
Immediate Sample Fixation Increases Circulating Tumour Cell (CTC) Capture and Preserves Phenotype in Head and Neck Squamous Cell Carcinoma: Towards a Standardised Approach to Microfluidic CTC Biomarker Discovery

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Patient and Healthy Donor Samples
2.3. Sample Collection and Processing
2.4. CTC Enrichment
2.5. RNA Sequencing
3. Results
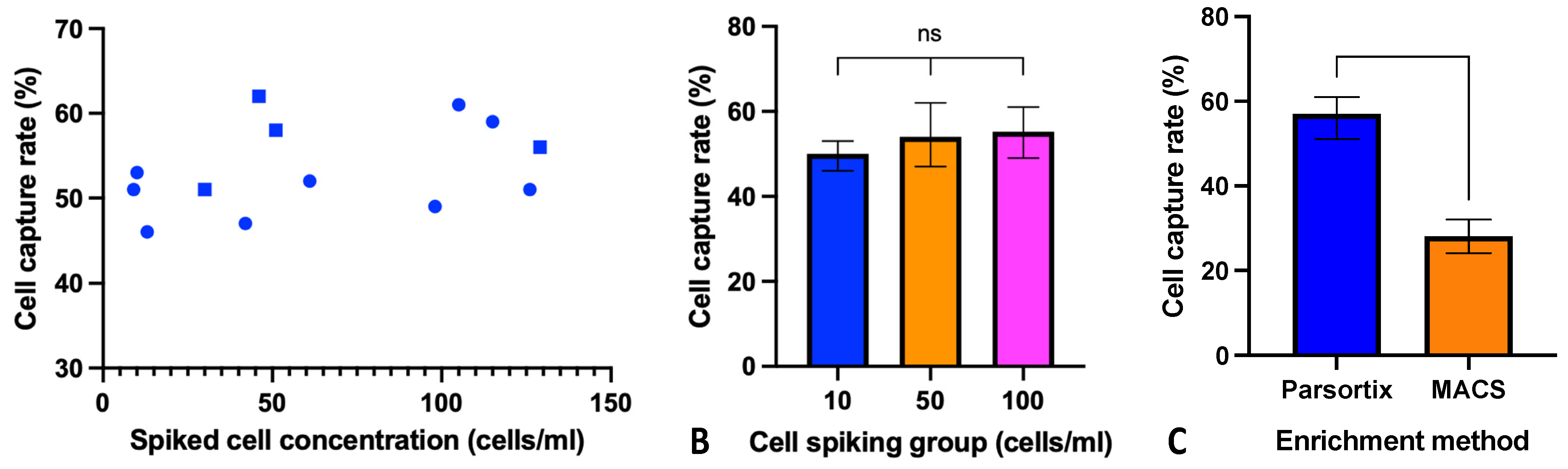
3.1. Parsortix CTC Enrichment in HNSCC Cell Line Model
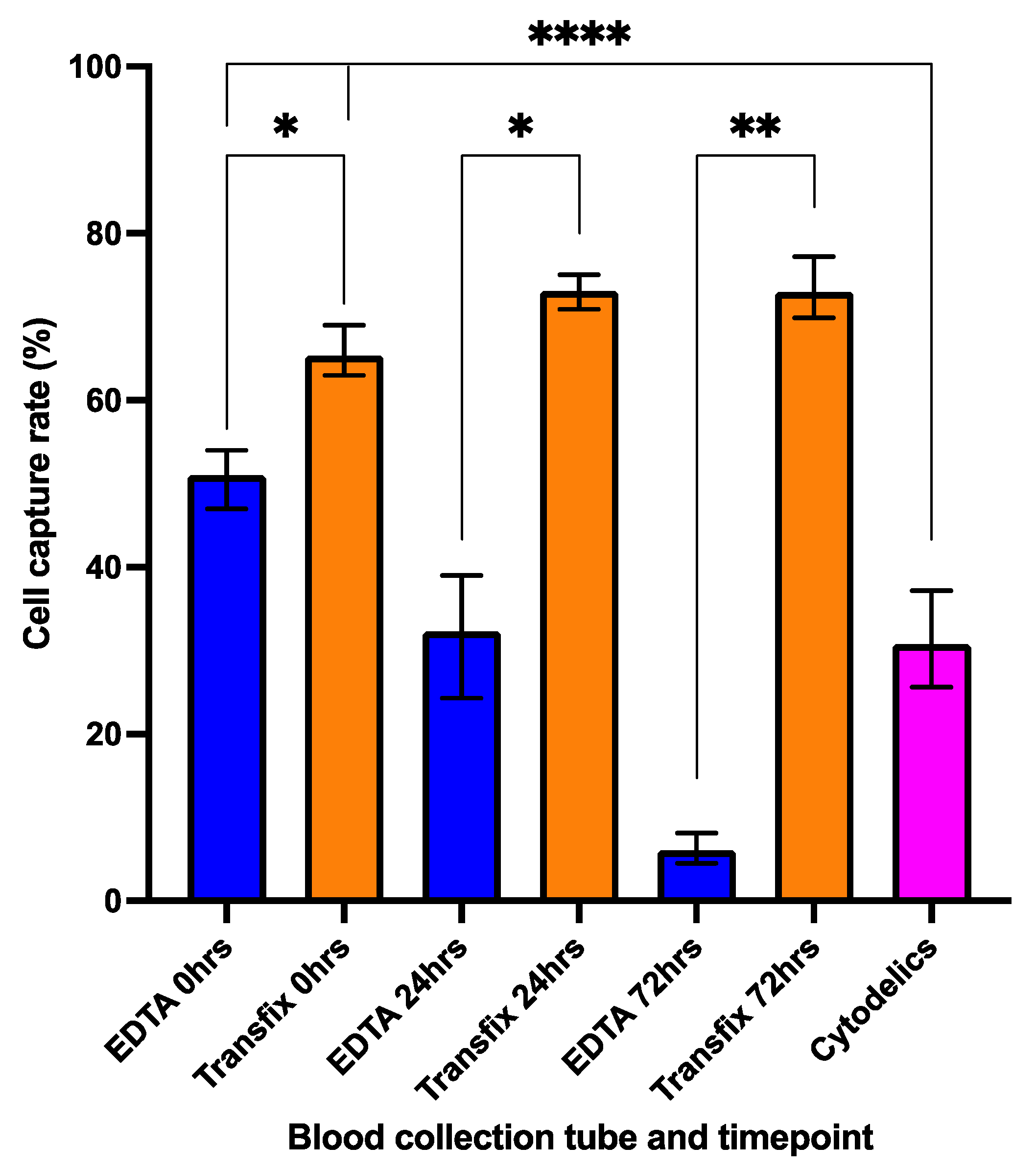
3.2. Cell Preservation and Sample Storage Time
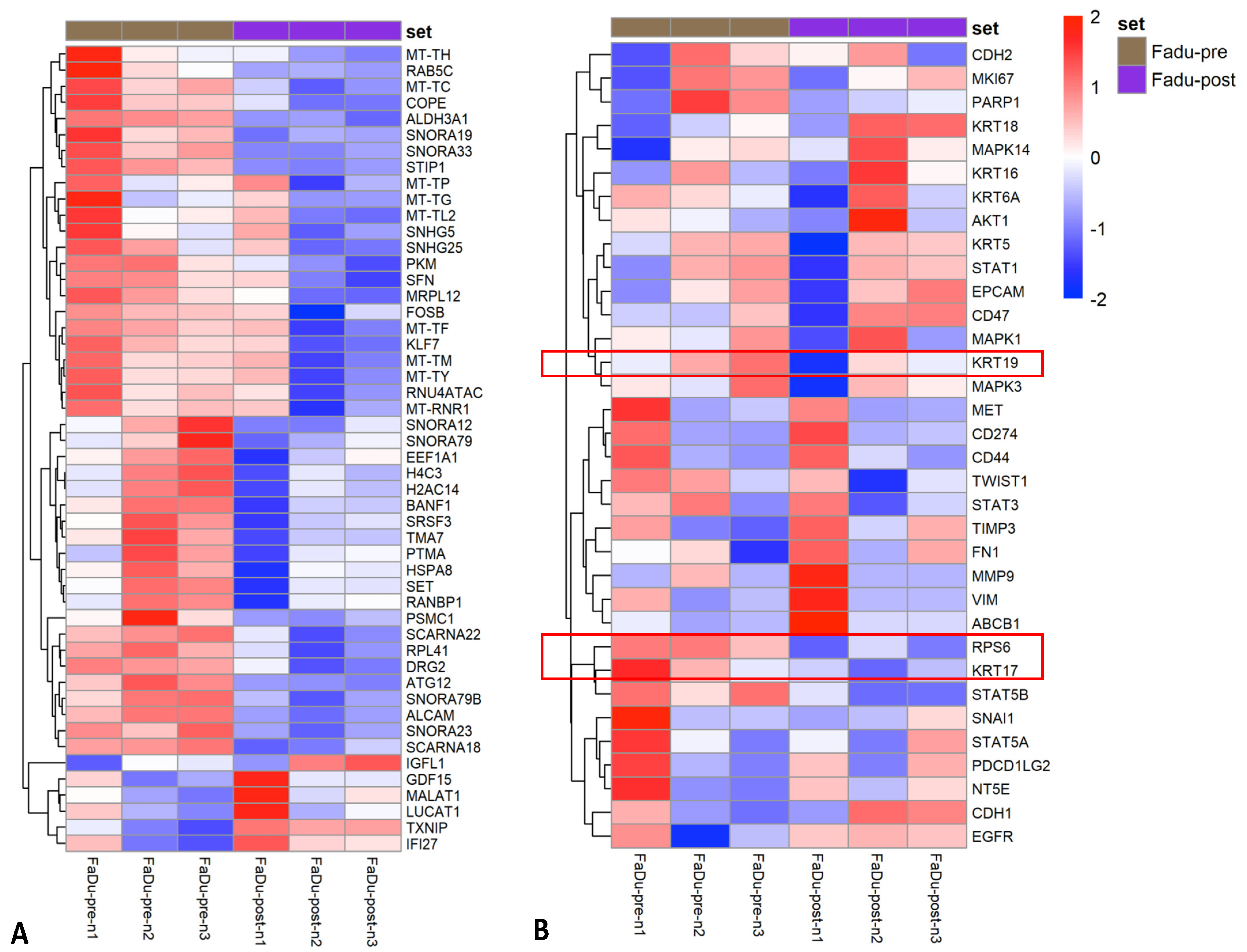
3.3. Impact of Parsortix Enrichment on Gene Expression Patterns of Unfixed HNSCC Cell Lines
3.4. RNA Extraction and Sequencing of Transfix Preserved Cells
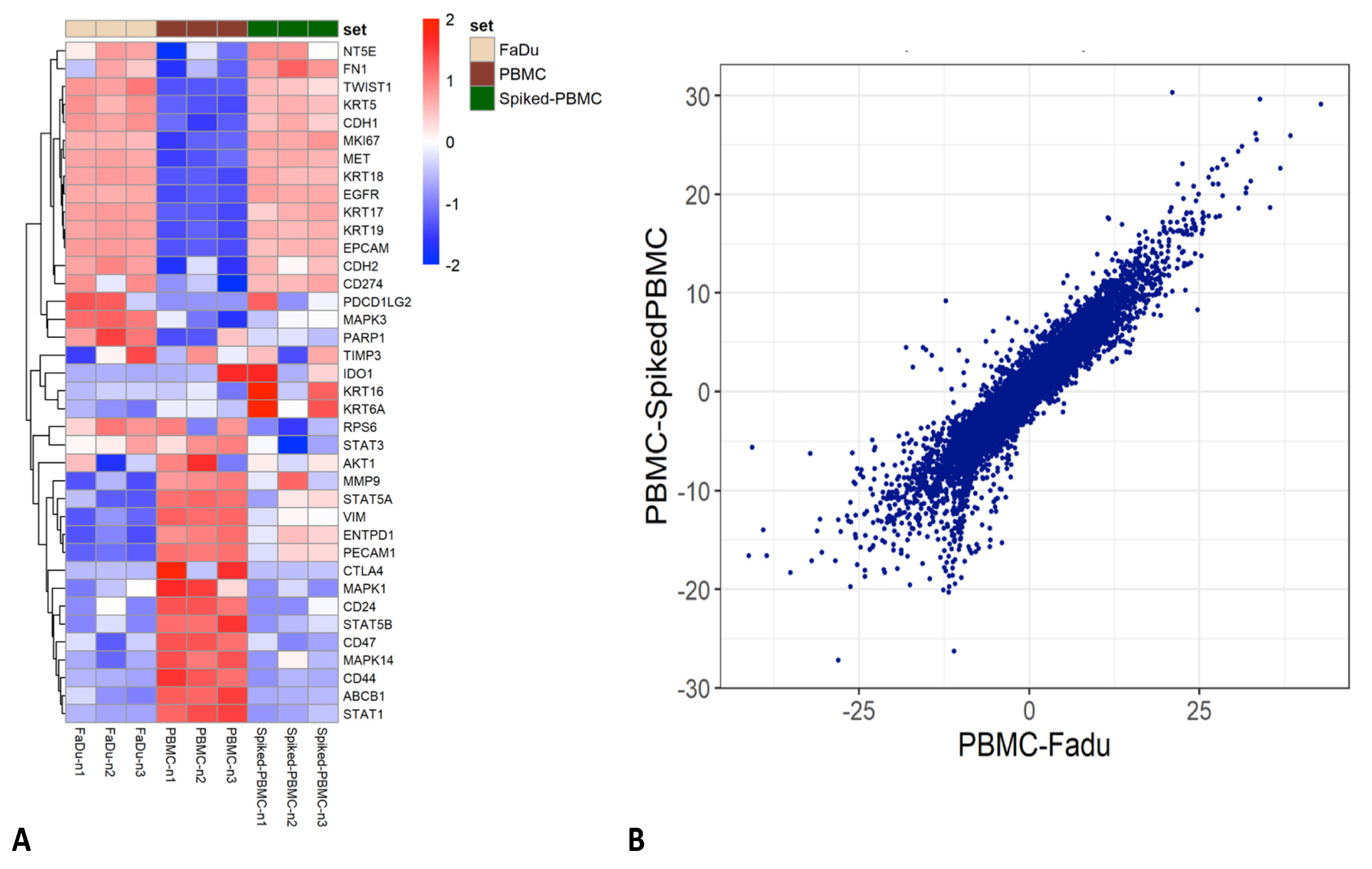
3.5. Enrichment of Transfix Blood Samples from HNSCC Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorges, T.M.; Pantel, K. Circulating tumor cells as therapy-related biomarkers in cancer patients. Cancer Immunol. Immunother. 2013, 62, 931–939. [Google Scholar] [CrossRef]
- Dawson, S.-J.; Tsui, D.W.Y.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.; Brooks, J.; Spruce, R.; Batis, N.; Taylor, G.; Nankivell, P.; Mehanna, H. Circulating Tumour Cell Biomarkers in Head and Neck Cancer: Current Progress and Future Prospects. Cancers 2019, 11, 1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Dong, Y.; Skelley, A.M.; Merdek, K.D.; Sprott, K.M.; Jiang, C.; Pierceall, W.E.; Lin, J.; Stocum, M.; Carney, W.P.; Smirnov, D.A. Microfluidics and circulating tumor cells. J. Mol. Diagn. 2013, 15, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, P.; Nel, I.; Hassenkamp, P.; Gauler, T. Detection of Circulating Tumor Cell Subpopulations in Patients with Head and Neck Squamous Cell Carcinoma ( HNSCC ). PLoS ONE 2014, 9, e113706. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, P.; Lang, J.C.; Jatana, K.R.; Miller, B.; Ozer, E.; Old, M.; Schuller, D.E.; Agrawal, A.; Teknos, T.N.; Summers, T.A.; et al. Multiparameter analysis, including EMT markers, on negatively enriched blood samples from patients with squamous cell carcinoma of the head and neck. PLoS ONE 2012, 7, e42048. [Google Scholar] [CrossRef]
- Kulasinghe, A.; Tran, T.H.P.; Blick, T.; O’Byrne, K.; Thompson, E.W.; Warkiani, M.E.; Nelson, C.; Kenny, L.; Punyadeera, C. Enrichment of circulating head and neck tumour cells using spiral microfluidic technology. Sci. Rep. 2017, 7, 42517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strati, A.; Koutsodontis, G.; Papaxoinis, G.; Angelidis, I.; Zavridou, M.; Economopoulou, P.; Kotsantis, I.; Avgeris, M.; Mazel, M.; Perisanidis, C.; et al. Prognostic significance of PD-L1 expression on circulating tumor cells in patients with head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Tu, Q.; Faure, G.; Gallet, P.; Kohler, C.; Bittencourt, M.D.C. Diagnostic and Prognostic Value of Circulating Tumor Cells in Head and Neck Squamous Cell Carcinoma: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 2010. [Google Scholar] [CrossRef] [Green Version]
- Kulasinghe, A.; Perry, C.; Kenny, L.; Warkiani, M.E.; Nelson, C.; Punyadeera, C. PD-L1 expressing circulating tumour cells in head and neck cancers. BMC Cancer 2017, 17, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Alt, J.R.; Hunsley, B.A.; Williams, T.L.; Fernando, M.R. Stabilization of circulating tumor cells in blood using a collection device with a preservative reagent. Cancer Cell Int. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Lee, M.; Kolatkar, A.; McCormick, M.; Dago, A.D.; Kendall, J.; Carlsson, N.A.; Bethel, K.; Greenspan, E.J.; Hwang, S.E.; Waitman, K.R.; et al. Effect of Blood Collection Tube Type and Time to Processing on the Enumeration and High-Content Characterization of Circulating Tumor Cells Using the High-Definition Single-Cell Assay. Arch. Pathol. Lab. Med. 2018, 142, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.; Joosse, S.A.; Schneegans, S.; Wilken, O.J.W.; Janning, M.; Loreth, D.; Müller, V.; Prieske, K.; Banys-Paluchowski, M.; Horst, L.J.; et al. Pre-Analytical and Analytical Variables of Label-Independent Enrichment and Automated Detection of Circulating Tumor Cells in Cancer Patients. Cancers 2020, 12, 442. [Google Scholar] [CrossRef] [Green Version]
- Hvichia, G.E.; Parveen, Z.; Wagner, C.; Janning, M.; Quidde, J.; Stein, A.; Müller, V.; Loges, S.; Neves, R.P.L.; Stoecklein, N.H.; et al. A novel microfluidic platform for size and deformability based separation and the subsequent molecular characterization of viable circulating tumor cells. Int. J. Cancer 2016, 138, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Chudziak, J.; Burt, D.J.; Mohan, S.; Rothwell, D.G.; Mesquita, B.; Antonello, J.; Dalby, S.; Ayub, M.; Priest, L.; Carter, L.; et al. Clinical evaluation of a novel microfluidic device for epitope-independent enrichment of circulating tumour cells in patients with small cell lung cancer. Analyst 2016, 141, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Batis, N.; Franke, A.C.; Clancey, G.; Hartley, M.; Ryan, G.; Brooks, J.; Southam, A.D.; Barnes, N.; Parish, J.; et al. Repurposed quinacrine synergizes with cisplatin, reducing the effective dose required for treatment of head and neck squamous cell carcinoma. Oncotarget 2019, 10, 5229–5244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisanti, S.; Almici, C.; Consoli, F.; Buglione, M.; Verardi, R.; Bolzoni-Villaret, A.; Bianchetti, A.; Ciccarese, C.; Mangoni, M.; Ferrari, L.; et al. Circulating tumor cells in patients with recurrent or metastatic head and neck carcinoma: Prognostic and predictive significance. PLoS ONE 2014, 9, e103918. [Google Scholar] [CrossRef] [PubMed]
- Jatana, K.; Balasubramanian, P.; Lang, J.; Yang, L.; Jatana, C.; White, E.; Agrawal, A.; Ozer, E.; Schuller, D.; Teknos, T.; et al. Significance of Circulating Tumor Cells in Patients With Squamous Cell Carcinoma of the Head and Neck: Initial Results. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 1274–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inhestern, J.; Oertel, K.; Stemmann, V.; Schmalenberg, H.; Dietz, A.; Rotter, N.; Veit, J.; Görner, M.; Sudhoff, H.; Junghanß, C.; et al. Prognostic Role of Circulating Tumor Cells during Induction Chemotherapy Followed by Curative Surgery Combined with Postoperative Radiotherapy in Patients with Locally Advanced Oral and Oropharyngeal Squamous Cell Cancer. PLoS ONE 2015, 10, e0132901. [Google Scholar] [CrossRef] [PubMed]
- Mcmullen, K.P.; Chalmers, J.J.; Lang, J.C.; Kumar, P.; Jatana, K.R. Circulating tumor cells in head and neck cancer: A review. World J. Otorhinolaryngol. Neck Surg. 2016, 2, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alber, A.B.; Suter, D.M. Dynamics of protein synthesis and degradation through the cell cycle. Cell Cycle 2019, 18, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.-P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol. Cancer Res. 2015, 13, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodie, T.M.; Tosevski, V.; Medová, M. OMIP-045: Characterizing human head and neck tumors and cancer cell lines with mass cytometry. Cytom. Part A 2018, 93, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Zoli, W.; Rigaud, M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: Characterization of cell subpopulations. Ann. Transl. Med. 2014, 2, 109. [Google Scholar] [CrossRef] [PubMed]
- Obermayr, E.; Agreiter, C.; Schuster, E.; Fabikan, H.; Weinlinger, C.; Baluchova, K.; Hamilton, G.; Hochmair, M.; Zeillinger, R. Molecular Characterization of Circulating Tumor Cells Enriched by A Microfluidic Platform in Patients with Small-Cell Lung Cancer. Cells 2019, 8, 880. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Xu, L.; Mao, X.; Grey, A.; Scandura, G.; Guo, T.; Burke, E.; Marzec, J.; Abdu, S.; Stankiewicz, E.; Davies, C.R.; et al. Noninvasive Detection of Clinically Significant Prostate Cancer Using Circulating Tumor Cells. J. Urol. 2019, 23, 73–82. [Google Scholar] [CrossRef]
- Künzel, J.; Gribko, A.; Lu, Q.; Stauber, R.H.; Wünsch, D. Nanomedical detection and downstream analysis of circulating tumor cells in head and neck patients. Biol. Chem. 2019, 400, 1465–1479. [Google Scholar] [CrossRef] [PubMed]
- Gribko, A.; Künzel, J.; Wünsch, D.; Lu, Q.; Nagel, S.M.; Knauer, S.K.; Stauber, R.H.; Ding, G.-B. Is small smarter? Nanomaterial-based detection and elimination of circulating tumor cells: Current knowledge and perspectives. Int. J. Nanomed. 2019, 14, 4187–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavridou, M.; Mastoraki, S.; Strati, A.; Koutsodontis, G.; Klinakis, A.; Psyrri, A.; Lianidou, E. Direct comparison of size-dependent versus EpCAM-dependent CTC enrichment at the gene expression and DNA methylation level in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 6551. [Google Scholar] [CrossRef] [Green Version]
- Kulasinghe, A.; Zhou, J.; Kenny, L.; Papautsky, I.; Punyadeera, C. Capture of Circulating Tumour Cell Clusters Using Straight Microfluidic Chips. Cancers 2019, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.H.K.; Tessier, S.N.; Miyamoto, D.T.; Miller, K.L.; Bookstaver, L.D.; Carey, T.R.; Stannard, C.J.; Thapar, V.; Tai, E.C.; Vo, K.D.; et al. Whole blood stabilization for the microfluidic isolation and molecular characterization of circulating tumor cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single Cell Profiling of Circulating Tumor Cells: Transcriptional Heterogeneity and Diversity from Breast Cancer Cell Lines. PLoS ONE 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, A.; Ma, Y.; Ding, P.; Young, F.; Chua, W.; Balakrishnar, B.; Dransfield, D.; de Souza, P.; Becker, T. CTC-mRNA (AR-V7) Analysis from Blood Samples—Impact of Blood Collection Tube and Storage Time. Int. J. Mol. Sci. 2017, 18, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, H.; Takahashi, H.; Kuwabara-Yokobori, Y.; Shino, M.; Chikamatsu, K. Molecular profiling of circulating tumor cells predicts clinical outcome in head and neck squamous cell carcinoma. Oral Oncol. 2020, 102, 104558. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.-J.; Hsieh, C.-H.; Hung, F.-C.; Wang, H.-M.; Chou, W.-P.; Wu, M.-H. The Integration of a Three-Dimensional Spheroid Cell Culture Operation in a Circulating Tumor Cell (CTC) Isolation and Purification Process: A Preliminary Study of the Clinical Significance and Prognostic Role of the CTCs Isolated from the Blood Samples of head and neck cancer patients. Cancers 2019, 11, 783. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Zamarchi, R. Single-Cell Analysis of Circulating Tumor Cells: How Far Have We Come in the -Omics Era? Front. Genet. 2019, 10, 958. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Phenotype Marker | Antibody | Host Species | Type | Clone | Flourochrome | Source |
|---|---|---|---|---|---|---|
| epithelial | EpCAM (CD326) | mouse | Monoclonal, conjugated | 9C4 | Alexa Fluor 488 | Biolegend |
| mesenchymal | N-cadherin (CD325) | mouse | Monoclonal, conjugated | 8C11 | APC | Biolegend |
| pan-leukocyte | CD45 | mouse | Monoclonal, conjugated | 2D1 | PE | Biolegend |
| nuclear dye | DAPI |
| Gene Ontology (GO) Category and Gene Set | Gene Set Size | Direction | p Value | q Value |
|---|---|---|---|---|
| GO Biological process | ||||
| GO:0016071 mRNA metabolic process | 750 | Down | 1.30 × 10−7 | 0.000694 |
| GO:0006402 mRNA catabolic process | 330 | Down | 4.70 × 10−7 | 0.000975 |
| GO:0006396 RNA processing | 884 | Down | 6.20 × 10−7 | 0.000975 |
| GO:0006412 translation | 560 | Down | 7.33 × 10−7 | 0.000975 |
| GO:0022613 ribonucleoprotein complex biogenesis | 423 | Down | 1.09 × 10−6 | 0.000999 |
| GO Cell component | ||||
| GO:1990904 ribonucleoprotein complex | 637 | Down | 2.79 × 10−10 | 0.000000194 |
| GO:0005730 nucleolus | 843 | Down | 0.000000324 | 0.000113 |
| GO:0005840 ribosome | 210 | Down | 0.000000945 | 0.000219 |
| GO:0044391 ribosomal subunit | 174 | Down | 0.00000153 | 0.000266 |
| GO:0022626 cytosolic ribosome | 99 | Down | 0.00000884 | 0.00123 |
| GO Molecular function | ||||
| GO:0003723 RNA binding | 1452 | Down | 2.20 × 10−14 | 1.98 × 10−11 |
| GO:0003735 structural constituent of ribosome | 151 | Down | 1.91 × 10−6 | 0.000779 |
| GO:0044877 protein-containing complex binding | 836 | Down | 2.60 × 10−6 | 0.000779 |
| GO:0005198 structural molecule activity | 374 | Down | 7.24 × 10−6 | 0.00162 |
| GO:0045296 cadherin binding | 291 | Down | 9.66 × 10−5 | 0.0174 |
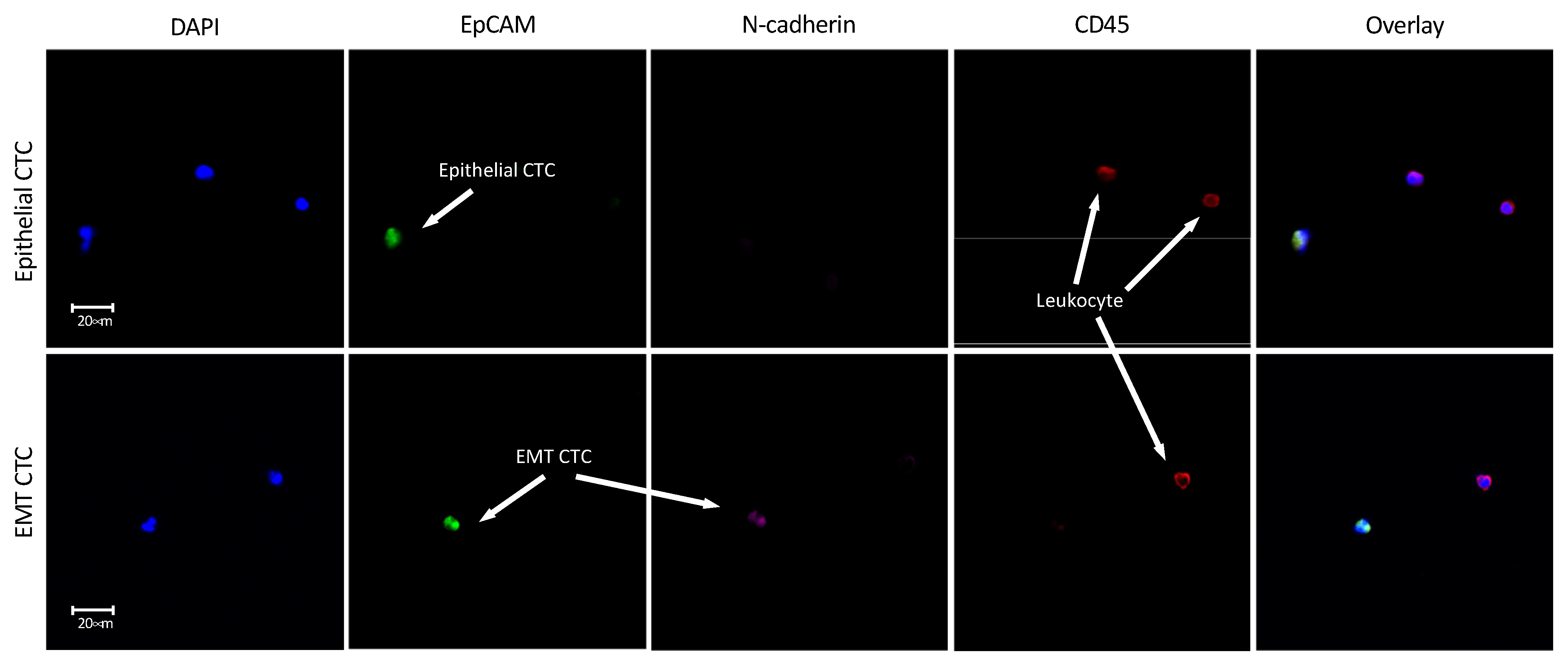
| Pt. ID | Age/Sex | Site | Stage | EpCAM+ CTC (DAPI+, CD45−) | EpCAM+/N-cadherin+ CTC (DAPI+, CD45−) |
|---|---|---|---|---|---|
| 1 | 84 F | Oral | T4a N2b M0 | + | + |
| 2 | 74 M | Laryngeal (recurrence) | T4a N0 M0 | − | − |
| 3 | 69 F | Oral | T4a N1 M0 | + | + |
| 4 | 73 M | Hypopharyngeal | T3 N0 M0 | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payne, K.; Brooks, J.M.; Taylor, G.S.; Batis, N.; Noyvert, B.; Pan, Y.; Nankivell, P.; Mehanna, H. Immediate Sample Fixation Increases Circulating Tumour Cell (CTC) Capture and Preserves Phenotype in Head and Neck Squamous Cell Carcinoma: Towards a Standardised Approach to Microfluidic CTC Biomarker Discovery. Cancers 2021, 13, 5519. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215519
Payne K, Brooks JM, Taylor GS, Batis N, Noyvert B, Pan Y, Nankivell P, Mehanna H. Immediate Sample Fixation Increases Circulating Tumour Cell (CTC) Capture and Preserves Phenotype in Head and Neck Squamous Cell Carcinoma: Towards a Standardised Approach to Microfluidic CTC Biomarker Discovery. Cancers. 2021; 13(21):5519. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215519
Chicago/Turabian StylePayne, Karl, Jill M. Brooks, Graham S. Taylor, Nikolaos Batis, Boris Noyvert, Yi Pan, Paul Nankivell, and Hisham Mehanna. 2021. "Immediate Sample Fixation Increases Circulating Tumour Cell (CTC) Capture and Preserves Phenotype in Head and Neck Squamous Cell Carcinoma: Towards a Standardised Approach to Microfluidic CTC Biomarker Discovery" Cancers 13, no. 21: 5519. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215519