
Type I Interferon Promotes Antitumor T Cell Response in CRPC by Regulating MDSC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Lines and Reagents
2.3. Myc-CaP Treatment In Vitro
2.4. MDSCs Differentiation from Bone Morrow Cells
2.5. Isolation of G-MDSCs
2.6. T Cell Inhibition by G-MDSCs
2.7. Isolation of Single Cells from Tumors
2.8. Flow Cytometry Analysis
2.9. Cytokine Production Analysis
2.10. RT-qPCR
2.11. Mouse CRPC Model and Immunotherapy
2.12. Statistical Analysis
3. Results
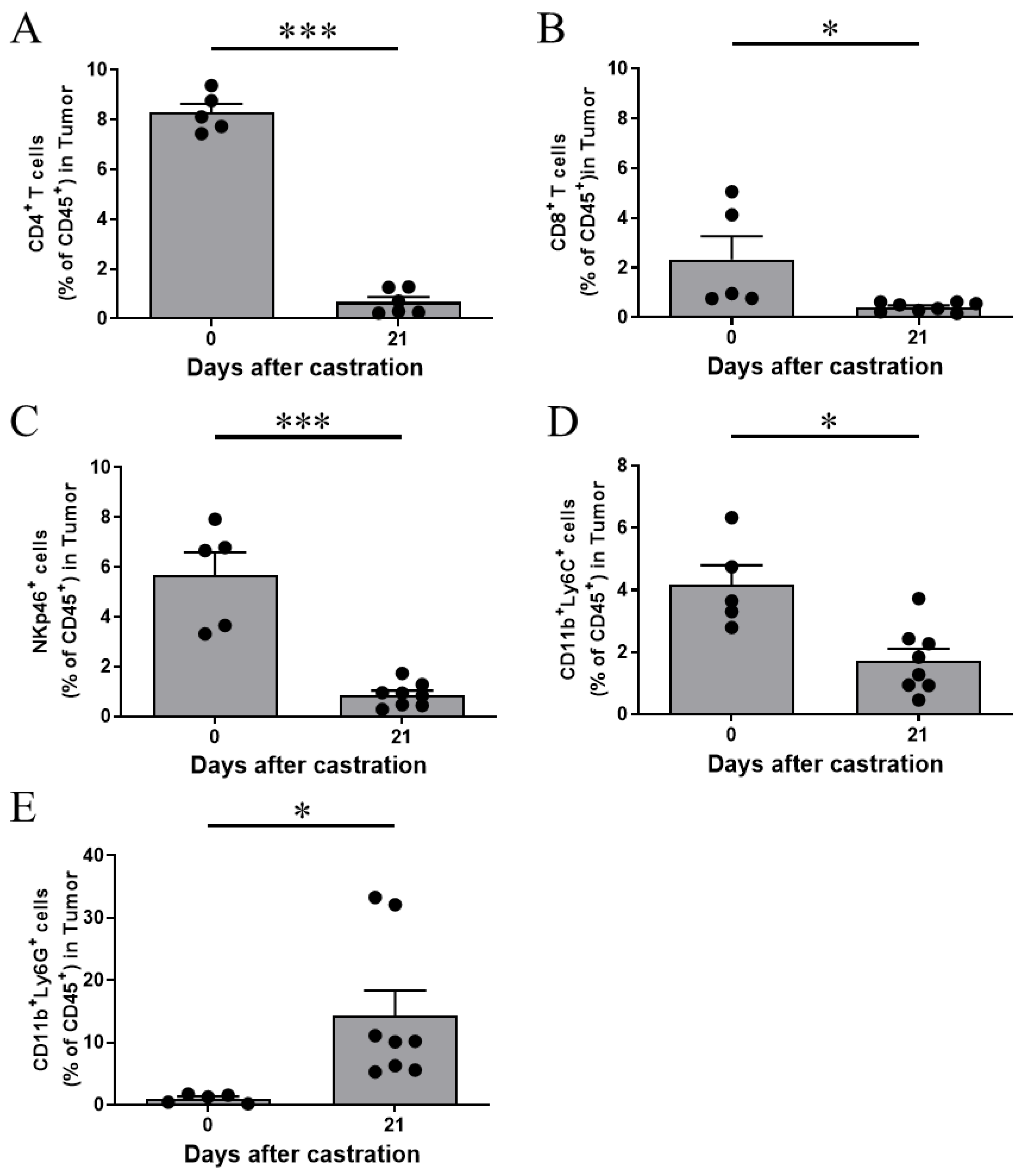
3.1. G-MDSCs Are Increased in Prostate TME Following Castration
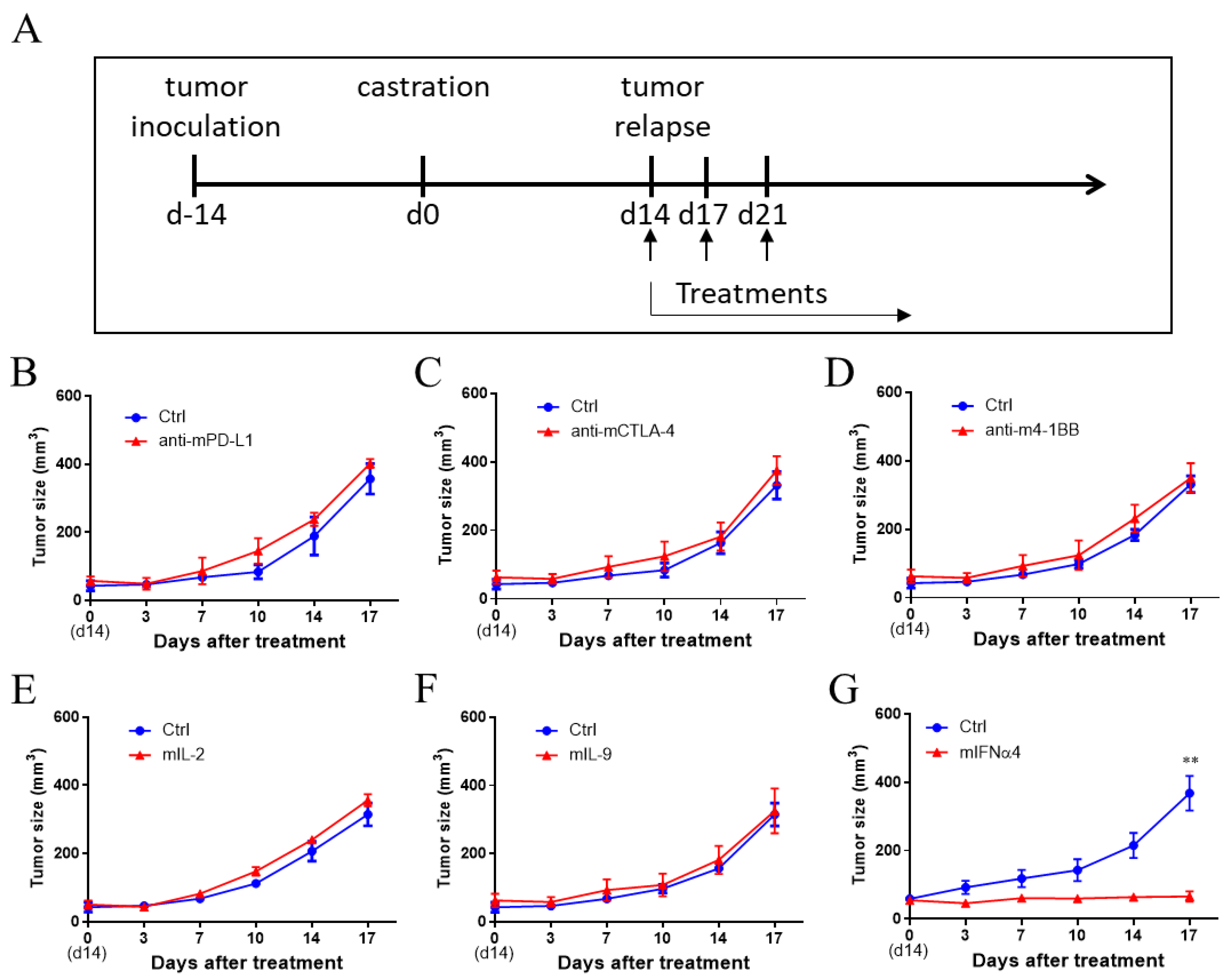
3.2. IFNα Was Effective in Controlling CRPC
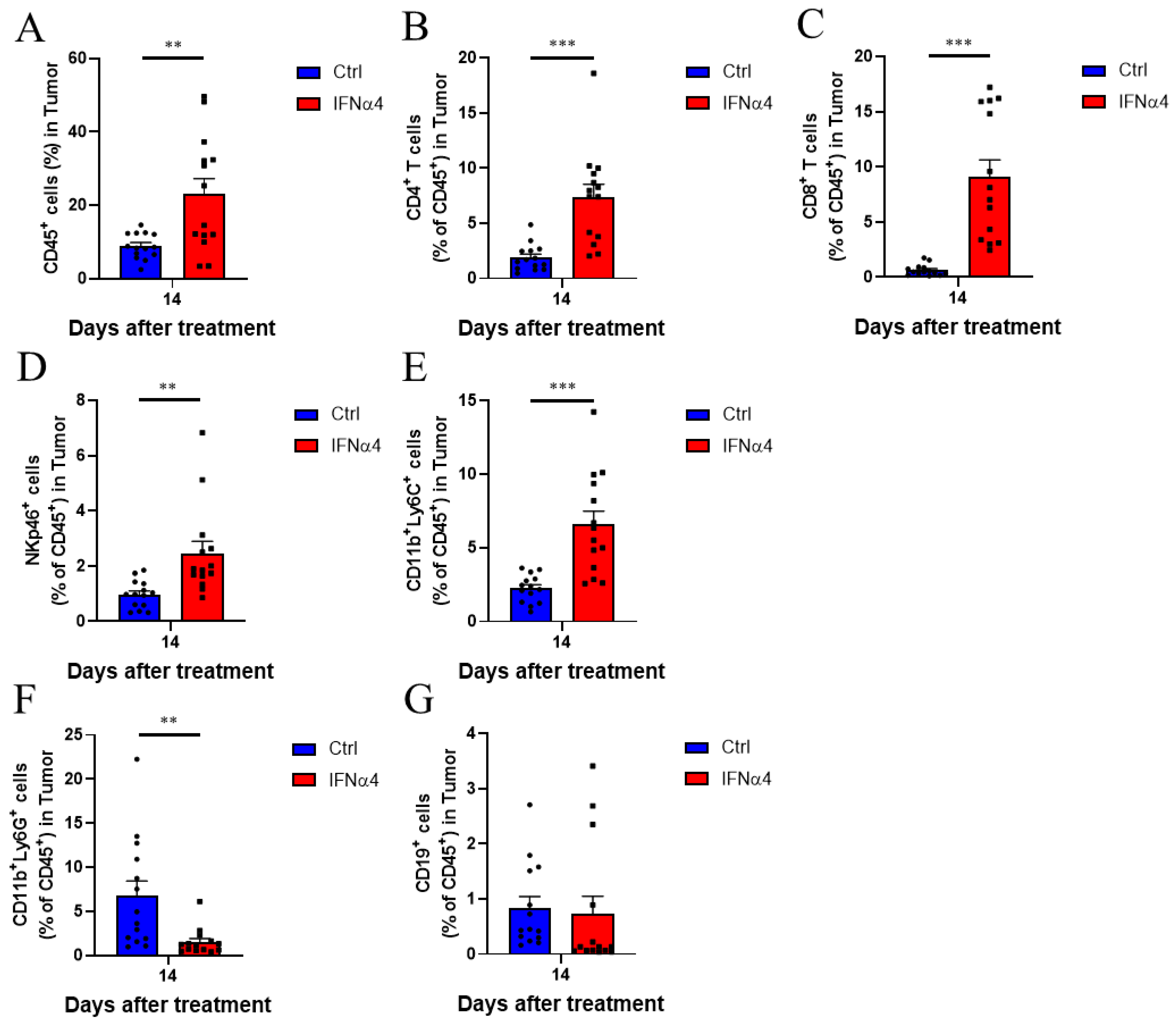
3.3. IFNα4 Reduced Immunosuppression in the TME
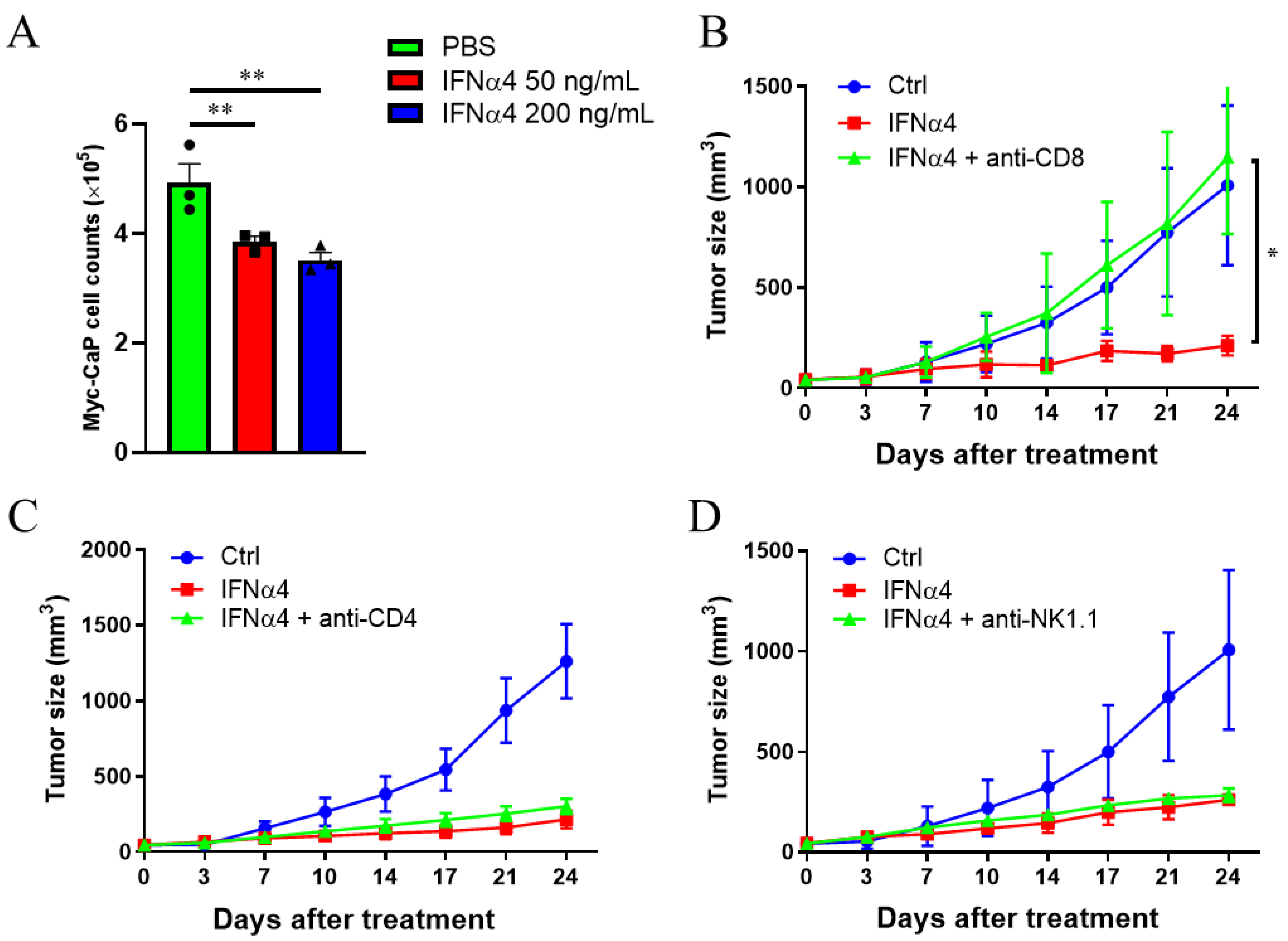
3.4. Cytotoxic T Cells Are Critical for IFNα-Mediated Therapeutic Effect on CRPC
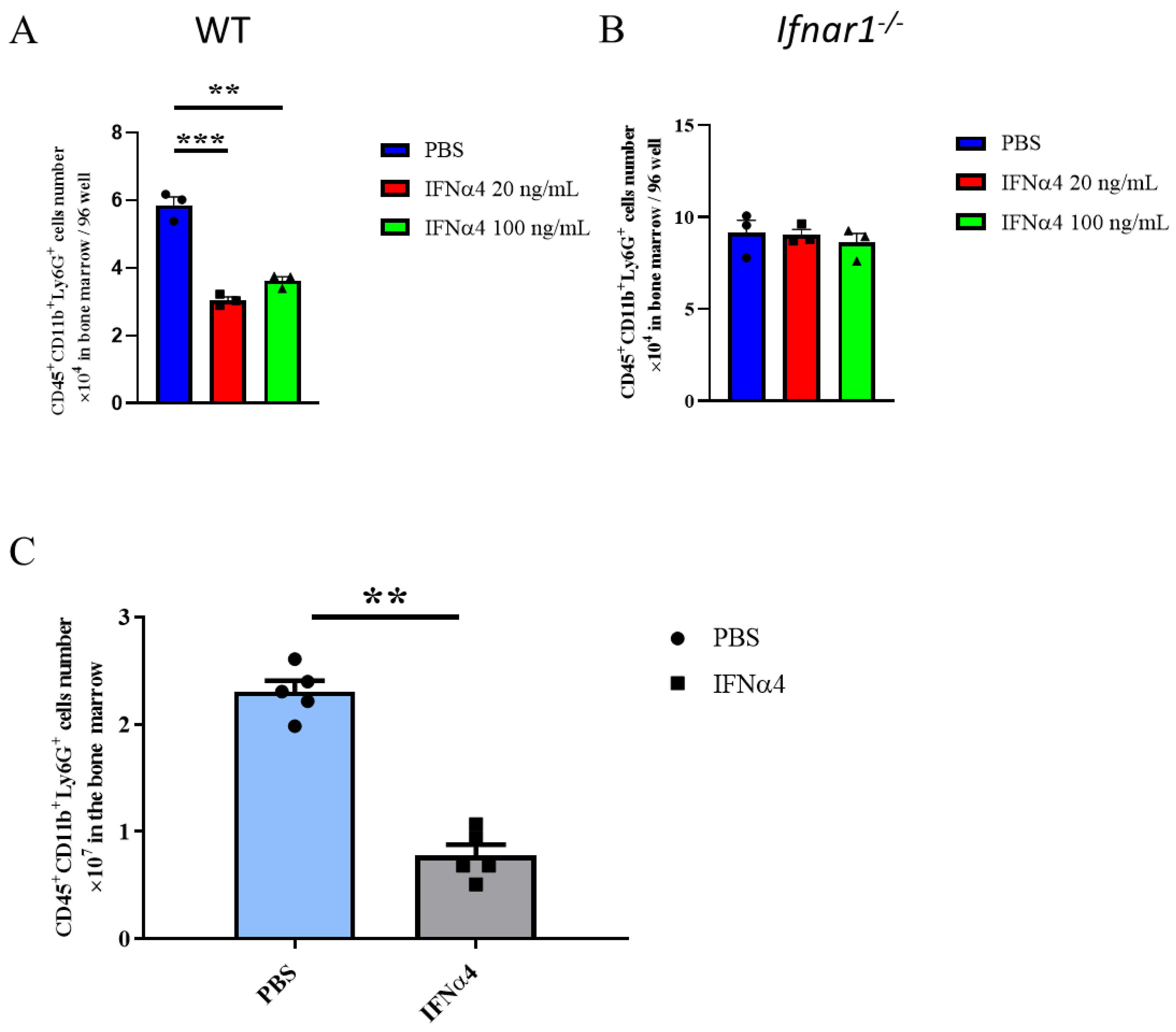
3.5. IFNα4 Inhibited the Differentiation or Proliferation of G-MDSCs
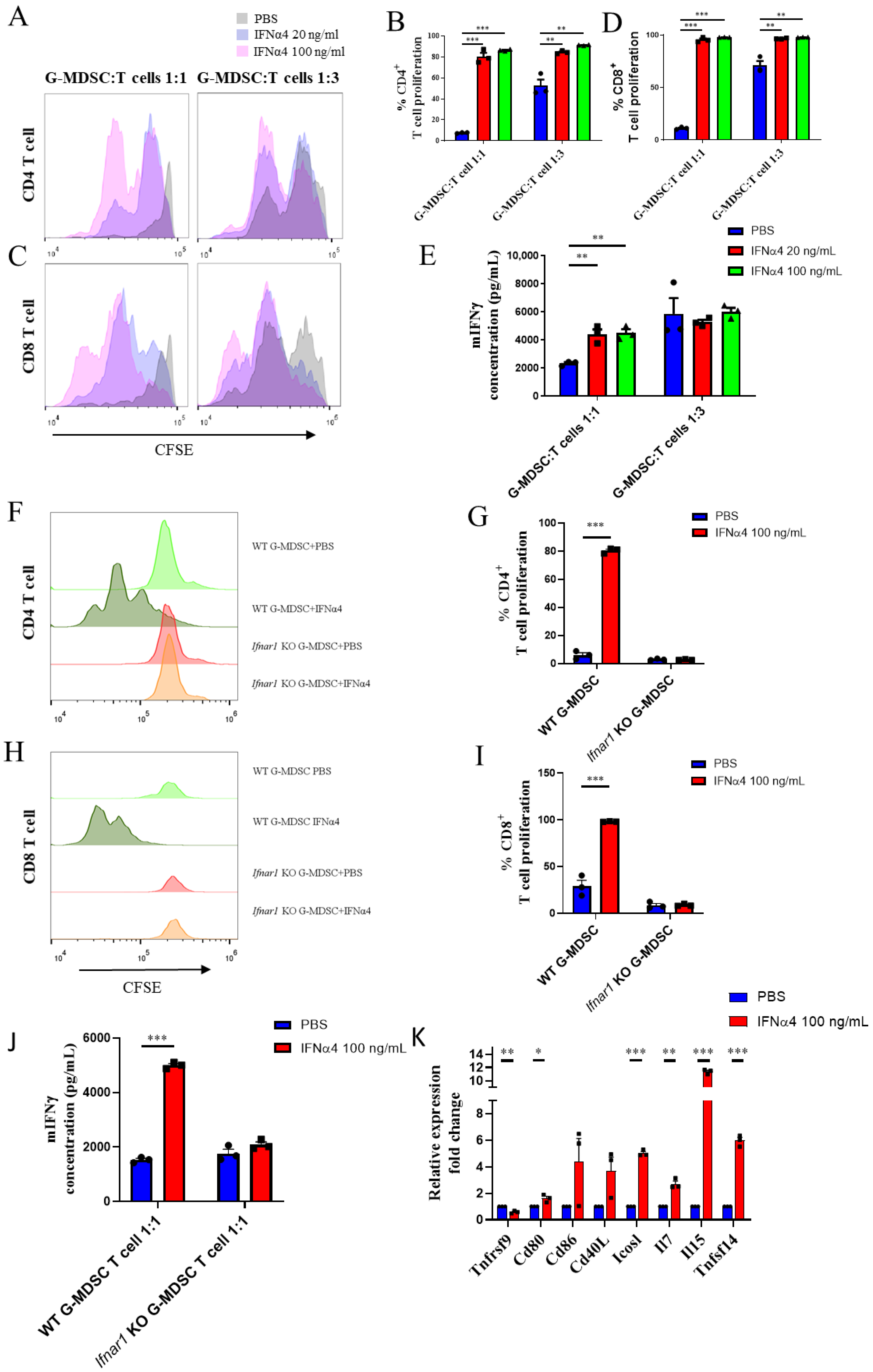
3.6. IFNα4 Inhibited the Immune Suppressive Function of G-MDSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Hsing, A.W.; Tsao, L.; Devesa, S.S. International trends and patterns of prostate cancer incidence and mortality. Int. J. Cancer 2000, 85, 60–67. [Google Scholar] [CrossRef]
- Wong, M.C.S.; Goggins, W.B.; Wang, H.H.X.; Fung, F.D.H.; Leung, C.; Wong, S.Y.S.; Ng, C.F.; Sung, J.J.Y. Global Incidence and Mortality for Prostate Cancer: Analysis of Temporal Patterns and Trends in 36 Countries. Eur. Urol. 2016, 70, 862–874. [Google Scholar] [CrossRef] [Green Version]
- Nam, R.K.; Cheung, P.; Herschorn, S.; Saskin, R.K.; Su, J.D.; Klotz, L.H.; Chang, M.; Kulkarni, G.S.; Lee, Y.; Kodama, R.T.; et al. Incidence of complications other than urinary incontinence or erectile dysfunction after radical prostatectomy or radiotherapy for prostate cancer: A population-based cohort study. Lancet Oncol. 2014, 15, 223–231. [Google Scholar] [CrossRef]
- Auchus, R.J.; Sharifi, N. Sex Hormones and Prostate Cancer. Annu. Rev. Med. 2020, 71, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Reid, A.H.M.; A’Hern, R.; Parker, C.; Oommen, N.B.; Folkerd, E.; Messiou, C.; Molife, L.R.; Maier, G.; Thompson, E.; et al. Selective Inhibition of CYP17 With Abiraterone Acetate Is Highly Active in the Treatment of Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2009, 27, 3742–3748. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Enzalutamide: A Review in Castration-Resistant Prostate Cancer. Drugs 2018, 78, 1913–1924. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I interferon receptors: Biochemistry and biological functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef] [Green Version]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef] [Green Version]
- Mowen, K.A.; Tang, J.; Zhu, W.; Schurter, B.T.; Shuai, K.; Herschman, H.R.; David, M. Arginine methylation of STAT1 modulates IFN alpha/beta-induced transcription. Cell 2001, 104, 731–741. [Google Scholar] [CrossRef]
- Lu, L.; Zhu, F.; Zhang, M.L.; Li, Y.G.; Drennan, A.C.; Kimpara, S.; Rumball, I.; Selzer, C.; Cameron, H.; Kellicut, A.; et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E498–E505. [Google Scholar] [CrossRef] [Green Version]
- Hobeika, A.C.; Subramaniam, P.S.; Johnson, H.M. IFNalpha induces the expression of the cyclin-dependent kinase inhibitor p21 in human prostate cancer cells. Oncogene 1997, 14, 1165–1170. [Google Scholar] [CrossRef]
- Hiroishi, K.; Tuting, T.; Lotze, M.T. IFN-alpha-expressing tumor cells enhance generation and promote survival of tumor-specific CTLs. J. Immunol. 2000, 164, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.J.; Wei, H.M.; Sun, R.; Tian, Z.G. IFN alpha regulates NK cell cytotoxicity through STAT1 pathway. Cytokine 2003, 23, 190–199. [Google Scholar] [CrossRef]
- Braun, D.; Caramalho, I.; Demengeot, J. IFN-alpha/beta enhances BCR-dependent B cell responses. Int. Immunol. 2002, 14, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 2019, 348, 217–262. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Huang, R.; Zong, X.; He, X.; Mo, W. INFalpha-2b inhibitory effects on CD4(+)CD25(+)FOXP3(+) regulatory T cells in the tumor microenvironment of C57BL/6 J mice with melanoma xenografts. BMC Cancer 2016, 16, 397. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Sun, S.Q.; Hwang, I.K.; Tough, D.F.; Sprent, J. Potent and selective stimulation of memory-phenotype CD8(+) T cells in vivo by IL-15. Immunity 1998, 8, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Jiang, P.; Wei, S.H.; Xu, X.F.; Wang, J.J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Toso, A.; Alimonti, A. Molecular Pathways: Targeting Tumor-Infiltrating Myeloid-Derived Suppressor Cells for Cancer Therapy. Clin. Cancer Res. 2015, 21, 3108–3112. [Google Scholar] [CrossRef] [Green Version]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Pal, S.; Nandi, M.; Dey, D.; Chakraborty, B.C.; Shil, A.; Ghosh, S.; Banerjee, S.; Santra, A.; Ahammed, S.K.M.; Chowdhury, A.; et al. Myeloid-derived suppressor cells induce regulatory T cells in chronically HBV infected patients with high levels of hepatitis B surface antigen and persist after antiviral therapy. Aliment. Pharmacol. Ther. 2019, 49, 1346–1359. [Google Scholar] [CrossRef]
- Huang, B.; Pan, P.Y.; Li, Q.S.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1(+)CD115(+) immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.F.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Pu, Y.; Xu, M.; Liang, Y.; Yang, K.; Guo, Y.; Yang, X.; Fu, Y.X. Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 2016, 8, 333ra347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Hassan, D.; Aldawsari, H.M.; Molugulu, N.; Shukla, R.; Kesharwani, P. Immune checkpoint inhibitors: A promising anticancer therapy. Drug Discov. Today 2020, 25, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Q.; Zhang, C.; Jin, S.J.; Gao, Z.H.; Cao, J.; Wang, A.Q.; Li, D.W.; Wang, Q.; Sun, X.; Bai, D.S. Progress of immune checkpoint therapy in the clinic. Oncol. Rep. 2019, 41, 3–14. [Google Scholar] [CrossRef]
- Tang, H.D.; Qiao, J.; Fu, Y.X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef]
- Chen, S.H.; Lee, L.F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.A.; et al. Combination of 4-1BB Agonist and PD-1 Antagonist Promotes Antitumor Effector/Memory CD8 T Cells in a Poorly Immunogenic Tumor Model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Qu, Q.X.; Zhu, X.Y.; Du, W.W.; Wang, H.B.; Shen, Y.; Zhu, Y.B.; Chen, C. 4-1BB Agonism Combined With PD-L1 Blockade Increases the Number of Tissue-Resident CD8+T Cells and Facilitates Tumor Abrogation. Front. Immunol. 2020, 11, 577. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Dechant, C.; Cui, X.Y.; Chongsathidkiet, P.; Wilkinson, D.; Waibl-Polania, J.; Sanchez-Perez, L.; Fecci, P.E. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin. Cancer Res. 2020, 26, 1349–1358. [Google Scholar] [CrossRef]
- Qi, X.Y.; Li, F.L.; Wu, Y.; Cheng, C.; Han, P.; Wang, J.Y.; Yang, X.M. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with Fc gamma R affinity. Nat. Commun. 2019, 10, 2141. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The First Effective Immunotherapy for Human Cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Zheng, N.B.; Lu, Y. Targeting the IL-9 pathway in cancer immunotherapy. Hum. Vaccines Immunother. 2020, 16, 2333–2340. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-alpha and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef]
- Shiota, M.; Yokomizo, A.; Eto, M. Taxane Chemotherapy for Hormone-naive Prostate Cancer with its expanding Role as Breakthrough Strategy. Front. Oncol. 2016, 5, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, V.; Gladney, W.; Plesa, G.; Vapiwala, N.; Carpenter, E.L.; Maude, S.L.; Lal, P.; Lacey, S.F.; Melenhorst, J.J.; Fraietta, J.; et al. A phase I clinical trial of PSMA-directed/TGF beta-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2020, 38. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.Z.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.Y.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.P.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [Green Version]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. 2013, 13, 5. [Google Scholar] [PubMed]
- Jiang, T.; Zhou, C.C.; Ren, S.X. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Zhu, Z.W.; Bai, Q.; Brady, T.J.; Xiao, H.P.; Wakefield, M.R.; Fang, Y.J. The Role of Interleukin-9 in Cancer. Pathol. Oncol. Res. 2020, 26, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Caraglia, M.; Marra, M.; Pelaia, G.; Maselli, R.; Caputi, M.; Marsico, S.A.; Abbruzzese, A. Alpha-interferon and its effects on signal transduction pathways. J. Cell. Physiol. 2005, 202, 323–335. [Google Scholar] [CrossRef]
- Maeda, S.; Wada, H.; Naito, Y.; Nagano, H.; Simmons, S.; Kagawa, Y.; Naito, A.; Kikuta, J.; Ishii, T.; Tomimaru, Y.; et al. Interferon-alpha acts on the S/G2/M phases to induce apoptosis in the G1 phase of an IFNAR2-expressing hepatocellular carcinoma cell line. J. Biol. Chem. 2014, 289, 23786–23795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tai, Y.T.; Ho, M.Z.G.; Qiu, L.G.; Anderson, K.C. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp. Hematol. Oncol. 2017, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.F.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Velho, P.I.; Antonarakis, E.S. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert Rev. Clin. Pharmcol. 2018, 11, 475–486. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, L.; Xu, G.; Cao, J.; Li, M.; Zhang, H.; Li, F.; Qi, X.; Zhang, X.; Li, Z.; Han, P.; et al. Type I Interferon Promotes Antitumor T Cell Response in CRPC by Regulating MDSC. Cancers 2021, 13, 5574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215574
Fan L, Xu G, Cao J, Li M, Zhang H, Li F, Qi X, Zhang X, Li Z, Han P, et al. Type I Interferon Promotes Antitumor T Cell Response in CRPC by Regulating MDSC. Cancers. 2021; 13(21):5574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215574
Chicago/Turabian StyleFan, Lilv, Guiliang Xu, Jingjing Cao, Min Li, Huihui Zhang, Fanlin Li, Xinyue Qi, Xiaoqing Zhang, Zeyu Li, Ping Han, and et al. 2021. "Type I Interferon Promotes Antitumor T Cell Response in CRPC by Regulating MDSC" Cancers 13, no. 21: 5574. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215574