 2; IM, immunomodulatory; ML, mesenchymal-like; MSL, mesenchymal stem-like; LAR, luminal androgen receptor.
Figure 1.
Association of different TNBC subtypes. TNBC gene expression subtypes are related to unique molecular characteristics and different TNBCs. Subtypes of TNBC: BL1, basal-like 1; BL2, basal-like 2; IM, immunomodulatory; ML, mesenchymal-like; MSL, mesenchymal stem-like; LAR, luminal androgen receptor.

Figure 2.
Schematic drawing presents the detail signaling pathways of breast carcinogenesis. All signaling pathways represented in this diagram have been found to be operative in the breast cancer cell context, whereas evidence to date supports a predominant role of HER2 in BRCA1/2 signaling and EGFR–PARP cross-talk in breast cancer cells. CR: cytokine receptor.
Figure 2.
Schematic drawing presents the detail signaling pathways of breast carcinogenesis. All signaling pathways represented in this diagram have been found to be operative in the breast cancer cell context, whereas evidence to date supports a predominant role of HER2 in BRCA1/2 signaling and EGFR–PARP cross-talk in breast cancer cells. CR: cytokine receptor.


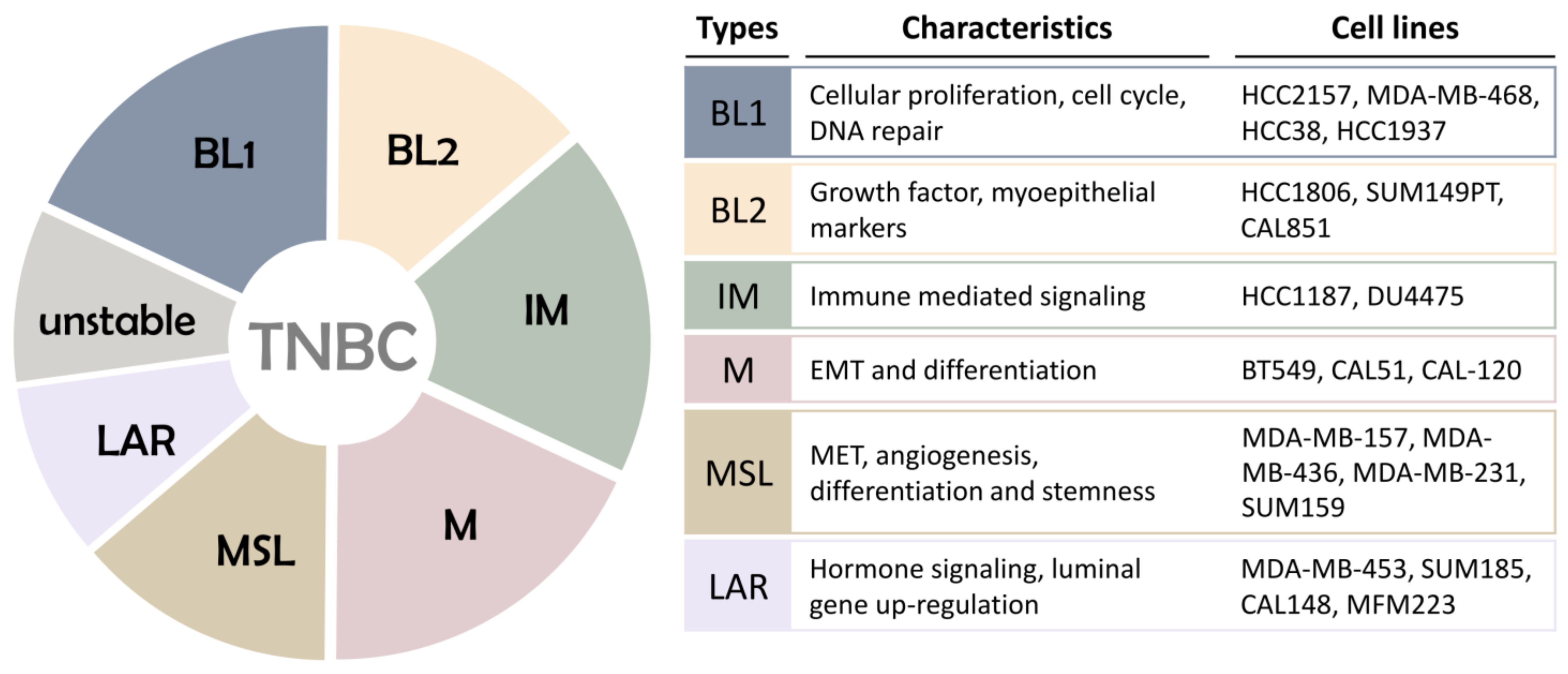{kind=link}
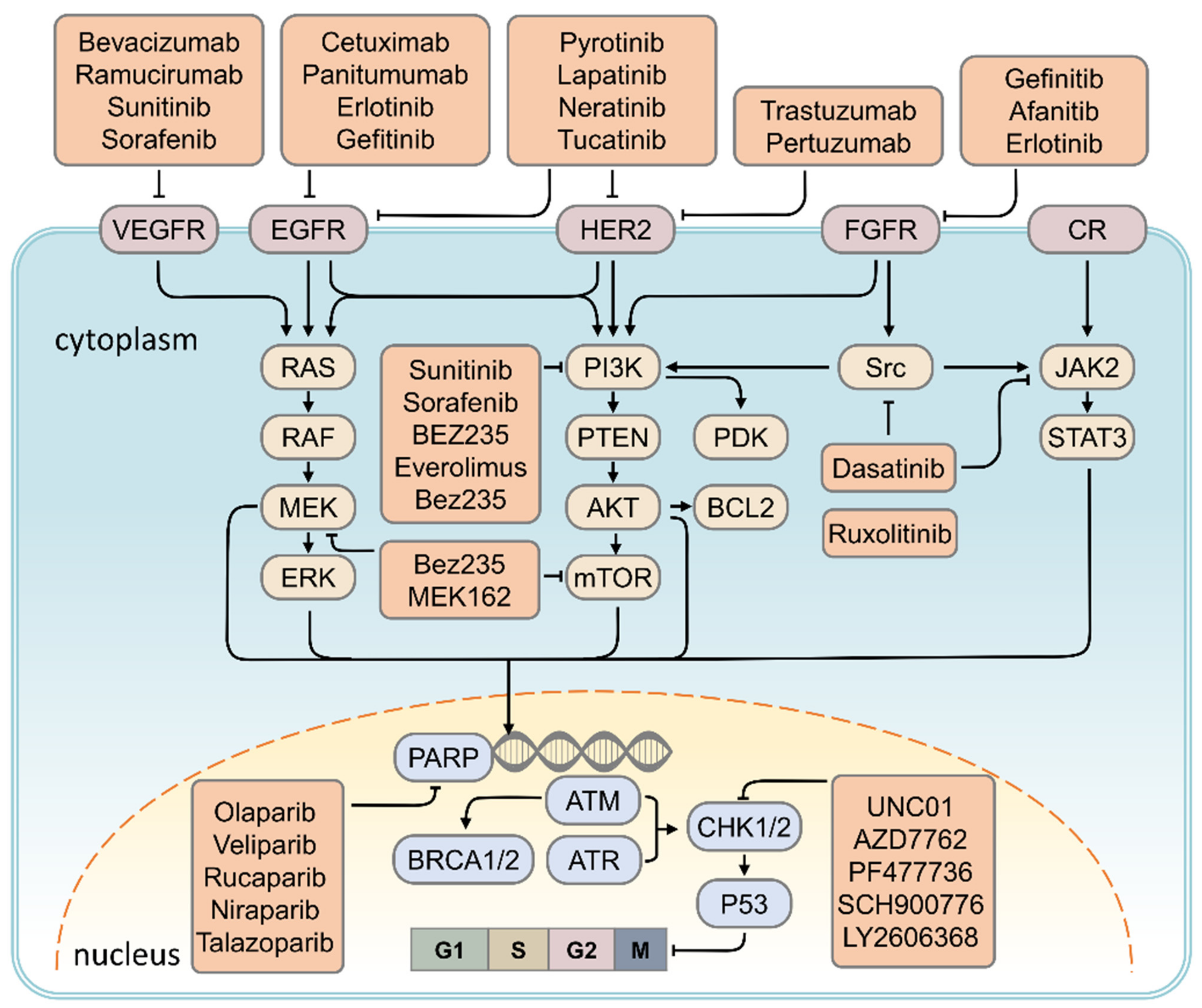{kind=link}
Table 1.
BI-RADS mammographic assessment categories.
| Category | Management | Risk of Cancer |
|---|---|---|
| Category 0: Need additional evaluation | Re-evaluation | |
| Category 1: Negative finding | Routine screening | Essentially 0% |
| Category 2: Benign lesion | Routine screening | Essentially 0% |
| Category 3: Probably benign lesion | Short-interval follow up | 0–2% |
| Category 4A: Low suspicion of malignancy | Tissue diagnosis | 2–10% |
| Category 4B: Moderate suspicion of malignancy | Tissue diagnosis | 10–50% |
| Category 4C: High suspicion of malignancy | Tissue diagnosis | 50–95% |
| Category 5: Highly suggestive of malignancy | Tissue diagnosis | ≥95% |
| Category 6: Proven malignancy | Surgical excision | - |
Table 2.
TNM system of breast cancer.
| When T | When N | When M | Clinical Stage |
|---|---|---|---|
| Tis | N0 | M0 | 0 |
| T1 | N0 | M0 | IA |
| T0 | N1mi | M0 | IB |
| T1 | N1mi | M0 | IB |
| T0 | N1 | M0 | IIA |
| T1 | N1 | M0 | IIA |
| T2 | N0 | M0 | IIA |
| T2 | N1 | M0 | IIB |
| T3 | N0 | M0 | IIB |
| T0 | N2 | M0 | IIIA |
| T1 | N2 | M0 | IIIA |
| T2 | N2 | M0 | IIIA |
| T3 | N1 | M0 | IIIA |
| T3 | N2 | M0 | IIIA |
| T4 | N0 | M0 | IIIB |
| T4 | N1 | M0 | IIIB |
| T4 | N2 | M0 | IIIB |
| Any T | N3 | M0 | IIIC |
| Any T | Any N | M1 | IV |
N1mi: nodal micrometastases.
Table 3.
Targets and targeted drugs for triple negative breast cancer.
| Signaling Pathway | Target | Target Drugs | Ref. |
|---|---|---|---|
| DNA repair pathway | PARP | Olaparib, Veliparib, Rucaparib, Niraparib, Talazoparib | [93] |
| EGF signaling | EGFR | Cetuximab, Panitumumab, Erlotinib, Gefitinib | [94,95] |
| Angiogenesis pathway | VEGF | Bevacizumab, Ramucirumab, Sunitinib, Sorafenib | [80,95,96] |
| PI3K/AKT/mTOR signaling | PDGF, PI3K, mTOR | Sunitinib, Sorafenib, BEZ235, Everolimus, Bez235 | [96,97] |
| MAPK signaling | ERK, mTOR | Bez235, MEK162 | [98] |
| JAK/STAT signaling | JAK2 | Ruxolitinib | [99] |
| Cell cycle pathway | CHK1 | UNC01, AZD7762, PF477736, SCH900776, LY2606368 | [92] |
| Src tyrosine kinase signaling | Src | Dasatinib | [100,101] |
| Human epidermal growth factor receptor 2 | HER2 | Lapatinib, Neratinib, Tucatinib, Poziotinib, Pyrotinib, Trastuzumab, Pertuzumab | [102,103,104,105,106] |
| Fibroblast growth factor receptor signaling | FGFR | Gefinitib, Afanitib, Erlotinib | [107,108,109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, C.-J.; Tzeng, Y.-D.T.; Chiu, Y.-H.; Lin, H.-Y.; Hou, M.-F.; Chu, P.-Y. Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer. Cancers 2021, 13, 2978. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122978
AMA Style
Li C-J, Tzeng Y-DT, Chiu Y-H, Lin H-Y, Hou M-F, Chu P-Y. Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer. Cancers. 2021; 13(12):2978. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122978
Chicago/Turabian StyleLi, Chia-Jung, Yen-Dun Tony Tzeng, Yi-Han Chiu, Hung-Yu Lin, Ming-Feng Hou, and Pei-Yi Chu. 2021. "Pathogenesis and Potential Therapeutic Targets for Triple-Negative Breast Cancer" Cancers 13, no. 12: 2978. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122978
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.