
Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethics Statement
2.3. Ralbp1 Knockout Mice
2.4. Generation of Rlip-MMTV-Erbb2 and Rlip-MMTV-PyVT GEM Mice
2.5. Genotyping
2.6. Tumor Growth and Tissue Processing
2.7. Dosing of Rlip Antisense Locked Nucleic Acid (Rlip-LNA)
2.8. Determination of mRNA Levels by Real-Time Polymerase Chain Reaction
2.9. Assessment of Angiogenesis, Proliferation, and Apoptotic Signaling
2.10. Statistical Analysis
3. Results
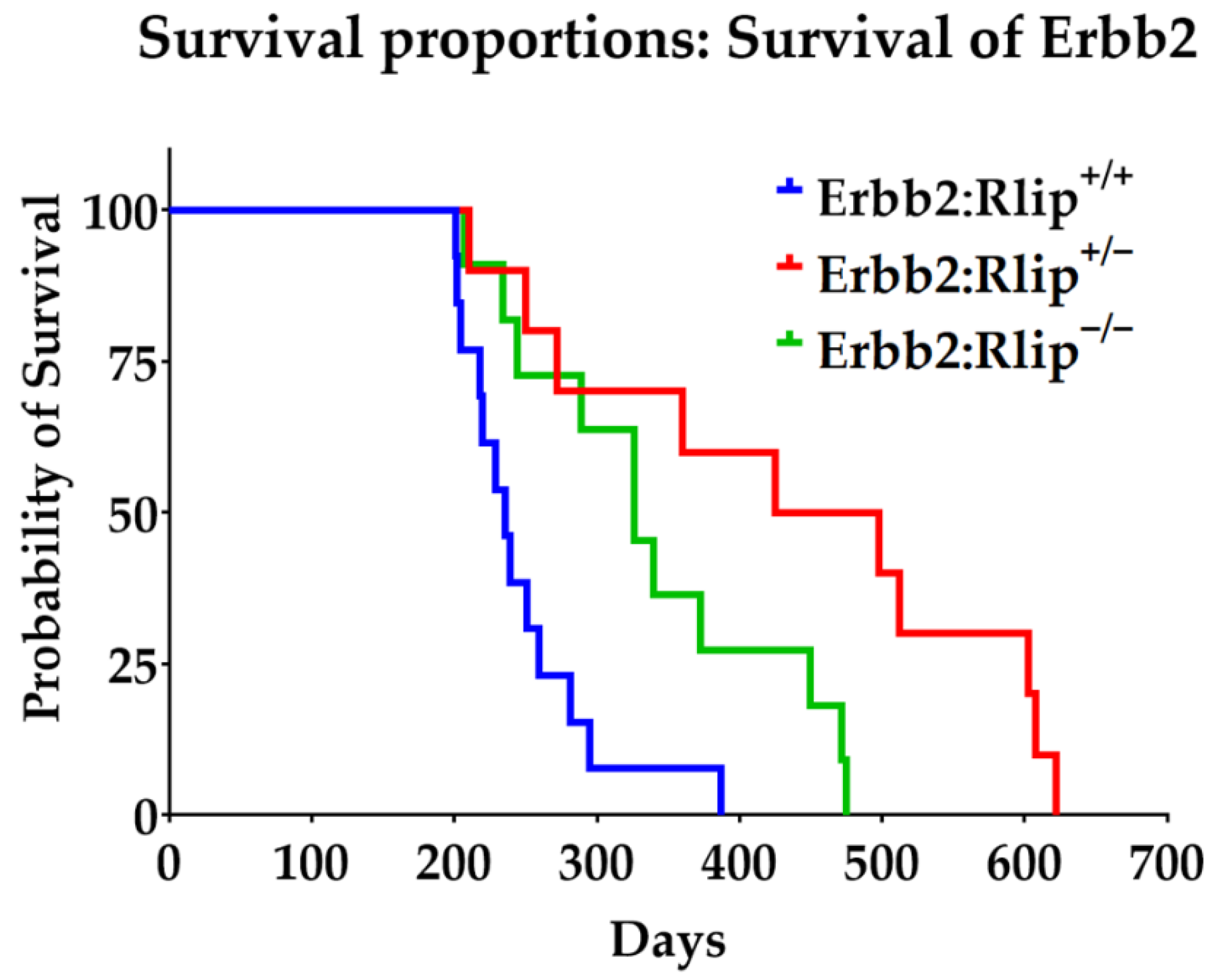
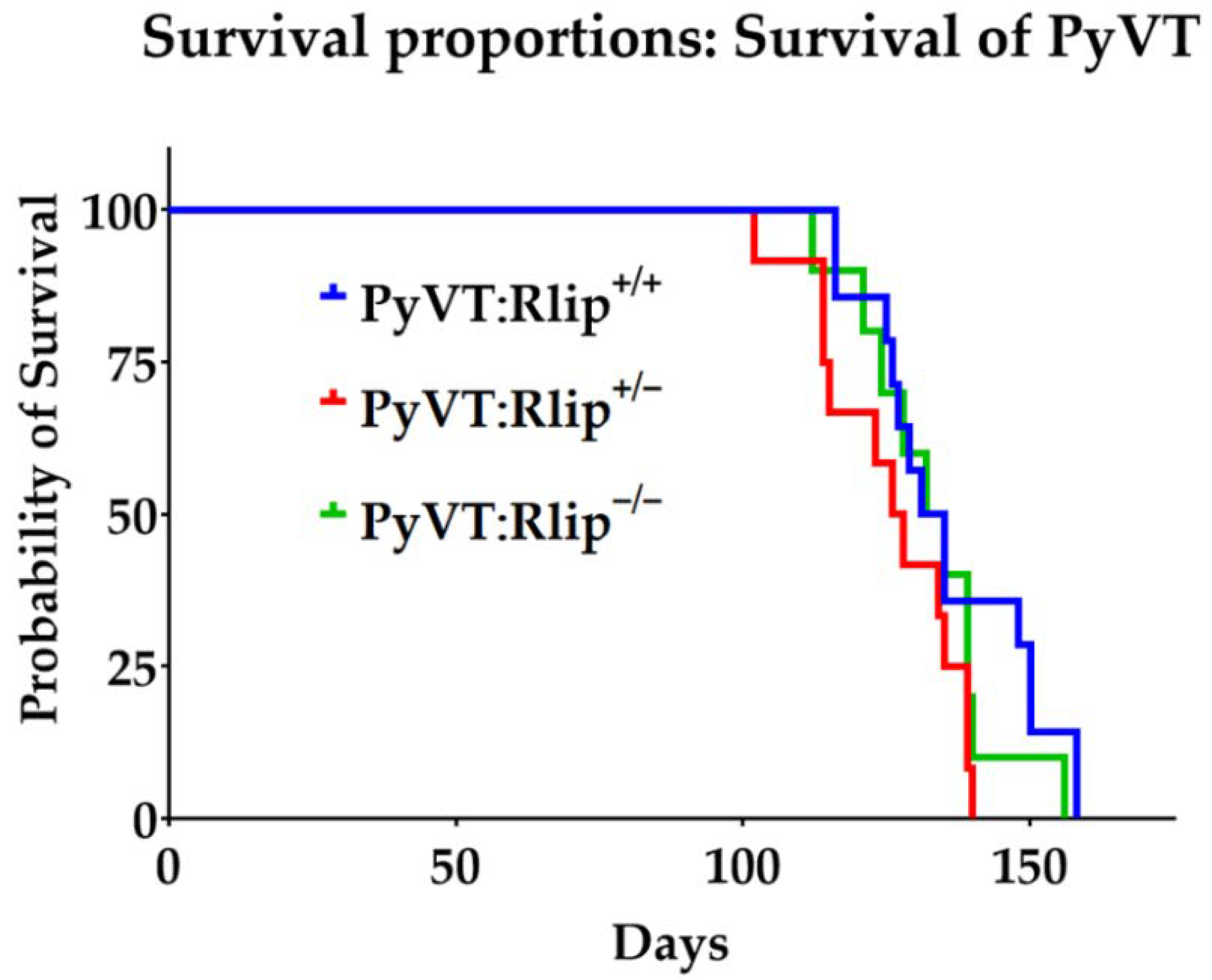
3.1. Effects of Rlip Depletion on Spontaneous Tumor Development
3.2. Congenital Rlip Deficiency Prevents Lung Metastasis
3.3. Effect of Rlip Depletion on Key Tumor Proteins Involved in Progression and Signaling
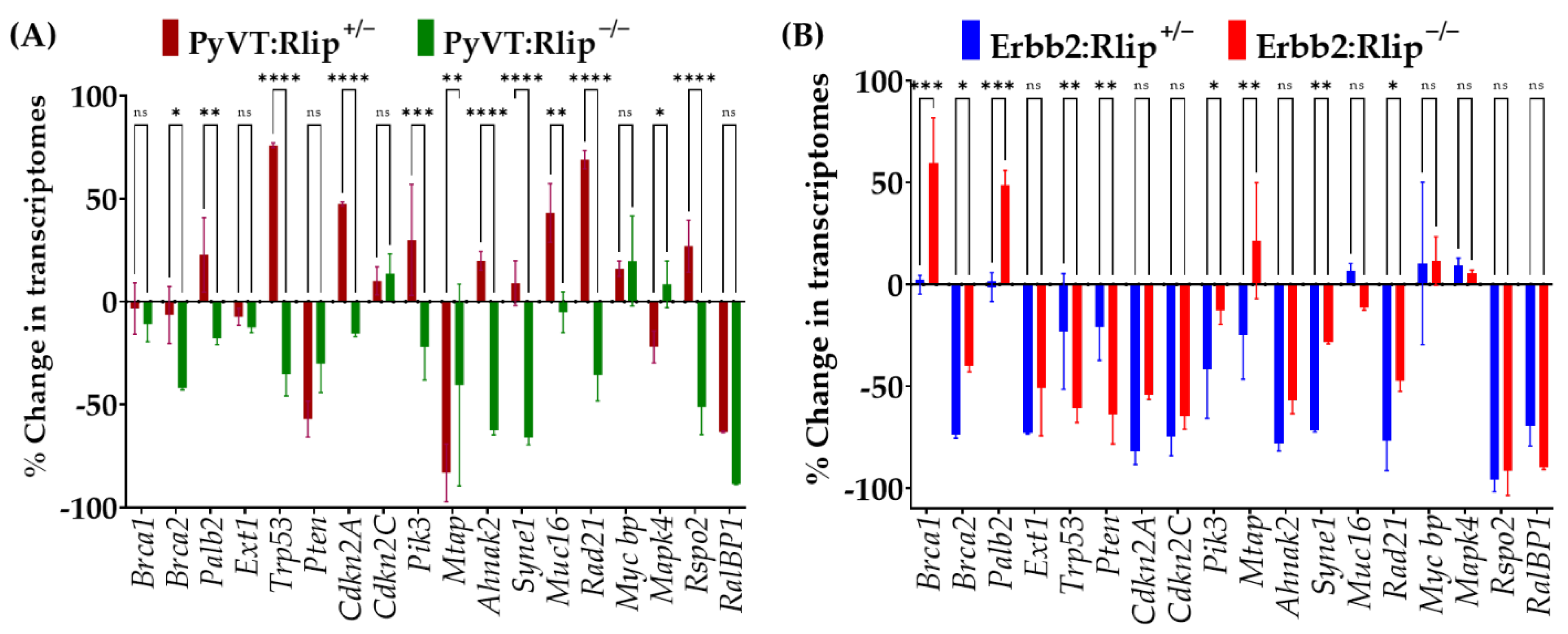
3.4. mRNA Expression of Key Breast Cancer-Related Genes in Tumor Biopsies
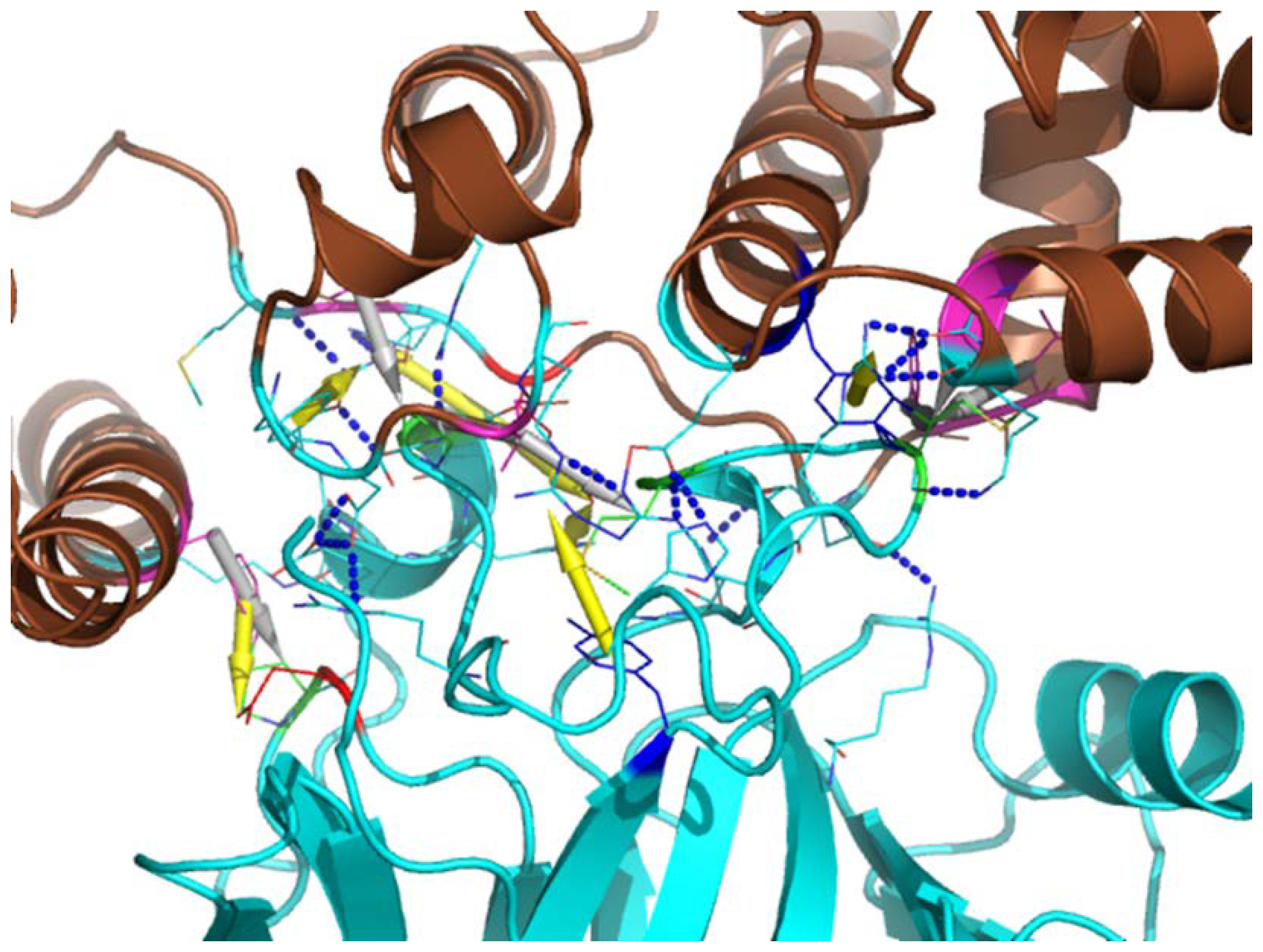
3.5. Computational Modeling of Rlip-p53 Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, K.; Ding, P.; Wu, Y.; Tian, W.; Pan, T.; Zhang, S. Global patterns and trends in the breast cancer incidence and mortality according to sociodemographic indices: An observational study based on the global burden of diseases. BMJ Open 2019, 9, e028461. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Eliyatkın, N.; Yalçın, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular classification of breast carcinoma: From Traditional, old-fashioned way to a new age, and a new way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farahmand, M.; Monavari, S.H.; Shoja, Z.; Ghaffari, H.; Tavakoli, M.; Tavakoli, A. Epstein-Barr virus and risk of breast cancer: A systematic review and meta-analysis. Future Oncol. 2019, 15, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.S.; Heng, B. Viruses and breast cancer. Cancers 2010, 2, 752. [Google Scholar] [CrossRef]
- Hsu, C.R.; Lu, T.M.; Chin, L.W.; Yang, C.C. Possible DNA viral factors of human breast cancer. Cancers 2010, 2, 498. [Google Scholar] [CrossRef] [Green Version]
- Hachana, M.; Trimeche, M.; Ziadi, S.; Amara, K.; Korbi, S. Evidence for a role of the Simian Virus 40 in human breast carcinomas. Breast Cancer Res. Treat. 2009, 113, 43–58. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Fedorova, O.; Daks, A.; Shuvalov, O.; Kizenko, A.; Petukhov, A.; Gnennaya, Y.; Barlev, N. Attenuation of p53 mutant as an approach for treatment Her2-positive cancer. Cell Death Discov. 2020, 6, 100. [Google Scholar] [CrossRef]
- Sen, F. Adjuvant systemic treatment in hormone receptor positive, HER2 negative breast cancer. Breast Cancer Surg. 2018. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.B.B.; Chow, K.H.M.; Xu, A.; Lam, K.S.L.; Liu, J.; Wong, N.-S.; Moon, R.T.; Shepherd, P.R.; Cooper, G.J.S.; Wang, Y. Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyVT mice by modulation of phosphatase and tensin homolog activities. PLoS ONE 2009, 4, e4968. [Google Scholar] [CrossRef]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Connelly, L.; Barham, W.; Onishko, H.M.; Sherrill, T.; Chodosh, L.A.; Blackwell, T.S.; Yull, F.E. Inhibition of NF-kappa B activity in mammary epithelium increases tumor latency and decreases tumor burden. Oncogene 2011, 30, 1402–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Muraoka-Cook, R.S.; Kurokawa, H.; Koh, Y.; Forbes, J.T.; Roebuck, L.R.; Barcellos-Hoff, M.H.; Moody, S.E.; Chodosh, L.A.; Arteaga, C.L. Conditional overexpression of active transforming growth factor β1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004, 64, 9002–9011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almholt, K.; Lund, L.R.; Rygaard, J.; Nielsen, B.S.; Danø, K.; Rømer, J.; Johnsen, M. Reduced metastasis of transgenic mammary cancer in urokinase-deficient mice. Int. J. Cancer 2005, 113, 525–532. [Google Scholar] [CrossRef]
- Cuevas, B.D.; Winter-Vann, A.M.; Johnson, N.L.; Johnson, G.L. MEKK1 controls matrix degradation and tumor cell dissemination during metastasis of polyoma middle-T driven mammary cancer. Oncogene 2006, 25, 4998–5010. [Google Scholar] [CrossRef] [Green Version]
- Wallis, J.; Katti, P.; Martin, A.M.; Hills, T.; Seymour, L.W.; Shenton, D.P.; Carlisle, R.C. A liposome-based cancer vaccine for a rapid and high-titre anti-ErbB-2 antibody response. Eur. J. Pharm. Sci. 2020, 152, 105456. [Google Scholar] [CrossRef]
- Pénzváltó, Z.; Chen, J.Q.; Tepper, C.G.; Davis, R.R.; Silvestrini, M.T.; Umeh-Garcia, M.; Sweeney, C.; Borowsky, A.D. A syngeneic ErbB2 mammary cancer model for preclinical immunotherapy trials. J. Mammary Gland Biol. Neoplas. 2019, 24, 149–162. [Google Scholar] [CrossRef]
- Meyers, N.; Gérard, C.; Lemaigre, F.P.; Jacquemin, P. Differential impact of the ERBB receptors EGFR and ERBB2 on the initiation of precursor lesions of pancreatic ductal adenocarcinoma. Sci. Rep. 2020, 10, 5241. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Rosen, J.M.; McMenamin-Balano, J.; Muller, W.J.; Perkins, A.S. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Mol. Cell. Biol. 1997, 17, 3155–3163. [Google Scholar] [CrossRef] [Green Version]
- Steffens Reinhardt, L.; Zhang, X.; Wawruszak, A.; Groen, K.; De Iuliis, G.N.; Avery-Kiejda, K.A. Good cop, bad cop: Defining the roles of Δ40p53 in cancer and aging. Cancers 2020, 12, 1659. [Google Scholar] [CrossRef] [PubMed]
- Agupitan, A.D.; Neeson, P.; Williams, S.; Howitt, J.; Haupt, S.; Haupt, Y. P53: A guardian of immunity becomes its saboteur through mutation. Int. J. Mol. Sci. 2020, 21, 3452. [Google Scholar] [CrossRef] [PubMed]
- Vieler, M.; Sanyal, S. p53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Tompkins, J.; Singhal, J.; Riggs, A.D.; Yadav, S.; Wu, X.; Singh, S.; Warden, C.; Liu, Z.; Wang, J.; et al. Rlip depletion prevents spontaneous neoplasia in TP53 null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 3918–3923. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Singhal, S.S.; Sharma, R.; Zimniak, P.; Awasthi, Y.C. Transport of glutathione conjugates and chemotherapeutic drugs by RLIP76 (RALBP1): A novel link between G-protein and tyrosine kinase signaling and drug resistance. Int. J. Cancer 2003, 106, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sharma, A.; Sharma, R.; Patrick, B.; Singhal, S.S.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. Cells preconditioned with mild, transient UVA irradiation acquire resistance to oxidative stress and UVA-induced apoptosis: Role of 4-hydroxynonenal in UVA-mediated signaling for apoptosis. J. Biol. Chem. 2003, 278, 41380–41388. [Google Scholar] [CrossRef] [Green Version]
- Cantor, S.B.; Urano, T.; Feig, L.A. Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol. Cell. Biol. 1995, 15, 4578–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jullien-Flores, V.; Dorseuil, O.; Romero, F.; Letourneur, F.; Saragosti, S.; Berger, R.; Tavitian, A.; Gacon, G.; Camonis, J.H. Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J. Biol. Chem. 1995, 270, 22473–22477. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Cheng, J.; Singhal, S.S.; Saini, M.K.; Pandya, U.; Pikula, S.; Bandorowicz-Pikula, J.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry 2000, 39, 9327–9334. [Google Scholar] [CrossRef]
- Awasthi, S.; Cheng, J.Z.; Singhal, S.S.; Pandya, U.; Sharma, R.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Functional reassembly of ATP-dependent xenobiotic transport by the n- and C-terminal domains of RLIP76 and identification of ATP binding sequences. Biochemistry 2001, 40, 4159–4168. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Pikula, S.; Piper, J.T.; Srivastava, S.K.; Torman, R.T.; Bandorowicz-Pikula, J.; Lin, J.T.; Singh, S.V.; Zimniak, P.; et al. ATP-Dependent human erythrocyte glutathione-conjugate transporter. II. Functional reconstitution of transport activity. Biochemistry 1998, 37, 5239–5248. [Google Scholar] [CrossRef]
- Awasthi, S.; Sharma, R.; Yang, Y.; Singhal, S.S.; Pikula, S.; Bandorowicz-Pikula, J.; Singh, S.V.; Zimniak, P.; Awasthi, Y.C. Transport functions and physiological significance of 76 kDa Ral-binding GTPase activating protein (RLIP76). Acta Biochim. Pol. 2002, 49, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Singhal, S.S.; Srivastava, S.K.; Torman, R.T.; Zimniak, P.; Bandorowicz-Pikula, J.; Singh, S.V.; Piper, J.T.; Awasthi, Y.C.; Pikula, S. ATP-Dependent human erythrocyte glutathione-conjugate transporter. I. Purification, photoaffinity labeling, and kinetic characteristics of ATPase activity. Biochemistry 1998, 37, 5231–5238. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Herzog, N.K.; Chaubey, M.; Awasthi, Y.C. Modulation of cisplatin cytotoxicity by sulphasalazine. Br. J. Cancer 1994, 70, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Singhal, S.S.; Yadav, S.; Singhal, J.; Vatsyayan, R.; Zajac, E.; Luchowski, R.; Borvak, J.; Gryczynski, K.; Awasthi, Y.C. A central role of RLIP76 in regulation of glycemic control. Diabetes 2010, 59, 714–725. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, Y.C.; Sharma, R.; Singhal, S.S. Human glutathione S-transferases. Int. J. Biochem. 1994, 26, 295–308. [Google Scholar] [CrossRef]
- Sharma, R.; Singhal, S.S.; Wickramarachchi, D.; Awasthi, Y.C.; Awasthi, S. RLIP76 (RALBP1)-mediated transport of leukotriene C4 (LTC4) in cancer cells: Implications in drug resistance. Int. J. Cancer 2004, 112, 934–942. [Google Scholar] [CrossRef]
- Nagaprashantha, L.; Vartak, N.; Awasthi, S.; Awasthi, S.; Singhal, S.S. Novel anti-cancer compounds for developing combinatorial therapies to target anoikis-resistant tumors. Pharm. Res. 2012, 29, 621–636. [Google Scholar] [CrossRef]
- Awasthi, S.; Singhal, S.S.; Awasthi, Y.C.; Martin, B.; Woo, J.H.; Cunningham, C.C.; Frankel, A.E. RLIP76 and cancer. Clin. Cancer Res. 2008, 14, 4372–4377. [Google Scholar] [CrossRef] [Green Version]
- Stuckler, D.; Singhal, J.; Singhal, S.S.; Yadav, S.; Awasthi, Y.C.; Awasthi, S. RLIP76 transports vinorelbine and mediates drug resistance in non-small cell lung cancer. Cancer Res. 2005, 65, 991–998. [Google Scholar] [PubMed]
- Jullien-Flores, V.; Mahe, Y.; Mirey, G.; Leprince, C.; Meunier-Bisceuil, B.; Sorkin, A.; Camonis, J.H. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: Involvement of the Ral pathway in receptor endocytosis. J. Cell Sci. 2000, 113, 2837–2844. [Google Scholar] [CrossRef]
- Morinaka, K.; Koyama, S.; Nakashima, S.; Hinoi, T.; Okawa, K.; Iwamatsu, A.; Kikuchi, A. Epsin binds to the EH domain of POB1 and regulates receptor-mediated endocytosis. Oncogene 1999, 18, 5915–5922. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, S.; Morinaka, K.; Koyama, S.; Ikeda, M.; Kishida, M.; Okawa, K.; Iwamatsu, A.; Kishida, S.; Kikuchi, A. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J. 1999, 18, 3629–3642. [Google Scholar] [CrossRef]
- Rosse, C.; L’Hoste, S.; Offner, N.; Picard, A.; Camonis, J. RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate epsin during the switch off of endocytosis in mitosis. J. Biol. Chem. 2003, 278, 30597–30604. [Google Scholar] [CrossRef] [Green Version]
- Tazat, K.; Harsat, M.; Goldshmid-Shagal, A.; Ehrlich, M.; Henis, Y.I. Dual effects of Ral-activated pathways on p27 localization and TGF-β signaling. Mol. Biol. Cell 2013, 24, 1812–1824. [Google Scholar] [CrossRef]
- Hu, Y.; Mivechi, N.F. HSF-1 interacts with Ral-binding protein 1 in a stress-responsive, multiprotein complex with HSP90 in vivo. J. Biol. Chem. 2003, 278, 17299–17306. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, D.F.; Lim, K.H.; Brady, D.C.; Pershing, N.L.; Cox, A.D.; Counter, C.M. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskalenko, S.; Henry, D.O.; Rosse, C.; Mirey, G.; Camonis, J.H.; White, M.A. The exocyst is a Ral effector complex. Nat. Cell Biol. 2002, 4, 66–72. [Google Scholar] [CrossRef]
- Park, S.H.; Weinberg, R.A. A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene 1995, 11, 2349–2355. [Google Scholar]
- Leake, K.; Singhal, J.; Nagaprashantha, L.D.; Awasthi, S.; Singhal, S.S. RLIP76 regulates PI3K/Akt signaling and chemo-radiotherapy resistance in pancreatic cancer. PLoS ONE 2012, 7, e34582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Awasthi, Y.C.; Awasthi, S. Regression of melanoma in a murine model by RLIP76 depletion. Cancer Res. 2006, 66, 2354–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Roth, C.; Leake, K.; Singhal, J.; Yadav, S.; Awasthi, S. Regression of prostate cancer xenografts by RLIP76 depletion. Biochem. Pharmacol. 2009, 77, 1074–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Singhal, J.; Yadav, S.; Dwivedi, S.; Boor, P.J.; Awasthi, Y.C.; Awasthi, S. Regression of lung and colon cancer xenografts by depleting or inhibiting RLIP76 (Ral-binding protein 1). Cancer Res. 2007, 67, 4382–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Singhal, J.; Yadav, S.; Sahu, M.; Awasthi, Y.C.; Awasthi, S. RLIP76: A target for kidney cancer therapy. Cancer Res. 2009, 69, 4244–4251. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Nagaprashantha, L.; Li, H.; Yuan, Y.C.; Liu, Z.; Berz, D.; Igid, H.; Green, W.C.; et al. Anticancer activity of 2’-hydroxyflavanone towards lung cancer. Oncotarget 2018, 9, 36202–36219. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Nagaprashantha, L.; Vatsyayan, R.; Awasthi, S.; Singhal, S.S. RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome. PLoS ONE 2011, 6, e24688. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Figarola, J.; Singhal, J.; Reddy, M.A.; Liu, X.; Berz, D.; Natarajan, R.; Awasthi, S. RLIP76 protein knockdown attenuates obesity due to a high-fat diet. J. Biol. Chem. 2013, 288, 23394–23406. [Google Scholar] [CrossRef] [Green Version]
- Shanzer, M.; Ricardo-Lax, I.; Keshet, R.; Reuven, N.; Shaul, Y. The polyomavirus middle T-antigen oncogene activates the Hippo pathway tumor suppressor Lats in a Src-dependent manner. Oncogene 2015, 34, 4190–4198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, C.; Yadav, S.; Singhal, S.S.; Singhal, J.; Hindle, A.; Lee, J.; Cheedella, N.K.S.; Rehman, S.; Rahman, R.L.; Jones, C.; et al. Rlip depletion suppresses growth of breast cancer. Cancers 2020, 12, 1446. [Google Scholar] [CrossRef]
- Bose, C.; Singh, S.P.; Igid, H.; Green, W.C.; Singhal, S.S.; Lee, J.; Palade, P.T.; Rajan, A.; Ball, S.; Tonk, V.; et al. Topical 2’-hydroxyflavanone for cutaneous melanoma. Cancers 2019, 11, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, C.; Awasthi, S.; Sharma, R.; Benes, H.; Hauer-Jensen, M.; Boerma, M.; Singh, S.P. Sulforaphane potentiates anticancer effects of doxorubicin and attenuates its cardiotoxicity in a breast cancer model. PLoS ONE 2018, 13, e0193918. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, S.; Chartoire, N.; Piguet, F.; Hérault, Y.; Pavlovic, G. Optimizing PCR for mouse genotyping: Recommendations for reliable, rapid, cost effective, robust and adaptable to high-throughput genotyping protocol for any type of mutation. Current Protoc. Mouse Biol. 2019, 9, e65. [Google Scholar] [CrossRef] [Green Version]
- Engle, M.R.; Singh, S.P.; Czernik, P.J.; Gaddy, D.; Montague, D.C.; Ceci, J.D.; Yang, Y.; Awasthi, S.; Awasthi, Y.C.; Zimniak, P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: Generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharmacol. 2004, 194, 296–308. [Google Scholar] [CrossRef]
- Singhal, J.; Chikara, S.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. 2′-Hydroxyflavanone inhibits in vitro and in vivo growth of breast cancer cells by targeting RLIP76. Mol. Carcinog. 2018, 57, 1751–1762. [Google Scholar] [CrossRef]
- Wang, Y. Modulation of breast cancer development in MMTV-PyVT mice by adiponectin: A perspective on tumor microenvironment. J. Clin. Oncol. 2015, 33, 34. [Google Scholar] [CrossRef]
- Shishido, S.N.; Delahaye, A.; Beck, A.; Nguyen, T.A. The anticancer effect of PQ1 in the MMTV-PyVT mouse model. Int. J. Cancer 2014, 134, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteva-Font, C.; Jin, B.-J.; Verkman, A.S. Aquaporin-1 gene deletion reduces breast tumor growth and lung metastasis in tumor-producing MMTV-PyVT mice. FASEB J. 2014, 28, 1446–1453. [Google Scholar] [CrossRef] [Green Version]
- Singhal, J.; Chikara, S.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. RLIP inhibition suppresses breast-to-lung metastasis. Cancer Lett. 2019, 447, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Yadav, S.; Nagaprashantha, L.D.; Vatsyayan, R.; Singhal, S.S.; Awasthi, S. Targeting p53-null neuroblastomas through RLIP76. Cancer Prev. Res. 2011, 4, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- RRajasekar, K.; Campbell, L.J.; Nietlispach, D.; Owen, D.; Mott, H.R. The structure of the RLIP76 RhoGAP-Ral binding domain dyad: Fixed position of the domains leads to dual engagement of small G proteins at the membrane. Structure 2013, 21, 2131–2142. [Google Scholar] [CrossRef] [Green Version]
- Lilyestrom, W.; Klein, M.G.; Zhang, R.; Joachimiak, A.; Chen, X.S. Crystal structure of SV40 large T-antigen bound to p53: Interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006, 20, 2373–2382. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, Z.; Xia, Y.; Lin, W.; Li, H. Investigation of the genetic variation in ACE2 on the structural recognition by the novel coronavirus (SARS-CoV-2). J. Transl. Med. 2020, 18, 321. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Salgia, R.; Singhal, S.; Horne, D.; Awasthi, S. RLIP: An existential requirement for breast carcinogenesis. Biochim. Biophys. Acta 2019, 1871, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Singhal, P.; Horne, D.; Salgia, R.; Awasthi, S.; Singhal, S.S. Metastasis of breast tumor cells to brain is suppressed by targeting RLIP alone and in combination with 2’-Hydroxyflavanone. Cancer Lett. 2018, 438, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Singhal, S.S.; Singhal, J.; Cheng, J.; Zimniak, P.; Awasthi, Y.C. Role of RLIP76 in lung cancer doxorubicin resistance: II. Doxorubicin transport in lung cancer by RLIP76. Int. J. Oncol. 2003, 22, 713–720. [Google Scholar] [CrossRef]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. RLIP76, a novel transporter catalyzing ATP-dependent efflux of xenobiotics. Drug Metab. Dispos. 2002, 30, 1300–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.S.; Singhal, J.; Cheng, J.; Pikula, S.; Sharma, R.; Zimniak, P.; Awasthi, Y.C.; Awasthi, S. Purification and functional reconstitution of intact ral-binding Gtpase activating protein, RLIP76, in artificial liposomes. Acta Biochim. Pol. 2001, 48, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Salgia, R.; Verma, N.; Horne, D.; Awasthi, S. RLIP controls receptor-ligand signaling by regulating clathrin-dependent endocytosis. Biochim. Biophys. Acta 2020, 1873, 188337. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Goldfinger, L.E. RLIP76 regulates HIF-1 activity, VEGF expression and secretion in tumor cells, and secretome transactivation of endothelial cells. FASEB J. 2014, 28, 4158–4168. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, A.; Yadav, S.; Awasthi, Y.C.; Basu, A.; Warden, C.; Awasthi, S. P300 regulates the human RLIP76 promoter activity and gene expression. Biochem. Pharmacol. 2013, 85, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Wurtzel, J.G.; Singhal, S.S.; Awasthi, S.; Goldfinger, L.E. RALBP1/RLIP76 depletion in mice suppresses tumor growth by inhibiting tumor neovascularization. Cancer Res. 2012, 72, 5165–5173. [Google Scholar] [CrossRef] [Green Version]
- Fry, E.A.; Taneja, P.; Inoue, K. Clinical applications of mouse models for breast cancer engaging HER2/neu. Integr. Cancer Sci. Ther. 2016, 3, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Suter, T.M.; Cook-Bruns, N.; Barton, C. Cardiotoxicity associated with trastuzumab (Herceptin) therapy in the treatment of metastatic breast cancer. Breast 2004, 13, 173–183. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Brca1 | CGAATCTGAGTCCCCTAAAGAGC | AAGCAACTTGACCTTGGGGTA |
| Brca2 | TGGTAGATGTTGCTAGTCCGC | ACCACTGGCTTTTCTCGTTGT |
| Palb2 | GGGAAGCCCCTCAGCTATG | CGAGCAAGTGTCCTGCTGTAT |
| Ext1 | TGGAGGCGTGCAGTTTAGG | GAAGCGGGGCCAGAAATGA |
| Trp53 | CTCTCCCCCGCAAAAGAAAAA | CGGAACATCTCGAAGCGTTTA |
| Pten | CCTTTTGAAGACCATAACCCACC | GAATTGCTGCAACATGATTGTCA |
| Cdkn2A | CGCAGGTTCTTGGTCACTGT | TGTTCACGAAAGCCAGAGCG |
| Cdkn2C | GGGGACCTAGAGCAACTTACT | AAATTGGGATTAGCACCTCTGAG |
| Pik3CA | CCACGACCATCTTCGGGTG | GGGGAGTAAACATTCCACTAGGA |
| Mtap | ACGGCGGTGAAGATTGGAATA | ATGGCTTGCCGAATGGAGTAT |
| Ahnak2 | CCACCCCAACTGGGACTTTG | CACTCCCCTGTAACTTGCCTG |
| Syne1 | AGACTGCGACTGCGATGTC | CTGTGCTGTGTTTCTCGATGT |
| Muc16 | ACTTCTCACCATTGGCTCGG | AACTGGGTACCATTGTGCGT |
| Rad21 | ATGTTCTACGCACATTTTGTCCT | CATGGGCTTTGGTTAGCTTCT |
| Mycbp | GCTGGACACGCTGACGAAA | TCTAGGCGAAGCAGCTCTATTT |
| Mapk4 | GCCAGCGTCTACGGGTATG | GCGTCACTCAGAACGATCTTCTT |
| Rspo2 | ACGCAATAAGCGAGCTAGTTATG | ACATCGGCTGCAACCATTGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.P.; Lee, J.; Bose, C.; Li, H.; Yuan, Y.-C.; Hindle, A.; Singhal, S.S.; Kopel, J.; Palade, P.T.; Jones, C.; et al. Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis. Cancers 2021, 13, 3329. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133329
Singh SP, Lee J, Bose C, Li H, Yuan Y-C, Hindle A, Singhal SS, Kopel J, Palade PT, Jones C, et al. Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis. Cancers. 2021; 13(13):3329. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133329
Chicago/Turabian StyleSingh, Sharda P., Jihyun Lee, Chhanda Bose, Hongzhi Li, Yate-Ching Yuan, Ashly Hindle, Sharad S. Singhal, Jonathan Kopel, Philip T. Palade, Catherine Jones, and et al. 2021. "Haploinsufficiency Interactions between RALBP1 and p53 in ERBB2 and PyVT Models of Mouse Mammary Carcinogenesis" Cancers 13, no. 13: 3329. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133329