
Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer

 , , , , , , and
, , , , , , and 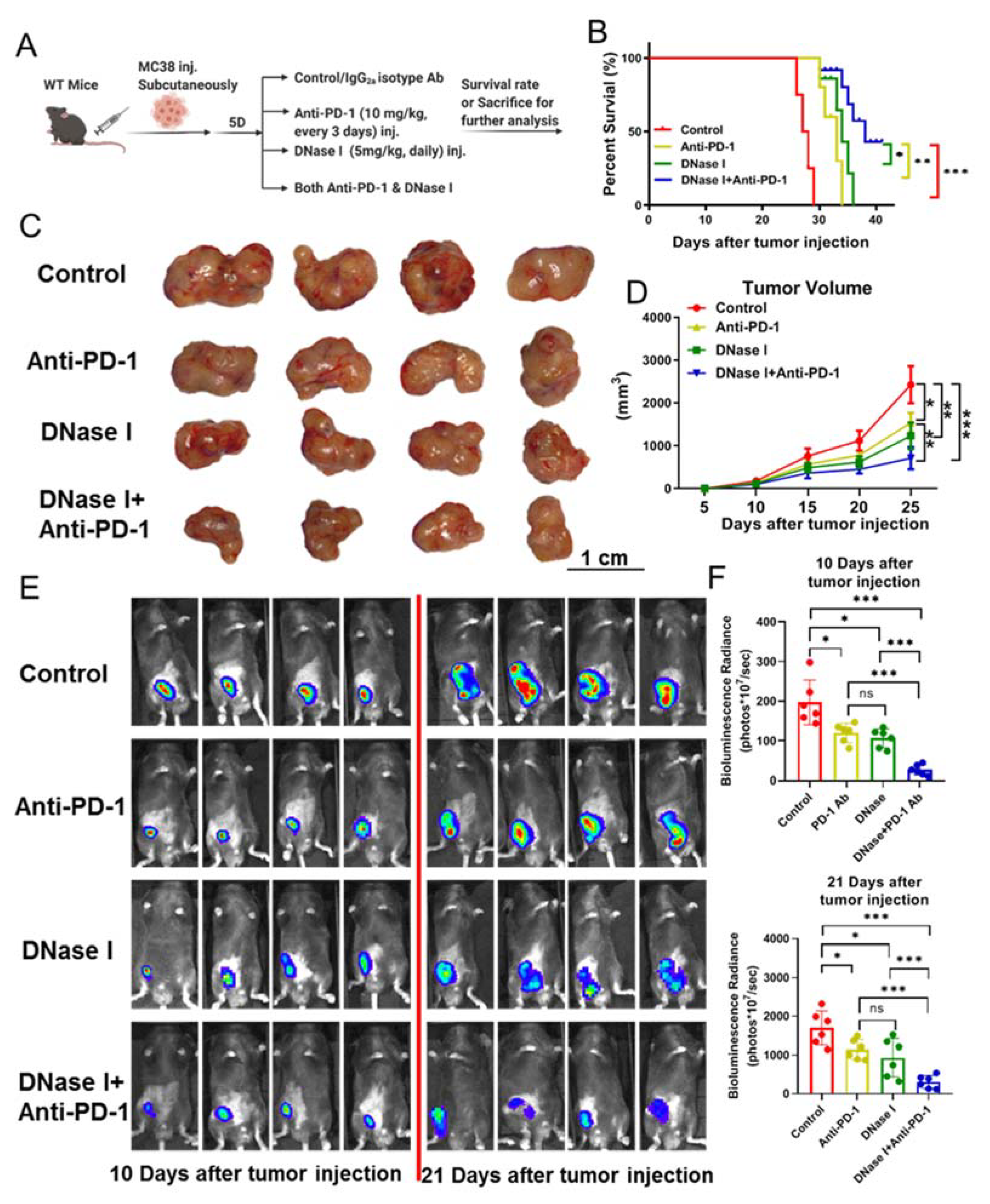{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Luciferase Transfection and Bioluminescence Imaging
2.3. Animals and Tumor Models
2.4. Neutrophil Isolation and In Vitro NET Formation
2.5. Quantification of NETs
2.6. Western Blotting
2.7. Flow Cytometry
2.8. In Vitro Cytotoxicity Assay
2.9. Quantitative Real-Time PCR
2.10. BioRender
2.11. Statistical Analysis
3. Results
3.1. Systemic Administration of DNase Improves the Efficacy of PD-1 Blockade to Reduce the Growth of Cancer
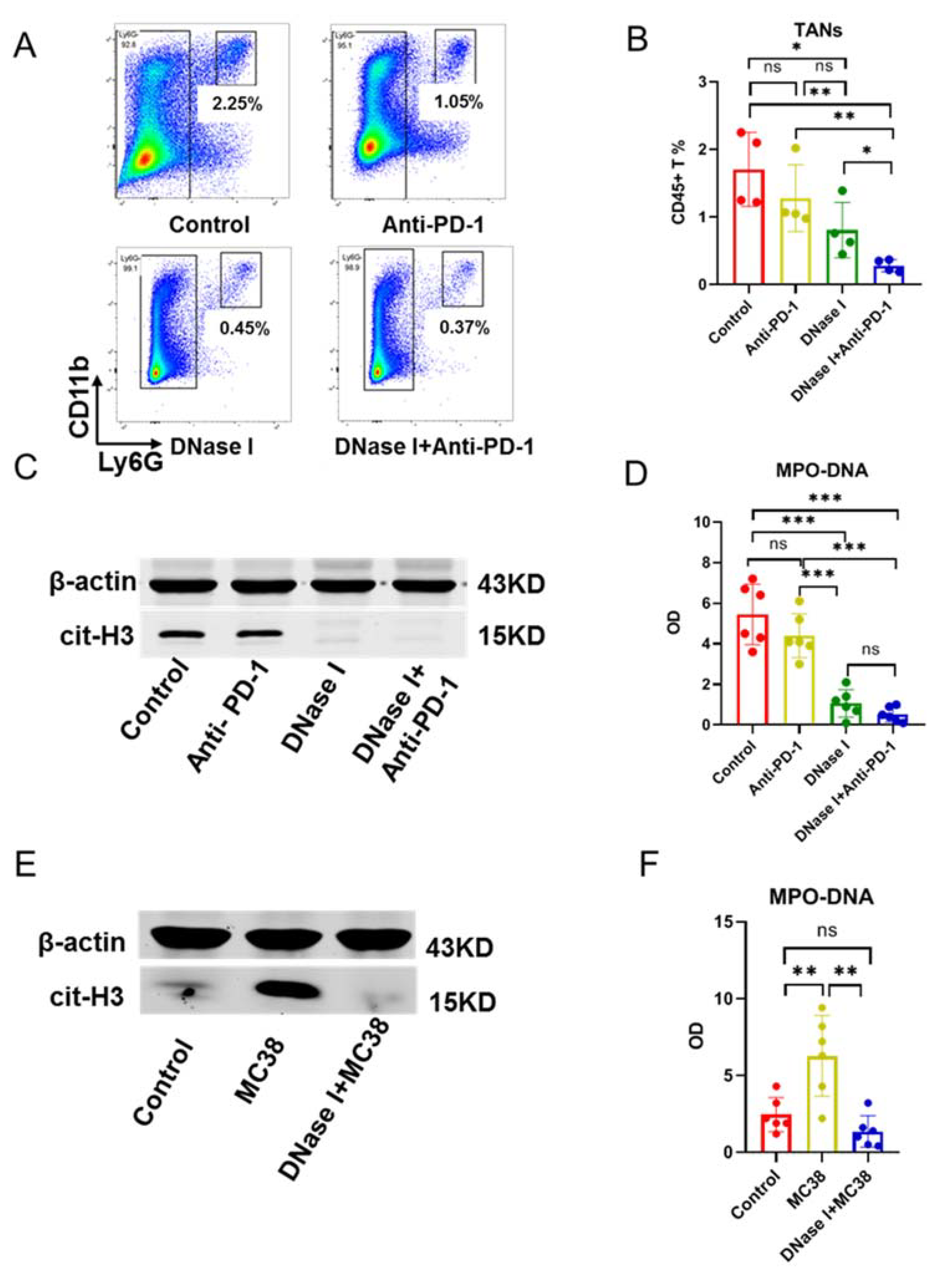
3.2. DNase I Inhibits MC38 Tumor Cell-Induced TANS to Form NETs in Tumors
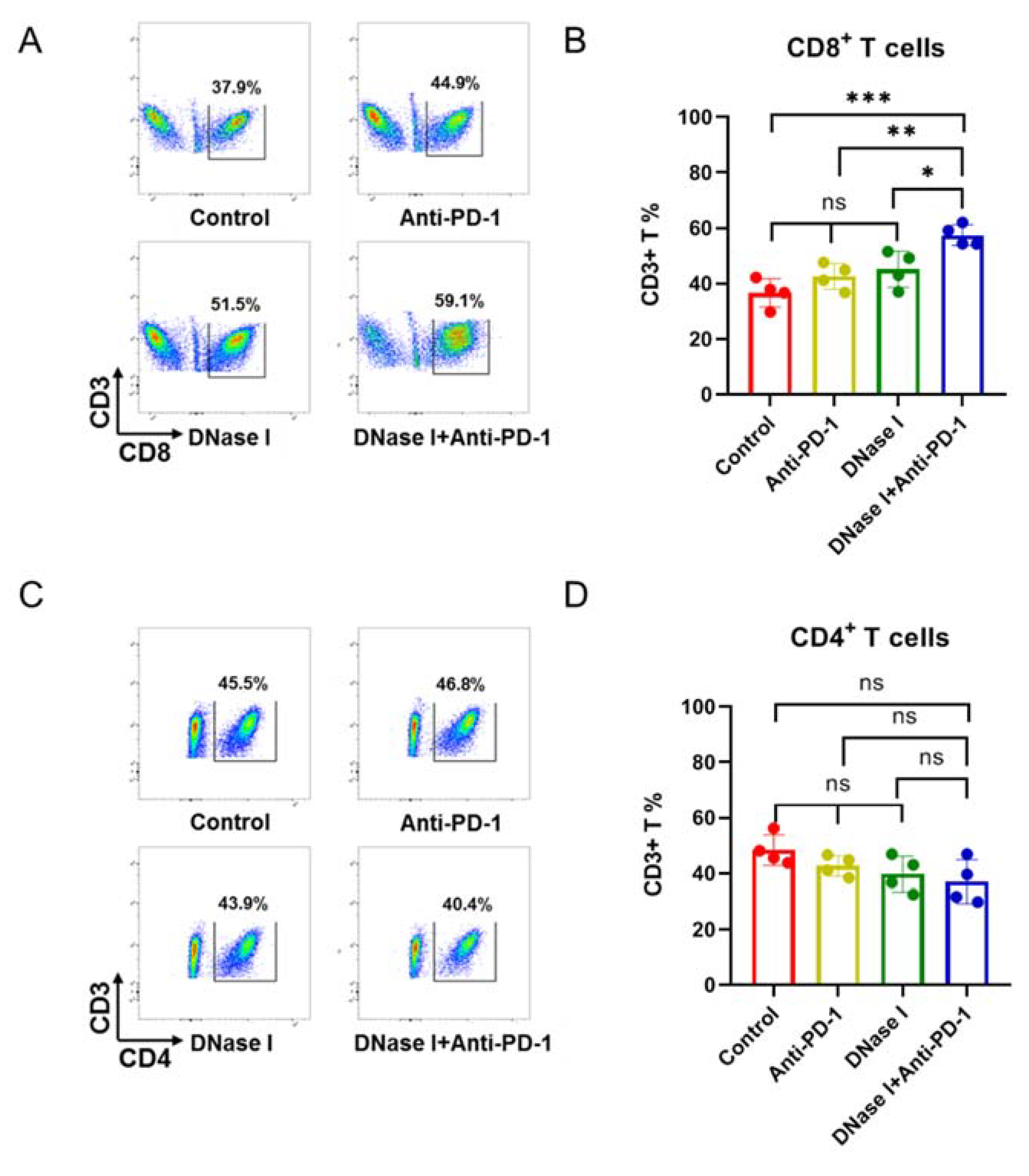
3.3. Inhibition of NETs Improves Response to Anti-PD-1 Therapy to Anti-PD-1, Which Is Associated with Lack of CD8+ T Cell Infiltration
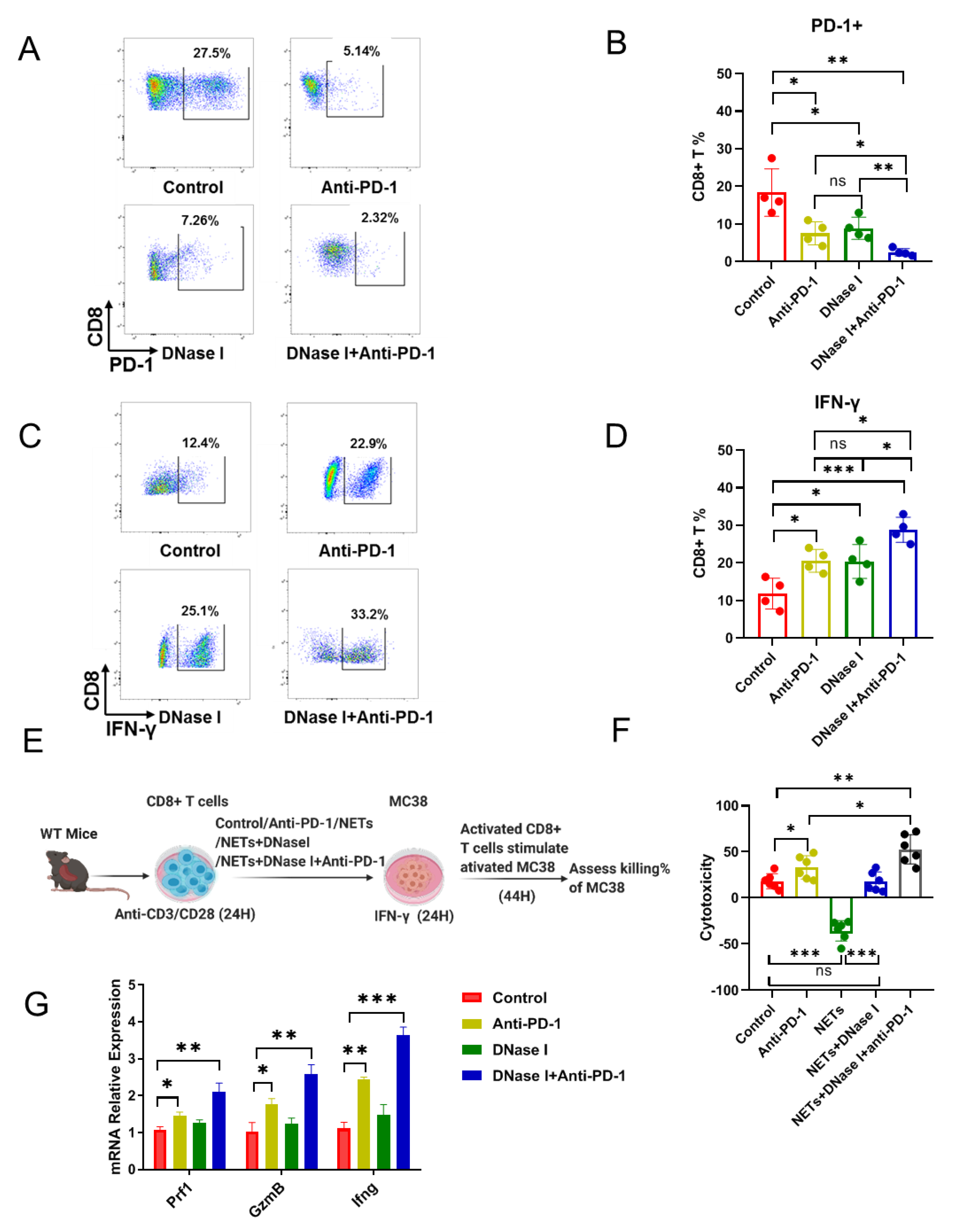
3.4. NETs Induce the Resistance to Anti-PD-1 via Predisposition to the Cytotoxicity of CD8+ T Cells
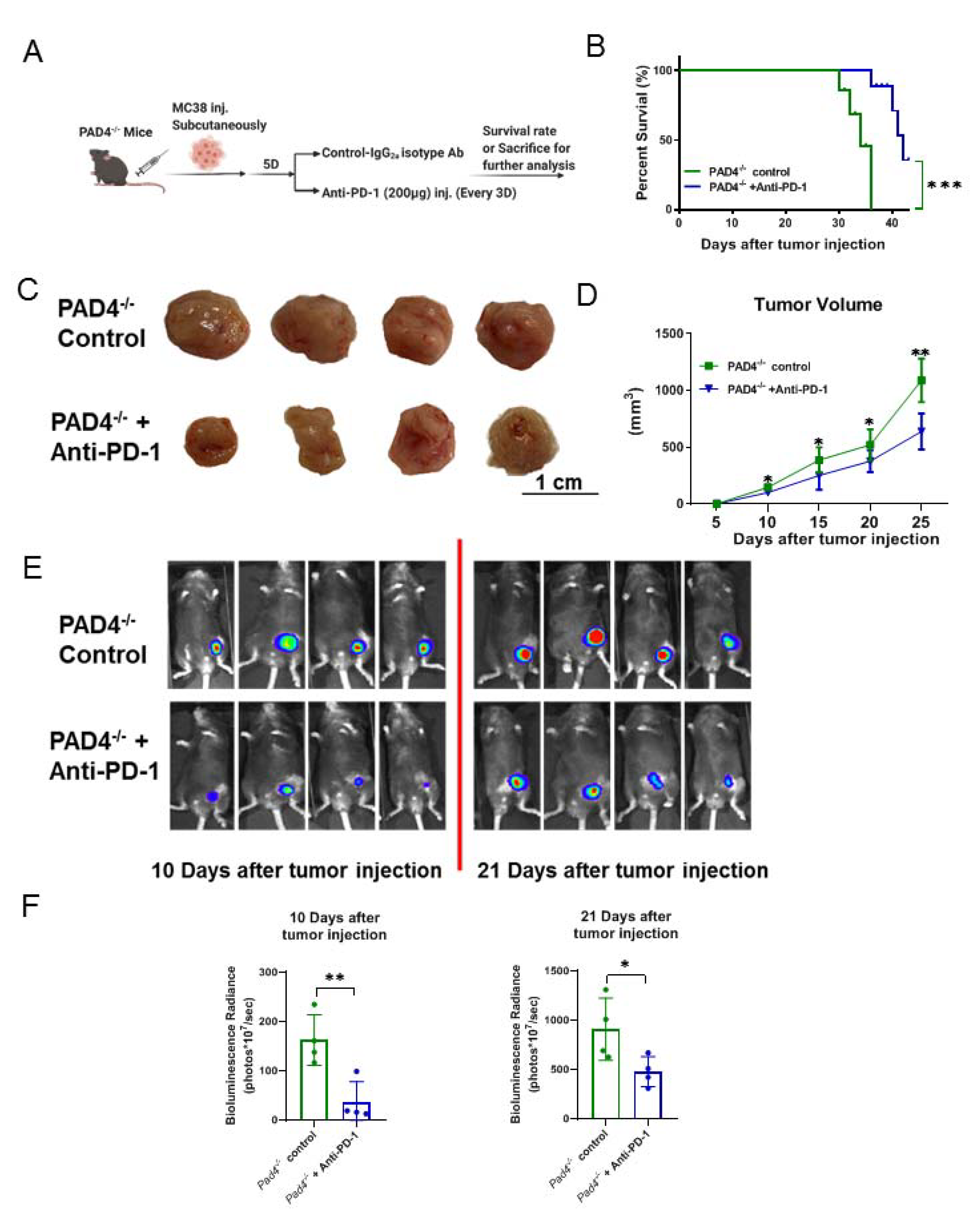
3.5. Being Genetically Incapable of NET Formation Improves the Efficacy of PD-1 Blockade to Reduce the Growth of Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Abril-Rodriguez, G.; Torrejon, D.Y.; Liu, W.; Zaretsky, J.M.; Nowicki, T.S.; Tsoi, J.; Puig-Saus, C.; Baselga-Carretero, I.; Medina, E.; Quist, M.J.; et al. PAK4 inhibition improves PD-1 blockade immunotherapy. Nat. Cancer 2020, 1, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Le, D.T.; Boland, P.M. Chapter Five—Immunotherapy in colorectal cancer. In Advances in Cancer Research; Berger, F.G., Boland, C.R., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 151, pp. 137–196. [Google Scholar]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Ther. Adv. Gastroenterol. 2020, 13, 1756284820917527. [Google Scholar] [CrossRef]
- Guimaraes-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceicao-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, H.; Onuma, A.E.; Tsung, A. Neutrophil Elastase and Neutrophil Extracellular Traps in the Tumor Microenvironment. In Tumor Microenvironment: State of the Science; Birbrair, A., Ed.; Springer: Cham, Switzerland, 2020; pp. 13–23. [Google Scholar] [CrossRef]
- Honda, M.; Kubes, P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Berger-Achituv, S.; Brinkmann, V.; Abed, U.A.; Kuhn, L.I.; Ben-Ezra, J.; Elhasid, R.; Zychlinsky, A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 2013, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; He, J.; Zhang, H.; Wang, H.; Tetz, G.; Maguire, C.A.; Wang, Y.; Onuma, A.; Genkin, D.; Tetz, V.; et al. AAV-mediated gene transfer of DNase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol. Oncol. 2020, 14, 2920–2935. [Google Scholar] [CrossRef]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Goswami, J.; Varley, P.; van der Windt, D.J.; Ren, J.; Loughran, P.; Yazdani, H.; Neal, M.D.; Simmons, R.L.; Zhang, J.; et al. Hepatic Surgical Stress Promotes Systemic Immunothrombosis That Results in Distant Organ Injury. Front. Immunol. 2020, 11, 987. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Huang, H.; Tohme, S.; Al-Khafaji, A.B.; Tai, S.; Loughran, P.; Chen, L.; Wang, S.; Kim, J.; Billiar, T.; Wang, Y.; et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 2015, 62, 600–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, T.; Ren, J.; Xia, Y.; Onuma, A.; Wang, Y.; He, J.; Wu, J.; Wang, H.; Hamad, A.; et al. Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nat. Metab. 2021, 3, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Haining, W.N.; Freeman, G.J.; Sharpe, A.H. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.D.; de Leenheer, E.; Fishman, S.; Siew, L.K.; Gross, G.; Wong, F.S. A reproducible method for the expansion of mouse CD8+ T lymphocytes. J. Immunol. Methods 2015, 417, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; He, J. Platelet TLR4-ERK5 Axis Facilitates NET-Mediated Capturing of Circulating Tumor Cells and Distant Metastasis after Surgical Stress. Cancer Res. 2021, 81, 2373–2385. [Google Scholar] [CrossRef]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, X.H.-F. Tumor-Associated Neutrophils and Macrophages—Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, H.O.; Roy, E. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Rev. 2019, 79, 5626–5639. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-Y.; Luo, Q.; Lu, L.; Zhu, W.-W.; Sun, H.-T.; Wei, R.; Lin, Z.-F.; Wang, X.-Y.; Wang, C.-Q.; Lu, M.; et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J. Hematol. Oncol. 2020, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, C.; Uekusa, Y.; Iwasaki, M.; Yamaguchi, N.; Mukai, T.; Gao, P.; Tomura, M.; Ono, S.; Tsujimura, T.; Fujiwara, H.; et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer Res. 2001, 61, 3399–3405. [Google Scholar]
- Bálint, Š.; Müller, S.; Fischer, R.; Kessler, B.M.; Harkiolaki, M.; Valitutti, S.; Dustin, M.L. Supramolecular attack particles are autonomous killing entities released from cytotoxic T cells. Science 2020, 368, 897–901. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germann, M.; Zangger, N.; Sauvain, M.O.; Sempoux, C.; Bowler, A.D.; Wirapati, P.; Kandalaft, L.E.; Delorenzi, M.; Tejpar, S.; Coukos, G.; et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGFβ. EMBO Mol. Med. 2020, 12, e10681. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Khan, U.; Chowdhury, S.; Billah, M.M. Neutrophil Extracellular Traps in Colorectal Cancer Progression and Metastasis. Int. J. Mol. Sci. 2021, 22, 7260. [Google Scholar] [CrossRef] [PubMed]
- Homa-Mlak, I.; Majdan, A.; Mlak, R.; Małecka-Massalska, T. Metastatic potential of NET in neoplastic disease. Postepy Hig. Med. Dosw. 2016, 70, 887–895. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, L.; Zhang, R.; Hong, J.; Wang, Y.; Wang, J.; Zuo, J.; Zhang, J.; Chen, J.; Hao, H. IL-8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J. Cancer 2020, 11, 4384–4396. [Google Scholar] [CrossRef]
- Alekseeva, L.A.; Sen’kova, A.V.; Zenkova, M.A.; Mironova, N.L. Targeting Circulating SINEs and LINEs with DNase I Provides Metastases Inhibition in Experimental Tumor Models. Mol. Ther. Nucleic Acids 2020, 20, 50–61. [Google Scholar] [CrossRef]
- Salganik, R.I.; Martynova, R.P.; Matienko, N.A.; Ronichevskaya, G.M. Effect of deoxyribonuclease on the course of lymphatic leukaemia in AKR mice. Nature 1967, 214, 100–102. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Agarwal, P.; Lins, D.C.; Mescher, M.F. Autocrine IFN-γ promotes naive CD8 T cell differentiation and synergizes with IFN-α to stimulate strong function. J. Immunol. 2012, 189, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Honjo, T. Combination therapy strategies for improving PD-1 blockade efficacy: A new era in cancer immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Kaufman, H.L.; Wigginton, J.M.; Wolchok, J.D. Combination immunotherapy: A road map. J. Immunother. Cancer 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-S.; Leng, C.-H.; Yeh, Y.-C.; Wu, C.-C.; Chen, H.-W.; Huang, H.-M.; Liu, S.-J. Toll-like receptor 9 agonist enhances anti-tumor immunity and inhibits tumor-associated immunosuppressive cells numbers in a mouse cervical cancer model following recombinant lipoprotein therapy. Mol. Cancer 2014, 13, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Xiao-Feng, H.; Guan-Jun, D.; Er-Ling, H.; Sheng, C.; Ting-Ting, W.; Qin-Gang, H.; Yan-Hong, N.; Ya-Yi, H. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2015, 1852, 2494–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Wang, Y.; Onuma, A.; He, J.; Wang, H.; Xia, Y.; Lal, R.; Cheng, X.; Kasumova, G.; Hu, Z.; et al. Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer. Cancers 2021, 13, 5333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215333
Zhang H, Wang Y, Onuma A, He J, Wang H, Xia Y, Lal R, Cheng X, Kasumova G, Hu Z, et al. Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer. Cancers. 2021; 13(21):5333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215333
Chicago/Turabian StyleZhang, Hongji, Yu Wang, Amblessed Onuma, Jiayi He, Han Wang, Yujia Xia, Rhea Lal, Xiang Cheng, Gyulnara Kasumova, Zhiwei Hu, and et al. 2021. "Neutrophils Extracellular Traps Inhibition Improves PD-1 Blockade Immunotherapy in Colorectal Cancer" Cancers 13, no. 21: 5333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215333