
Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Macropahge Generation and Culture
2.3. Isolation of Exosomes/EVs
2.4. Nanoparticle Tracking Analysis
2.5. Transmission Electron Microscopy
2.6. Confocal Microscopy
2.7. Flow Cytometry
2.8. Antibodies
2.9. Immunoblotting/Western Blot
2.10. Exosome Shaving
2.11. Xenograft Tumor Generation
2.12. Human Tumor Samples
2.13. Immunohistochemistry
2.14. Mass-Spectrometry
3. Results
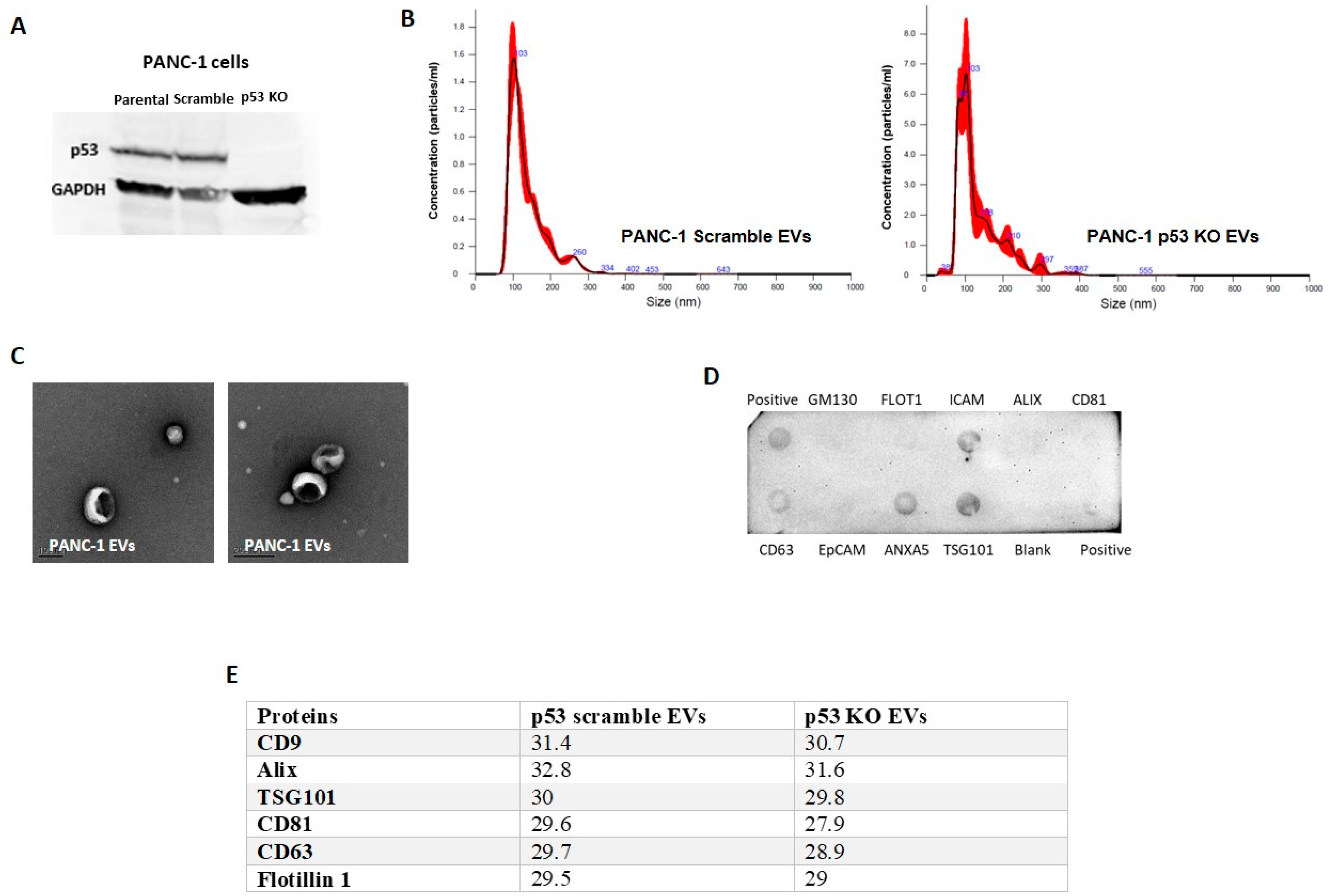
3.1. GOF Mutant p53 Proteins Are Sorted into EVs
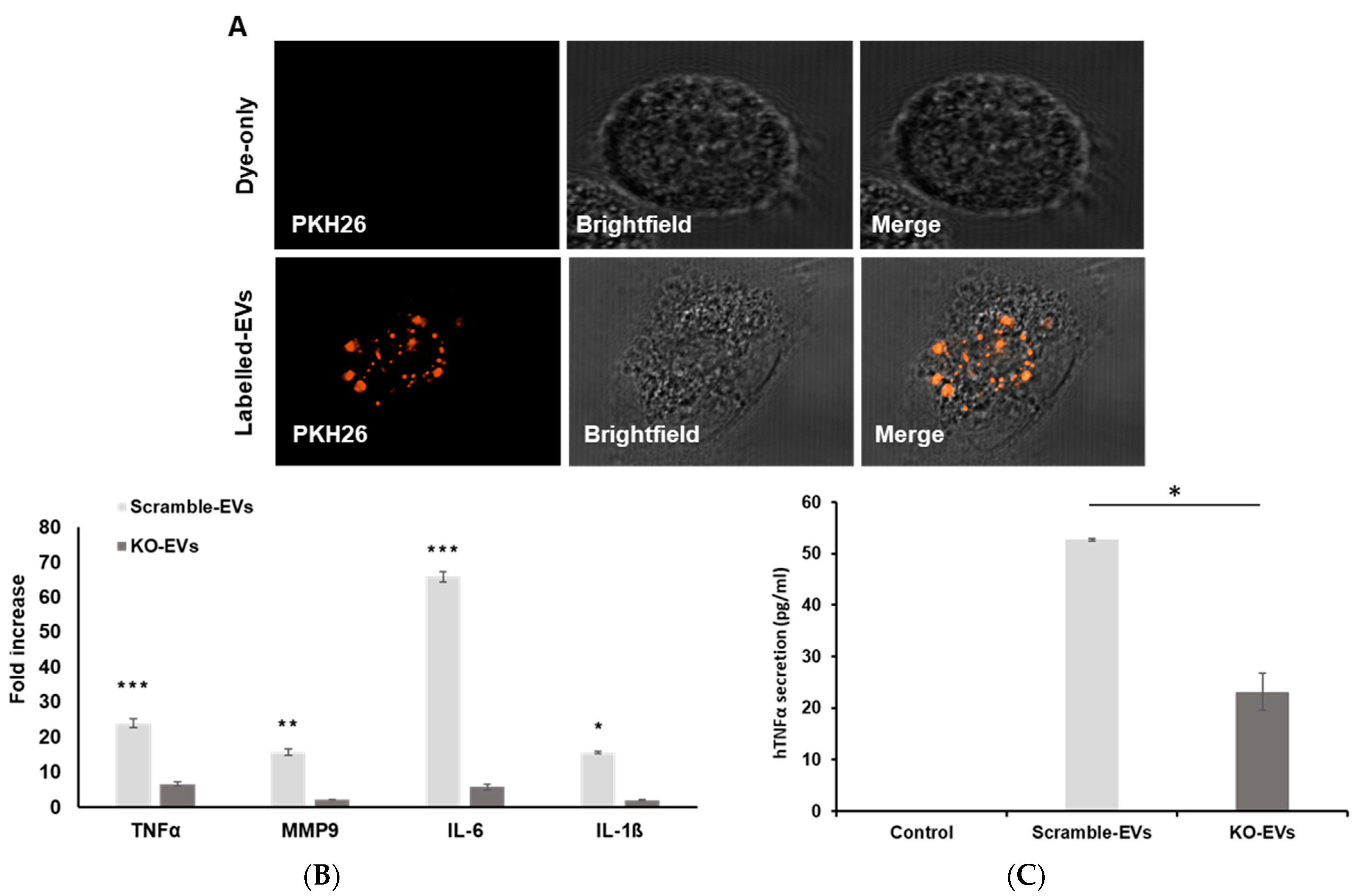
3.2. Vesicular Mutant p53 Proteins Are Taken Up by Neighboring Cancer Cells
3.3. EVs Enriched with GOF Mutant p53 Proteins Can Modulate Macrophage Phenotypes
3.4. Non-Epithelial Staining of Mutant p53 in Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurywchak, P.; Tavormina, J.; Kalluri, R. The emerging roles of exosomes in the modulation of immune responses in cancer. Genome Med. 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanini, F.; Fabbri, M. Cancer-derived exosomic microRNAs shape the immune system within the tumor microenvironment: State of the art. Semin. Cell Dev. Biol. 2017, 67, 23–28. [Google Scholar] [CrossRef]
- Choi, D.; Lee, T.H.; Spinelli, C.; Chennakrishnaiah, S.; D’Asti, E.; Rak, J. Extracellular vesicle communication pathways as regulatory targets of oncogenic transformation. Semin. Cell Dev. Biol. 2017, 67, 11–22. [Google Scholar] [CrossRef]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Haupt, Y.; Blandino, G. Editorial: Human Tumor-Derived p53 Mutants: A Growing Family of Oncoproteins. Front. Oncol. 2016, 6, 170. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.C.; Richardson, D.M.; Hardy, S.G.; Cookson, R.M.; Mackenzie, R.S.; Greenberg, M.R.; Glenn-Porter, B.; Kane, B.G. Computer-based reminder system effectively impacts physician documentation. Am. J. Emerg. Med. 2014, 32, 104–106. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Solomon, H.; Dinowitz, N.; Pateras, I.S.; Cooks, T.; Shetzer, Y.; Molchadsky, A.; Charni, M.; Rabani, S.; Koifman, G.; Tarcic, O.; et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene 2018, 37, 1669–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; MacFarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, C.; Ma, B.; Xu, M.; Xu, S.; Liu, J.; Tian, Y.; Fu, Y.; Luo, Y. Mutant p53 Drives Cancer Metastasis via RCP-Mediated Hsp90alpha Secretion. Cell Rep. 2020, 32, 107879. [Google Scholar] [CrossRef]
- Hussain, S.P.; Raja, K.; Amstad, P.A.; Sawyer, M.; Trudel, L.J.; Wogan, G.N.; Hofseth, L.J.; Shields, P.G.; Billiar, T.R.; Trautwein, C.; et al. Increased p53 mutation load in nontumorous human liver of Wilson disease and hemochromatosis: Oxyradical overload diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 12770–12775. [Google Scholar] [CrossRef] [Green Version]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef]
- Campbell, I.G.; Qiu, W.; Polyak, K.; Haviv, I. Breast-cancer stromal cells with TP53 mutations. N. Engl. J. Med. 2008, 358, 1634–1635. [Google Scholar] [CrossRef]
- Hwang, L.-A.; Phang, B.H.; Liew, O.W.; Iqbal, J.; Koh, X.H.; Koh, X.Y.; Othman, R.; Xue, Y.; Richards, A.M.; Lane, D.P.; et al. Monoclonal Antibodies against Specific p53 Hotspot Mutants as Potential Tools for Precision Medicine. Cell Rep. 2018, 22, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; McGuire, M.H.; Mangala, L.S.; Lee, S.; Stur, E.; Hu, W.; Bayraktar, E.; Villar-Prados, A.; Ivan, C.; Wu, S.Y.; et al. Gain-of-function p53 protein transferred via small extracellular vesicles promotes conversion of fibroblasts to a cancer-associated phenotype. Cell Rep. 2021, 34, 108726. [Google Scholar] [CrossRef]
- Ji, S.; Zhu, L.; Gao, Y.; Zhang, X.; Yan, Y.; Cen, J.; Li, R.; Zeng, R.; Liao, L.; Hou, C.; et al. Baf60b-mediated ATM-p53 activation blocks cell identity conversion by sensing chromatin opening. Cell Res. 2017, 27, 642–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, S.; Pagliarini, R.; Bunz, F.; Rago, C.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. USA 2009, 106, 3964–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappa, G.; Santos, M.F.; Green, T.M.; Karbanová, J.; Hassler, J.; Bai, Y.; Barsky, S.H.; Corbeil, D.; Lorico, A. Nuclear transport of cancer extracellular vesicle-derived biomaterials through nuclear envelope invagination-associated late endosomes. Oncotarget 2017, 8, 14443–14461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, J.; Ingram, A.; Al Saleh, H.A.; Platko, K.; Gabriel, K.; Kapoor, A.; Pinthus, J.; Majeed, F.; Qureshi, T.; Al-Nedawi, K. Nuclear transportation of exogenous epidermal growth factor receptor and androgen receptor via extracellular vesicles. Eur. J. Cancer 2017, 70, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, B.; Cooks, T. Reshaping the tumor microenvironment: Extracellular vesicles as messengers of cancer cells. Carcinogenesis 2020, 41, 1461–1470. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer therapeutic targeting using mutant-p53-specific siRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Pavlakis, E.; Neumann, M.; Stiewe, T. Extracellular Vesicles: Messengers of p53 in Tumor-Stroma Communication and Cancer Metastasis. Int. J. Mol. Sci. 2020, 21, 9648. [Google Scholar] [CrossRef]
- Caponnetto, F.; Manini, I.; Skrap, M.; Palmai-Pallag, T.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D.; Ferrari, E. Size-dependent cellular uptake of exosomes. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1011–1020. [Google Scholar] [CrossRef]
- Huyan, T.; Du, Y.; Huang, Q.; Huang, Q.; Li, Q. Uptake Characterization of Tumor Cell-derived Exosomes by Natural Killer Cells. Iran. J. Public Health 2018, 47, 803–813. [Google Scholar] [PubMed]
- Stary, V.; Unterleuthner, D.; Wolf, B.; Talic, M.; Strobl, J.; Beer, A.; Dolznig, H.; Bergmann, M. Irradiated cancer exosomes promote M1-like polarization of macrophages and enhance their anti-tumoral responses. Eur. J. Cancer 2019, 110, S32–S33. [Google Scholar] [CrossRef]
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization. Cells 2020, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Chow, A.; Zhou, W.; Liu, L.; Fong, M.Y.; Champer, J.; Van Haute, D.; Chin, A.R.; Ren, X.; Gugiu, B.G.; Meng, Z.; et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci. Rep. 2014, 4, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta 2015, 1853, 89–100. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer | Reverse Primer |
|---|---|---|
| TNFα | CACTTTGGAGTGATCGGCCC | AGCTTGAGGGTTTGCTACAAC |
| IL-6 | CACTCACCTCTTCAGAACGAAT | GCTGCTTTCACACATGTTACTC |
| IL-1β | CCTTAGGGTAGTGCTAAGAGGA | AAGTGAGTAGGAGAGGTGAGAG |
| MMP9 | GGCACCACCACAACATCACC | GATACCCGTCTCCGTGCTCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatta, B.; Luz, I.; Krueger, C.; Teo, F.X.; Lane, D.P.; Sabapathy, K.; Cooks, T. Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment. Cancers 2021, 13, 2985. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122985
Bhatta B, Luz I, Krueger C, Teo FX, Lane DP, Sabapathy K, Cooks T. Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment. Cancers. 2021; 13(12):2985. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122985
Chicago/Turabian StyleBhatta, Bibek, Ishai Luz, Christian Krueger, Fanny Xueting Teo, David P. Lane, Kanaga Sabapathy, and Tomer Cooks. 2021. "Cancer Cells Shuttle Extracellular Vesicles Containing Oncogenic Mutant p53 Proteins to the Tumor Microenvironment" Cancers 13, no. 12: 2985. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13122985