
In Vivo and In Vitro Models of Hepatocellular Carcinoma: Current Strategies for Translational Modeling

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Hepatocellular Carcinoma: Worldwide Trends and Mechanisms
1.1. Epidemiology and Contributing Factors
1.2. Hepatocarcinogenesis
2. In Vivo Models of HCC
2.1. Syngeneic and Xenograft Mouse Models
2.2. Chemical-Induced Rodent Models
2.2.1. Diethylnitrosamine (DEN)
2.2.2. Carbon Tetrachloride (CCl4)
2.2.3. Thioacetamide (TAA)
2.2.4. Phenobarbital (PB)
2.2.5. Resistant Hepatocyte Model
2.2.6. Aflatoxin B1
2.2.7. Miscellaneous Chemicals
2.2.8. Impact of Genetic Background and Sex
2.3. Diet-Induced Rodent Models
2.3.1. NAFLD-Associated HCC Models
2.3.2. ALD-Associated HCC Models
2.4. Genetically Engineered Mouse Models
2.4.1. Hepatitis Virus Transgenic Mice
2.4.2. Other Gene Expression Systems
2.5. Humanized Mouse Models
3. In Vitro Models of HCC
3.1. Primary Hepatocytes
3.2. Hepatic Cell Lines
3.3. Co-Cultures
3.4. Stem Cell-Derived Models
3.5. Spheroid and Organoid Models
3.6. Precision-Cut Liver Slices
4. Therapeutic Relevance of the HCC Models
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Greten, T.F.; Papendorf, F.; Bleck, J.S.; Kirchhoff, T.; Wohlberedt, T.; Kubicka, S.; Klempnauer, J.; Galanski, M.; Manns, M.P. Survival rate in patients with hepatocellular carcinoma: A retrospective analysis of 389 patients. Br. J. Cancer 2005, 92, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Op den Winkel, M.; Nagel, D.; Sappl, J.; op den Winkel, P.; Lamerz, R.; Zech, C.J.; Straub, G.; Nickel, T.; Rentsch, M.; Stieber, P.; et al. Prognosis of Patients with Hepatocellular Carcinoma. Validation and Ranking of Established Staging-Systems in a Large Western HCC-Cohort. PLoS ONE 2012, 7, e45066. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Kim, W.R.; Coelho, R.; Mettler, T.A.; Benson, J.T.; Sanderson, S.O.; Therneau, T.M.; Kim, B.; Roberts, L.R. Cirrhosis Is Present in Most Patients With Hepatitis B and Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baecker, A.; Liu, X.; La Vecchia, C.; Zhang, Z.-F. Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors. Eur. J. Cancer Prev. 2018, 27, 205–212. [Google Scholar] [CrossRef]
- Makarova-rusher, O.V.; Altekruse, S.F.; Mcneel, T.S.; Ulahannan, S.; Duffy, A.G.; Graubard, B.I.; Greten, T.F.; Mcglynn, K.A. Population Attributable Fractions of Risk Factors for Hepatocellular Carcinoma in the United States. Cancer 2016, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Bravi, F.; Bosetti, C.; Tavani, A.; Gallus, S.; La Vecchia, C. Coffee Reduces Risk for Hepatocellular Carcinoma: An Updated Meta-analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 1413–1421.e1. [Google Scholar] [CrossRef] [PubMed]
- Bravi, F.; Tavani, A.; Bosetti, C.; Boffetta, P.; La Vecchia, C. Coffee and the risk of hepatocellular carcinoma and chronic liver disease: A systematic review and meta-analysis of prospective studies. Eur. J. Cancer Prev. 2017, 26, 368–377. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Coleman, W.B. Mechanisms of Human Hepatocarcinogenesis. Curr. Mol. Med. 2003, 3, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, L.; Guan, X.-Y. The genetic and epigenetic alterations in human hepatocellular carcinoma: A recent update. Protein Cell 2014, 5, 673–691. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Zhang, B.; Sun, H.; Xu, Q.; Tan, Y.; Wang, G.; Luo, Q.; Xu, W.; Yang, S.; Li, J.; et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015, 6, 20160–20176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Sun, F.X.; Tang, Z.Y.; Liu, K.D.; Ye, S.L.; Xue, Q.; Gao, D.M.; Ma, Z.C. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. Int. J. Cancer 1996, 66, 239–243. [Google Scholar] [CrossRef]
- Genda, T.; Sakamoto, M.; Ichida, T.; Asakura, H.; Kojiro, M.; Narumiya, S.; Hirohashi, S. Cell motility mediated by rho and rho-associated protein kinase plays a critical role in intrahepatic metastasis of human hepatocellular carcinoma. Hepatology 1999, 30, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Blumer, T.; Fofana, I.; Matter, M.S.; Wang, X.; Montazeri, H.; Calabrese, D.; Coto-Llerena, M.; Boldanova, T.; Nuciforo, S.; Kancherla, V.; et al. Hepatocellular Carcinoma Xenografts Established From Needle Biopsies Preserve the Characteristics of the Originating Tumors. Hepatol. Commun. 2019, 3, 971–986. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz- scid IL2R γ null Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [Green Version]
- Aruga, A.; Takasaki, K.; Hanyu, F. Establishment and characterization of liver metastatic model of human hepatoma in nude mice. Int. Hepatol. Commun. 1993, 1, 138–145. [Google Scholar] [CrossRef]
- Yan, M.; Li, H.; Zhao, F.; Zhang, L.; Ge, C.; Yao, M.; Li, J. Establishment of NOD/SCID mouse models of human hepatocellular carcinoma via subcutaneous transplantation of histologically intact tumor tissue. Chinese J. Cancer Res. 2013, 25, 289–298. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans: Chemical Agents and Related Occu-pations. IARC Monogr. Eval. Carcinog. Risks Chem. Man. 2012, 10F, 1–628.
- Tricker, A.R.; Kubacki, S.J. Review of the occurrence and formation of non-volatile N -nitroso compounds in foods. Food Addit. Contam. 1992, 9, 39–69. [Google Scholar] [CrossRef]
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res. Toxicol. Environ. Mutagen. 1999, 443, 129–138. [Google Scholar] [CrossRef]
- Tsuda, S. DNA Damage Induced by Red Food Dyes Orally Administered to Pregnant and Male Mice. Toxicol. Sci. 2001, 61, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, S.S.; Granby, K.; Duedahl-Olesen, L. Formation and mitigation of N-nitrosamines in nitrite preserved cooked sausages. Food Chem. 2015, 174, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Wanibuchi, H.; Morimura, K.; Gonzalez, F.J.; Fukushima, S. Role of CYP2E1 in Diethylnitrosamine-Induced Hepatocarcinogenesis in vivo. Cancer Res. 2007, 67, 11141–11146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, Z.; Wang, G.-J.; Zhang, H.-X.; Gao, N.; Wang, J.; Wang, C.-E.; Chang, Z.; Fang, Y.; Zhang, Y.-F.; et al. Higher CYP2E1 Activity Correlates with Hepatocarcinogenesis Induced by Diethylnitrosamine. J. Pharmacol. Exp. Ther. 2018, 365, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verna, L. N-Nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Dyroff, M.C.; Bedell, M.A.; Popp, J.A.; Huh, N.; Kirstein, U.; Rajewsky, M.F. O4-ethyldeoxythymidine, but not O6-ethyldeoxyguanosine, accumulates in hepatocyte DNA of rats exposed continuously to diethylnitrosamine. Proc. Natl. Acad. Sci. USA 1984, 81, 1692–1695. [Google Scholar] [CrossRef] [Green Version]
- Aleksic, K.; Lackner, C.; Geigl, J.B.; Schwarz, M.; Auer, M.; Ulz, P.; Fischer, M.; Trajanoski, Z.; Otte, M.; Speicher, M.R. Evolution of genomic instability in diethylnitrosamine-induced hepatocarcinogenesis in mice. Hepatology 2011, 53, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Connor, F.; Rayner, T.F.; Aitken, S.J.; Feig, C.; Lukk, M.; Santoyo-Lopez, J.; Odom, D.T. Mutational landscape of a chemically-induced mouse model of liver cancer. J. Hepatol. 2018, 69, 840–850. [Google Scholar] [CrossRef]
- Aparicio-Bautista, D.I.; Pérez-Carreón, J.I.; Gutiérrez-Nájera, N.; Reyes-Grajeda, J.P.; Arellanes-Robledo, J.; Vásquez-Garzón, V.R.; Jiménez-García, M.N.; Villa-Treviño, S. Comparative proteomic analysis of thiol proteins in the liver after oxidative stress induced by diethylnitrosamine. Biochim. Biophys. Acta-Proteins Proteom. 2013, 1834, 2528–2538. [Google Scholar] [CrossRef]
- Ogawa, K. Molecular pathology of early stage chemically induced hepatocarcinogenesis. Pathol. Int. 2009, 59, 605–622. [Google Scholar] [CrossRef]
- Diwan, B.A.; Rice, J.M.; Ohshima, M.; Ward, J.M. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCr MTV- and DBA/2NCr mice. Carcinogenesis 1986, 7, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Vesselinovitch, S.D.; Koka, M.; Mihailovich, N.; Rao, K.V.N. Carcinogenicity of diethylnitrosamine in newborn, infant, and adult mice. J. Cancer Res. Clin. Oncol. 1984, 108, 60–65. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Weghorst, T.R.; Weghorst, C.M. Liver tumor promoting ability of corn oil gavage in B6C3F1 male mice. Cancer Lett. 1990, 50, 215–219. [Google Scholar] [CrossRef]
- Kushida, M.; Kamendulis, L.M.; Peat, T.J.; Klaunig, J.E. Dose-Related Induction of Hepatic Preneoplastic Lesions by Diethylnitrosamine in C57BL/6 Mice. Toxicol. Pathol. 2011, 39, 776–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaunig, J.E.; Pereira, M.A.; Ruch, R.J.; Weghorst, C.M. Dose-Response Relationship of Diethylnitrosamine-Initiated Tumors in Neonatal Balb/c Mice: Effect of Phenobarbital Promotion. Toxicol. Pathol. 1988, 16, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weghorst, C.M.; Pereira, M.A.; Klaunig, E. Strain differences in hepatic tumor promotion by phenobarbital in diethylnitrosamine- and dimethylnitrosamine-initiated infant male mice. Carcinogenesis 1989, 10, 1409–1412. [Google Scholar] [CrossRef]
- Goldsworthy, T.L.; Fransson-Steen, R. Quantitation of the Cancer Process in C57BL/6J, B6C3F1 and C3H/HeJ Mice. Toxicol. Pathol. 2002, 30, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Ainslie, G.R.; Kutanzi, K.; Pogribny, I.P.; Muskhelishvili, L.; Izawa, T.; Yamate, J.; Kosyk, O.; Shymonyak, S.; Bradford, B.U.; et al. Molecular Mechanisms of Fibrosis-Associated Promotion of Liver Carcinogenesis. Toxicol. Sci. 2013, 132, 53–63. [Google Scholar] [CrossRef]
- Memon, A.; Pyao, Y.; Jung, Y.; Lee, J.I.; Lee, W.K. A Modified Protocol of Diethylnitrosamine Administration in Mice to Model Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 5461. [Google Scholar] [CrossRef]
- Pereira, M.A.; Herren-Freund, S.L.; Long, R.E. Dose-response relationship of phenobarbital promotion of diethylnitrosamine initiated tumors in rat liver. Cancer Lett. 1986, 32, 305–311. [Google Scholar] [CrossRef]
- Sugie, S.; Okamoto, K.; Watanabe, T.; Tanaka, T.; Mori, H. Suppressive effect of irsogladine maleate on N-methyl-N-nitro-N-nitrosoguanidine (MNNG)-initiated and glyoxal-promoted gastric carcinogenesis in rats. Toxicology 2001, 166, 53–61. [Google Scholar] [CrossRef]
- Tan, Y.; Yin, P.; Tang, L.; Xing, W.; Huang, Q.; Cao, D.; Zhao, X.; Wang, W.; Lu, X.; Xu, Z.; et al. Metabolomics Study of Stepwise Hepatocarcinogenesis From the Model Rats to Patients: Potential Biomarkers Effective for Small Hepatocellular Carcinoma Diagnosis. Mol. Cell. Proteom. 2012, 11, M111.010694. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wu, Z.; Wei, Y.; Shu, L.; Peng, Y. Hepatic inflammation-fibrosis-cancer axis in the rat hepatocellular carcinoma induced by diethylnitrosamine. J. Cancer Res. Clin. Oncol. 2017, 143, 821–834. [Google Scholar] [CrossRef]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clément, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romualdo, G.R.; Prata, G.B.; da Silva, T.C.; Fernandes, A.A.H.; Moreno, F.S.; Cogliati, B.; Barbisan, L.F. Fibrosis-associated hepatocarcinogenesis revisited: Establishing standard medium-term chemically-induced male and female models. PLoS ONE 2018, 13, e0203879. [Google Scholar] [CrossRef]
- Zalatnai, A.; Lapis, K. Decreased hepatocarcinogenic effect of diethylnitrosamine in experimentally induced liver cirrhosis in rat: Delay or inhibition? Cancer Lett. 1994, 79, 1–7. [Google Scholar] [CrossRef]
- Romualdo, G.R.; Grassi, T.F.; Goto, R.L.; Tablas, M.B.; Bidinotto, L.T.; Fernandes, A.A.H.; Cogliati, B.; Barbisan, L.F. An integrative analysis of chemically-induced cirrhosis-associated hepatocarcinogenesis: Histological, biochemical and molecular features. Toxicol. Lett. 2017, 281, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Park, T.J.; Kim, H.S.; Byun, K.H.; Jang, J.J.; Lee, Y.S.; Lim, I.K. Sequential changes in hepatocarcinogenesis induced by diethylnitrosamine plus thioacetamide in Fischer 344 rats: Induction of gankyrin expression in liver fibrosis, pRB degradation in cirrhosis, and methylation ofp16INK4A exon 1 in hepatocellular carcinom. Mol. Carcinog. 2001, 30, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Solt, D.B.; Cayama, E.; Tsuda, H.; Enomoto, K.; Lee, G.; Farber, E. Promotion of liver cancer development by brief exposure to dietary 2-acetylaminofluorene plus partial hepatectomy or carbon tetrachloride. Cancer Res. 1983, 43, 188–191. [Google Scholar]
- Vesselinovitch, S.D.; Hacker, H.J.; Bannasch, P. Histochemical characterization of focal hepatic lesions induced by single diethylnitrosamine treatment in infant mice. Cancer Res. 1985, 45, 2774–2780. [Google Scholar] [PubMed]
- Septer, S.; Edwards, G.; Gunewardena, S.; Wolfe, A.; Li, H.; Daniel, J.; Apte, U. Yes-associated protein is involved in proliferation and differentiation during postnatal liver development. Am. J. Physiol. Liver Physiol. 2012, 302, G493–G503. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Tanaka, G.; Hamada, S.; Namiki, C.; Suzuki, T.; Nakajima, M.; Furihata, C. Dose-dependent alterations in gene expression in mouse liver induced by diethylnitrosamine and ethylnitrosourea and determined by quantitative real-time PCR. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 673, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bannasch, P.; Klimek, F.; Mayer, D. Early bioenergetic changes in hepatocarcinogenesis: Preneoplastic phenotypes mimic responses to insulin and thyroid hormone. J. Bioenerg. Biomembr. 1997, 29, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Bannasch, P. Relevance of Hepatic Preneoplasia for Human Hepatocarcinogenesis. Toxicol. Pathol. 2003, 31, 126–133. [Google Scholar] [CrossRef]
- Lahm, H.; Gittner, K.; Krebs, O.; Sprague, L.; Deml, E.; Oesterle, D.; Hoeflich, A.; Wanke, R.; Wolf, E. Diethylnitrosamine induces long-lasting re-expression of insulin-like growth factor II during early stages of liver carcinogenesis in mice. Growth Horm. IGF Res. 2002, 12, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Bauer-Hofmann, R.; Klimek, F.; Buchmann, A.; Müller, O.; Bannasch, P.; Schwarz, M. Role of mutations at codon 61 of the c-Ha-ras gene during diethylnitrosamine-induced hepatocarcinogenesis in C3H/He mice. Mol. Carcinog. 1992, 6, 60–67. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tanaka, H.; Xin, B.; Nishikawa, Y.; Yamazaki, K.; Shimizu, K.; Ogawa, K. Role of the Braf V637E mutation in hepatocarcinogenesis induced by treatment with diethylnitrosamine in neonatal B6C3F1 mice. Mol. Carcinog. 2017, 56, 478–488. [Google Scholar] [CrossRef]
- Cast, A.; Valanejad, L.; Wright, M.; Nguyen, P.; Gupta, A.; Zhu, L.; Shin, S.; Timchenko, N. C/EBPα-dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology 2018, 67, 1857–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, G.; Kutanzi, K.; Uehara, T.; Tryndyak, V.; Hong, H.-H.; Hoenerhoff, M.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Genetic and epigenetic changes in fibrosis-associated hepatocarcinogenesis in mice. Int. J. Cancer 2014, 134, 2778–2788. [Google Scholar] [CrossRef] [Green Version]
- Marrone, A.K.; Shpyleva, S.; Chappell, G.; Tryndyak, V.; Uehara, T.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Differentially Expressed MicroRNAs Provide Mechanistic Insight into Fibrosis-Associated Liver Carcinogenesis in Mice. Mol. Carcinog. 2016, 55, 808–817. [Google Scholar] [CrossRef]
- He, Q.; Wang, F.; Honda, T.; Lindquist, D.M.; Dillman, J.R.; Timchenko, N.A.; Redington, A.N. Intravenous miR-144 inhibits tumor growth in diethylnitrosamine-induced hepatocellular carcinoma in mice. Tumor Biol. 2017, 39, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E9879–E9888. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yamamoto, M.; Fujii, K.; Nagahama, Y.; Ooshio, T.; Xin, B.; Okada, Y.; Furukawa, H.; Nishikawa, Y. Differential reactivation of fetal/neonatal genes in mouse liver tumors induced in cirrhotic and non-cirrhotic conditions. Cancer Sci. 2015, 106, 972–981. [Google Scholar] [CrossRef]
- Yim, S.Y.; Shim, J.-J.; Shin, J.-H.; Jeong, Y.S.; Kang, S.-H.; Kim, S.-B.; Eun, Y.G.; Lee, D.J.; Conner, E.A.; Factor, V.M.; et al. Integrated Genomic Comparison of Mouse Models Reveals Their Clinical Resemblance to Human Liver Cancer. Mol. Cancer Res. 2018, 16, 1713–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Chu, I.-S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Liu, Y.-F.; Zha, B.-S.; Zhang, H.-L.; Zhu, X.-J.; Li, Y.-H.; Zhu, J.; Guan, X.-H.; Feng, Z.-Q.; Zhang, J.-P. Characteristic gene expression profiles in the progression from liver cirrhosis to carcinoma induced by diethylnitrosamine in a rat model. J. Exp. Clin. Cancer Res. 2009, 28, 107. [Google Scholar] [CrossRef] [Green Version]
- Moreno, F.S.; Rizzi, M.B.S.L.; Dagli, M.L.Z.; Penteado, M.V.C. Inhibitory effects of β-carotene on preneoplastic lesions induced in Wistar rats by the resistant hepatocyte model. Carcinogenesis 1991, 12, 1817–1822. [Google Scholar] [CrossRef]
- Romualdo, G.R.; Goto, R.L.; Henrique Fernandes, A.A.; Cogliati, B.; Barbisan, L.F. Dietary zinc deficiency predisposes mice to the development of preneoplastic lesions in chemically-induced hepatocarcinogenesis. Food Chem. Toxicol. 2016, 96, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romualdo, G.R.; Prata, G.B.; da Silva, T.C.; Evangelista, A.F.; Reis, R.M.; Vinken, M.; Moreno, F.S.; Cogliati, B.; Barbisan, L.F. The combination of coffee compounds attenuates early fibrosis-associated hepatocarcinogenesis in mice: Involvement of miRNA profile modulation. J. Nutr. Biochem. 2020, 85, 108479. [Google Scholar] [CrossRef]
- Miranda, M.L.P.; Furtado, K.S.; de Oliveira Andrade, F.; Heidor, R.; da Cruz, R.S.; Nogueira, M.S.; de Castro, I.A.; Purgatto, E.; Barbisan, L.F.; Moreno, F.S. β-ionone inhibits nonalcoholic fatty liver disease and its association with hepatocarcinogenesis in male Wistar rats. Chem. Biol. Interact. 2019, 308, 377–384. [Google Scholar] [CrossRef]
- De Conti, A.; Tryndyak, V.; Heidor, R.; Jimenez, L.; Moreno, F.S.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Butyrate-containing structured lipids inhibit RAC1 and epithelial-to-mesenchymal transition markers: A chemopreventive mechanism against hepatocarcinogenesis. J. Nutr. Biochem. 2020, 86, 108496. [Google Scholar] [CrossRef]
- Sarmiento-Machado, L.M.; Romualdo, G.R.; Zapaterini, J.R.; Tablas, M.B.; Fernandes, A.A.H.; Moreno, F.S.; Barbisan, L.F. Protective Effects of Dietary Capsaicin on the Initiation Step of a Two-Stage Hepatocarcinogenesis Rat Model. Nutr. Cancer 2021, 73, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and Mechanism of Action of Haloalkanes: Carbon Tetrachloride as a Toxicological Model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate. Nat. Publ. Gr. 2017. [Google Scholar] [CrossRef]
- Cho, K.-J.; Jang, J.-J. Effects of carbon tetrachloride, ethanol and acetaldehyde on diethylnitrosamine-induced hepatocarcinogenesis in rats. Cancer Lett. 1993, 70, 33–39. [Google Scholar] [CrossRef]
- Hajovsky, H.; Hu, G.; Koen, Y.; Sarma, D.; Cui, W.; Moore, D.S.; Staudinger, J.L.; Hanzlik, R.P. Metabolism and Toxicity of Thioacetamide and Thioacetamide S -Oxide in Rat Hepatocytes. Chem. Res. Toxicol. 2012, 25, 1955–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatematsu, M.; Tsuda, H.; Shirai, T.; Masui, T.; Ito, N. Placental Glutathione S-Transferase (GST-P) as a New Marker for Hepatocarcinogenesis: In Vivo Short-Term Screening for Hepatocarcinogens. Toxicol. Pathol. 1987, 15, 60–68. [Google Scholar] [CrossRef]
- Ito, S.; Tateno, C.; Tuda, M.; Yoshitake, A. Immunohistochemical Demonstration of the Gap Junctional Protein Connexin 32 and Proliferating Cell Nuclear Antigen in Glutathione S-Transferase Placental Form-Negative Lesions of Rat Liver Induced by Diethylnitrosamine and Clofibrate. Toxicol. Pathol. 1996, 24, 690–695. [Google Scholar] [CrossRef]
- Kimura, M.; Fujii, Y.; Yamamoto, R.; Yafune, A.; Hayashi, S.; Suzuki, K.; Shibutani, M. Involvement of multiple cell cycle aberrations in early preneoplastic liver cell lesions by tumor promotion with thioacetamide in a two-stage rat hepatocarcinogenesis model. Exp. Toxicol. Pathol. 2013, 65, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, T.; Wang, L.; Yafune, A.; Kimura, M.; Ohishi, T.; Suzuki, K.; Mitsumori, K.; Shibutani, M. Disruptive cell cycle regulation involving epigenetic downregulation of Cdkn2a (p16Ink4a) in early-stage liver tumor-promotion facilitating liver cell regeneration in rats. Toxicology 2012, 299, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, S.; Yafune, A.; Watanabe, Y.; Nakajima, K.; Jin, M.; Yoshida, T.; Shibutani, M. Identification of epigenetically downregulated Tmem70 and Ube2e2 in rat liver after 28-day treatment with hepatocarcinogenic thioacetamide showing gene product downregulation in hepatocellular preneoplastic and neoplastic lesions produced by tumor promoti. Toxicol. Lett. 2017, 266, 13–22. [Google Scholar] [CrossRef]
- Uehara, T.; Hirode, M.; Ono, A.; Kiyosawa, N.; Omura, K.; Shimizu, T.; Mizukawa, Y.; Miyagishima, T.; Nagao, T.; Urushidani, T. A toxicogenomics approach for early assessment of potential non-genotoxic hepatocarcinogenicity of chemicals in rats. Toxicology 2008, 250, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Omura, K.; Uehara, T.; Morikawa, Y.; Hayashi, H.; Mitsumori, K.; Minami, K.; Kanki, M.; Yamada, H.; Ono, A.; Urushidani, T. Detection of initiating potential of non-genotoxic carcinogens in a two-stage hepatocarcinogenesis study in rats. J. Toxicol. Sci. 2014, 39, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Diwan, B.A.; Rice, J.M.; Ward, J.M.; Ohshima, M.; Lynch, P.H. Inhibition by phenobarbital and lack of effect of amobarbital on the development of liver tumors induced by N-nitrosodiethylamine in juvenile B6C3F1 mice. Cancer Lett. 1984, 23, 223–234. [Google Scholar] [CrossRef]
- Greaves, P.; Irisarri, E.; Monro, A.M. Hepatic foci of cellular and enzymatic alteration and nodules in rats treated with clofibrate or diethylnitrosamine followed by phenobarbital: Their rate of onset and their reversibility. J. Natl. Cancer Inst. 1986, 76, 475–484. [Google Scholar]
- Jang, J.-J.; Henneman, J.R.; Kurata, Y.; Uno, H.; Ward, J.M. Alterations in populations of GST-p-immunoreactive single hepatocytes and hepatocellular foci after a single injection of N-nitrosodiethlyamine with or without phenobarbital promotion in male F344NCr rats. Cancer Lett. 1993, 71, 89–95. [Google Scholar] [CrossRef]
- Aydinlik, H.; Nguyen, T.D.; Moennikes, O.; Buchmann, A.; Schwarz, M. Selective pressure during tumor promotion by phenobarbital leads to clonal outgrowth of β-catenin-mutated mouse liver tumors. Oncogene 2001, 20, 7812–7816. [Google Scholar] [CrossRef] [Green Version]
- Lee, G. Review Article: Paradoxical Effects of Phenobarbital on Mouse Hepatocarcinogenesis. Toxicol. Pathol. 2000, 28, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Braeuning, A.; Schwarz, M. Is the question of phenobarbital as potential liver cancer risk factor for humans really resolved? Arch. Toxicol. 2016, 90, 1525–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiub, C.A.F.; Gadermaier, G.; Oliveira, I.; Felzenszwalb, I.; Ferreira, F.; Pinto, L.F.R.; Eckl, P. N-Nitrosodiethylamine genotoxicity in primary rat hepatocytes: Effects of cytochrome P450 induction by phenobarbital. Toxicol. Lett. 2011, 206, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Moore, R.; Goldsworthy, T.L.; Negishi, M.; Maronpot, R.R. The Orphan Nuclear Receptor Constitutive Active/Androstane Receptor Is Essential for Liver Tumor Promotion by Phenobarbital in Mice. Cancer Res. 2004, 64, 7197–7200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braeuning, A.; Heubach, Y.; Knorpp, T.; Kowalik, M.A.; Templin, M.; Columbano, A.; Schwarz, M. Gender-Specific Interplay of Signaling through β-Catenin and CAR in the Regulation of Xenobiotic-Induced Hepatocyte Proliferation. Toxicol. Sci. 2011, 123, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moennikes, O.; Buchmann, A.; Romualdi, A.; Ott, T.; Werringloer, J.; Willecke, K.; Schwarz, M. Lack of phenobarbital-mediated promotion of hepatocarcinogenesis in connexin32-null mice. Cancer Res. 2000, 60, 5087–5091. [Google Scholar] [PubMed]
- Marx-Stoelting, P.; Mahr, J.; Knorpp, T.; Schreiber, S.; Templin, M.F.; Ott, T.; Buchmann, A.; Schwarz, M. Tumor Promotion in Liver of Mice with a Conditional Cx26 Knockout. Toxicol. Sci. 2008, 103, 260–267. [Google Scholar] [CrossRef]
- Pereira, M.A.; Klaunig, J.E.; Herren-Freund, S.L.; Ruch, R.J. Effect of Phenobarbital on the Development of Liver Tumors in Juvenile and Adult Mice. JNCI J. Natl. Cancer Inst. 1986, 77, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Tamano, S.; Merlino, G.T.; Ward, J.M. Rapid development of hepatic tumors in transforming growth factor α transgenic mice associated with increased cell proliferation in precancerous hepatocellular lesions initiated by N -nitrosodiethylamine and promoted by phenobarbital. Carcinogenesis 1994, 15, 1791–1798. [Google Scholar] [CrossRef]
- Lee, C.-C.; Liu, J.-Y.; Lin, J.-K.; Chu, J.-S.; Shew, J.-Y. p53 point mutation enhanced by hepatic regeneration in aflatoxin B1-induced rat liver tumors and preneoplastic lesions. Cancer Lett. 1998, 125, 1–7. [Google Scholar] [CrossRef]
- Ray, J.S.; Harbison, M.L.; McClain, R.M.; Goodman, J.I. Alterations in the methylation status and expression of theraf oncogene in phenobarbital-induced and spontaneous B6C3F1 mouse liver tumors. Mol. Carcinog. 1994, 9, 155–166. [Google Scholar] [CrossRef]
- Phillips, J.M.; Burgoon, L.D.; Goodman, J.I. Phenobarbital Elicits Unique, Early Changes in the Expression of Hepatic Genes that Affect Critical Pathways in Tumor-Prone B6C3F1 Mice. Toxicol. Sci. 2009, 109, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Vorce, R.L.; Goodman, J.I. Hypomethylation of ras oncogenes in chemically induced and spontaneous b6c3f1 mouse liver tumors. J. Toxicol. Environ. Health 1991, 34, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Maronpot, R.R. Biological Basis of Differential Susceptibility to Hepatocarcinogenesis among Mouse Strains. J. Toxicol. Pathol. 2009, 22, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Solt, D.; Farber, E. New principle for the analysis of chemical carcinogenesis. Nature 1976, 263, 701–703. [Google Scholar] [CrossRef]
- Solt, D.B.; Medline, A.; Farber, E. Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 1977, 88, 595–618. [Google Scholar] [PubMed]
- Tsuda, H.; Lee, G.; Farber, E. Induction of resistant hepatocytes as a new principle for a possible short-term in vivo test for carcinogens. Cancer Res. 1980, 40, 1157–1164. [Google Scholar]
- Semple-Roberts, E.; Hayes, M.A.; Armstrong, D.; Becker, R.A.; Racz, W.J.; Farser, E. Alternative methods of selecting rat hepatocellular noduli resistant to 2-acetylaminofluorene. Int. J. Cancer 1987, 40, 643–645. [Google Scholar] [CrossRef]
- Higgins, G.F.; Anderson, R.M.; Higgins, G.M.; Anderson, R.M. Experimental pathology of liver: Restoration of the liver of the white rat following partial surgical removal. Arch. Pathol. 1931, 12, 186–202. [Google Scholar]
- Naves, M.M.V.; Silveira, E.R.; Dagli, M.L.Z.; Moreno, F.S. Effects of β-carotene and vitamin A on oval cell proliferation and connexin 43 expression during hepatic differentiation in the rat11This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP—process no. 1996/7566–. J. Nutr. Biochem. 2001, 12, 685–692. [Google Scholar] [CrossRef]
- Potter, V.R. Use of two sequential applications of initiators in the production of hepatomas in the rat: An examination of the Solt-Farber protocol. Cancer Res. 1984, 44, 2733–2736. [Google Scholar]
- Mazzantini, R.P.; de Conti, A.; Moreno, F.S. Persistent and remodeling hepatic preneoplastic lesions present differences in cell proliferation and apoptosis, as well as in p53, Bcl-2 and NF-κB pathways. J. Cell. Biochem. 2008, 103, 538–546. [Google Scholar] [CrossRef]
- Faris, R.A.; Monfils, B.A.; Dunsford, H.A.; Hixson, D.C. Antigenic relationship between oval cells and a subpopulation of hepatic foci, nodules, and carcinomas induced by the “resistant hepatocyte” model system. Cancer Res. 1991, 51, 1308–1317. [Google Scholar]
- Sell, S.; Dunsford, H.A. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am. J. Pathol. 1989, 134, 1347–1363. [Google Scholar]
- Andersen, J.B.; Loi, R.; Perra, A.; Factor, V.M.; Ledda-Columbano, G.M.; Columbano, A.; Thorgeirsson, S.S. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010, 51, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Perra, A.; Kowalik, M.A.; Ghiso, E.; Ledda-Columbano, G.M.; Di Tommaso, L.; Angioni, M.M.; Raschioni, C.; Testore, E.; Roncalli, M.; Giordano, S.; et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J. Hepatol. 2014, 61, 1088–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrelli, A.; Perra, A.; Cora, D.; Sulas, P.; Menegon, S.; Manca, C.; Migliore, C.; Kowalik, M.A.; Ledda-Columbano, G.M.; Giordano, S.; et al. MicroRNA/gene profiling unveils early molecular changes and nuclear factor erythroid related factor 2 (NRF2) activation in a rat model recapitulating human hepatocellular carcinoma (HCC). Hepatology 2014, 59, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Newberne, P.M.; Wogan, G.N. Sequential morphologic changes in aflatoxin B carcinogenesis in the rat. Cancer Res. 1968, 28, 770–781. [Google Scholar] [PubMed]
- Butler, W.H.; Greenblatt, M.; Lijinsky, W. Carcinogenesis in rats by aflatoxins B1, G1, and B2. Cancer Res. 1969, 29, 2206–2211. [Google Scholar]
- Wogan, G.N.; Paglialunga, S.; Newberne, P.M. Carcinogenic effects of low dietary levels of aflatoxin B1 in rats. Food Cosmet. Toxicol. 1974, 12, 681–685. [Google Scholar] [CrossRef]
- Nixon, J.E.; Hendricks, J.D.; Pawloswki, N.E.; Loveland, P.M.; Sinnhuber, R.O. Carcinogenicity of Aflatoxlcol in Fischer 344 Rats2, 3, 4. JNCI J. Natl. Cancer Inst. 1981, 66, 1159–1163. [Google Scholar] [CrossRef]
- Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflatoxicosis in developing countries: A review of toxicology, exposure, potential health consequences, and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar] [CrossRef]
- Aguilar, F.; Hussain, S.P.; Cerutti, P. Aflatoxin B1 induces the transversion of G-->T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 8586–8590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulla, J.E.; Chen, Z.Y.; Eaton, D.L. Aflatoxin B1-induced rat hepatic hyperplastic nodules do not exhibit a site-specific mutation within the p53 gene. Cancer Res. 1993, 53, 9–11. [Google Scholar] [PubMed]
- Shen, H.M.; Shi, C.Y.; Lee, H.P.; Ong, C.N. Aflatoxin B1-Induced Lipid Peroxidation in Rat Liver. Toxicol. Appl. Pharmacol. 1994, 127, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Maurya, B.K.; Trigun, S.K. Activation of oxidative stress and inflammatory factors could account for histopathological progression of aflatoxin-B1 induced hepatocarcinogenesis in rat. Mol. Cell. Biochem. 2015, 401, 185–196. [Google Scholar] [CrossRef]
- Shi, J.; He, J.; Lin, J.; Sun, X.; Sun, F.; Ou, C.; Jiang, C. Distinct response of the hepatic transcriptome to Aflatoxin B1 induced hepatocellular carcinogenesis and resistance in rats. Sci. Rep. 2016, 6, 31898. [Google Scholar] [CrossRef] [PubMed]
- Columbano, A.; Rajalakshmi, S.; Sarma, D.S. Requirement of cell proliferation for the initiation of liver carcinogenesis as assayed by three different procedures. Cancer Res. 1981, 41, 2079–2083. [Google Scholar]
- Sakai, H.; Tsukamoto, T.; Yamamoto, M.; Shirai, N.; Lidaka, T.; Yanai, T.; Masegi, T.; Tatematsu, M. Differential Effects of Partial Hepatectomy and Carbon Tetrachloride Administration on Induction of Liver Cell Foci in a Model for Detection of Initiation Activity. Japanese J. Cancer Res. 2001, 92, 1018–1025. [Google Scholar] [CrossRef]
- Ramos Caetano, B.F.; Baptista Tablas, M.; Ribeiro Romualdo, G.; Marchesan Rodrigues, M.A.; Barbisan, L.F. Early molecular events associated with liver and colon sub-acute responses to 1,2-dimethylhydrazine: Potential implications on preneoplastic and neoplastic lesion development. Toxicol. Lett. 2020, 329, 67–79. [Google Scholar] [CrossRef]
- Punvittayagul, C.; Chariyakornkul, A.; Chewonarin, T.; Jarukamjorn, K.; Wongpoomchai, R. Augmentation of diethylnitrosamine–induced early stages of rat hepatocarcinogenesis by 1,2-dimethylhydrazine. Drug Chem. Toxicol. 2019, 42, 641–648. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Peitta, G.; Seddaiu, M.A.; Feo, F.; Calvisi, D.F. Experimental Models to Define the Genetic Predisposition to Liver Cancer. Cancers 2019, 11, 1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilger, A.; Bennett, L.M.; Carabeo, R.A.; Chiaverotti, T.A.; Dvorak, C.; Liss, K.M.; Schadewald, S.A.; Pitot, H.C.; Drinkwater, N.R. A Potent Modifier of Liver Cancer Risk on Distal Mouse Chromosome 1 This article is dedicated to the memory of our late colleague, Kristin M. Liss. Genetics 2004, 167, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Drinkwater, N.R. Genetic Control of Hepatocarcinogenesis In C3H Mice. Drug Metab. Rev. 1994, 26, 201–208. [Google Scholar] [CrossRef]
- Hanigan, M.H.; Kemp, C.J.; Ginsler, J.J.; Drinkwater, N.R. Rapid growth of preneoplastic lesions in hepatocarcinogen-Sensitive C3H/HeJ male mice relative to C57BL/6J male mice. Carcinogenesis 1988, 9, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Gariboldi, M.; Manenti, G.; Canzian, F.; Falvella, F.S.; Pierotti, M.A.; Della Porta, G.; Binelli, G.; Dragani, T.A. Chromosome mapping of murine susceptibility loci to liver carcinogenesis. Cancer Res. 1993, 53, 209–211. [Google Scholar]
- Lee, G.H.; Bennett, L.M.; Carabeo, R.A.; Drinkwater, N.R. Identification of hepatocarcinogen-resistance genes in DBA/2 mice. Genetics 1995, 139, 387–395. [Google Scholar] [CrossRef]
- Dragani, T.A.; Manenti, G.; Gariboldi, M.; De Gregorio, L.; Pierotti, M.A. Genetics of liver tumor susceptibility in mice. Toxicol. Lett. 1995, 82–83, 613–619. [Google Scholar] [CrossRef]
- Manenti, G.; Binelli, G.; Gariboldi, M.; Canzian, F.; De Gregorio, L.; Falvella, F.S.; Dragani, T.A.; Pierotti, M.A. Multiple Loci Affect Genetic Predisposition to Hepatocarcinogenesis in Mice. Genomics 1994, 23, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Manenti, G.; Galvan, A.; Falvella, F.S.; Pascale, R.M.; Spada, E.; Milani, S.; Neira, A.G.; Feo, F.; Dragani, T.A. Genetic control of resistance to hepatocarcinogenesis by the mouse Hpcr3 locus. Hepatology 2008, 48, 617–623. [Google Scholar] [CrossRef]
- De Miglio, M.R.; Pascale, R.M.; Simile, M.M.; Muroni, M.R.; Virdis, P.; Kwong, K.M.T.; Wong, L.K.L.; Bosinco, G.M.; Pulina, F.R.; Calvisi, D.F.; et al. Polygenic control of hepatocarcinogenesis in Copenhagen × F344 rats. Int. J. Cancer 2004, 111, 9–16. [Google Scholar] [CrossRef]
- De Miglio, M.R.; Virdis, P.; Calvisi, D.F.; Frau, M.; Muroni, M.R.; Simile, M.M.; Daino, L.; Careddu, G.M.; Sanna-Passino, E.; Pascale, R.M.; et al. Mapping a Sex Hormone–Sensitive Gene Determining Female Resistance to Liver Carcinogenesis in a Congenic F344.BN- Hcs4 Rat. Cancer Res. 2006, 66, 10384–10390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y. Resistance of DRH strain rats to chemical carcinogenesis of liver: Genetic analysis of later progression stage. Carcinogenesis 2002, 23, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, K.; Denda, A.; Higashi, T.; Hiai, H. Genetic resistance to chemical hepatocarcinogenesis in the DRH rat strain. Comp. Med. 2004, 54, 373–377. [Google Scholar]
- Ghisletti, S.; Meda, C.; Maggi, A.; Vegeto, E. 17β-Estradiol Inhibits Inflammatory Gene Expression by Controlling NF-κB Intracellular Localization. Mol. Cell. Biol. 2005, 25, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lu, Y.; Xu, Y.; Xu, L.; Zheng, W.; Wu, Y.; Li, L.; Shen, P. Estrogen represses hepatocellular carcinoma (HCC) Growth via Inhibiting Alternative Activation of Tumor-associated Macrophages (TAMs). J. Biol. Chem. 2012, 287, 40140–40149. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.-J.; Levasseur, M.-P.; Dufour, C.R.; Perry, M.-C.; Giguere, V. Loss of estrogen-related receptor promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc. Natl. Acad. Sci. USA 2013, 110, 17975–17980. [Google Scholar] [CrossRef] [Green Version]
- Kalra, M.; Mayes, J.; Assefa, S.; Kaul, A.K.; Kaul, R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5945. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.-X.; Yang, Y.; Zhang, Y.; Wang, H.-Y.; Zheng, X.F.S. Significance and mechanism of androgen receptor overexpression and androgen receptor/mechanistic target of rapamycin cross-talk in hepatocellular carcinoma. Hepatology 2018, 67, 2271–2286. [Google Scholar] [CrossRef]
- Ma, W.; Hsu, C.; Wu, M.; Wu, C.; Wu, C.; Lai, J.; Jou, Y.; Chen, C.; Yeh, S.; Chang, C. Androgen Receptor Is a New Potential Therapeutic Target for the Treatment of Hepatocellular Carcinoma. Gastroenterology 2008, 135, 947–955.e5. [Google Scholar] [CrossRef] [Green Version]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Almind, K.; Kahn, C.R. Genetic Determinants of Energy Expenditure and Insulin Resistance in Diet-Induced Obesity in Mice. Diabetes 2004, 53, 3274–3285. [Google Scholar] [CrossRef] [Green Version]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Dowman, J.K.; Hopkins, L.J.; Reynolds, G.M.; Nikolaou, N.; Armstrong, M.J.; Shaw, J.C.; Houlihan, D.D.; Lalor, P.F.; Tomlinson, J.W.; Hübscher, S.G.; et al. Development of Hepatocellular Carcinoma in a Murine Model of Nonalcoholic Steatohepatitis Induced by Use of a High-Fat/Fructose Diet and Sedentary Lifestyle. Am. J. Pathol. 2014, 184, 1550–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessitore, A.; Cicciarelli, G.; Del Vecchio, F.; Gaggiano, A.; Verzella, D.; Fischietti, M.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Barnabei, R.; et al. MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: Study on C57BL/6J mice. BMC Cancer 2016, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC Development Is Associated to Peripheral Insulin Resistance in a Mouse Model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef]
- Takakura, K.; Koido, S.; Fujii, M.; Hashiguchi, T.; Shibazaki, Y.; Yoneyama, H.; Katagi, H.; Kajihara, M.; Misawa, T.; Homma, S.; et al. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res. 2014, 34, 4849–4855. [Google Scholar]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- De Conti, A.; Ortega, J.F.; Tryndyak, V.; Dreval, K.; Moreno, F.S.; Rusyn, I.; Beland, F.A.; Pogribny, I.P. MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget 2017, 8, 88517–88528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of Streptozotocin. Mutat. Res. Mutat. Res. 2002, 512, 121–134. [Google Scholar] [CrossRef]
- Guo, S.; Mao, X.; Yan, Y.; Zhang, Y.; Ming, L. Changes of liver transcriptome profiles following oxidative stress in streptozotocin-induced diabetes in mice. PeerJ 2020, 8, e8983. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.; Hazim, A.; He, Y.; Peyressatre, M.; Kim, D.-Y.; Song, X.; Beretta, L. Proteomic and Lipidomic Signatures of Lipid Metabolism in NASH-Associated Hepatocellular Carcinoma. Cancer Res. 2013, 73, 4722–4731. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, B.; Ekström, T. The effects of long-term oral administration of ethanol on Sprague-Dawley rats — A condensed report. Toxicology 1995, 96, 133–145. [Google Scholar] [CrossRef]
- Beland, F.A.; Benson, R.W.; Mellick, P.W.; Kovatch, R.M.; Roberts, D.W.; Fang, J.-L.; Doerge, D.R. Effect of ethanol on the tumorigenicity of urethane (ethyl carbamate) in B6C3F1 mice. Food Chem. Toxicol. 2005, 43, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Hirota, Y.; Kuriyama, M.; Nishiguchi, S.; Otani, S. Cessation of Long-term Alcohol Administration and Two-day Cycling of Exposure Respectively Promote and Inhibit Hepatocarcinogenesis in Rats. Asian Pac. J. Cancer Prev. 2000, 1, 325–328. [Google Scholar]
- Wanibuchi, H.; Zhang, Y.; Kinoshita, A.; Wei, M.; Kang, J.S.; Fukushima, S. Effects of cessation of alcohol exposure on rat hepatocarcinogenesis. Asian Pac. J. Cancer Prev. 2006, 7, 122–126. [Google Scholar] [PubMed]
- Kato, H.; Naiki-Ito, A.; Naiki, T.; Suzuki, S.; Yamashita, Y.; Sato, S.; Sagawa, H.; Kato, A.; Kuno, T.; Takahashi, S. Connexin 32 dysfunction promotes ethanol-related hepatocarcinogenesis via activation of Dusp1-Erk axis. Oncotarget 2016, 7, 2009–2021. [Google Scholar] [CrossRef]
- Kushida, M.; Wanibuchi, H.; Morimura, K.; Kinoshita, A.; Kang, J.S.; Puatanachokchai, R.; Wei, M.; Funae, Y.; Fukushima, S. Dose-dependence of promotion of 2-amino-3,8-dimethylimidazo [4,5-f] quinoxaline-induced rat hepatocarcinogenesis by ethanol: Evidence for a threshold. Cancer Sci. 2005, 96, 747–757. [Google Scholar] [CrossRef]
- Pires, P.W.; Furtado, K.S.; Justullin, L.A.; Rodrigues, M.A.M.; Felisbino, S.L.; Barbisan, L.F. Chronic ethanol intake promotes double gluthatione S-transferase/transforming growth factor-α-positive hepatocellular lesions in male Wistar rats. Cancer Sci. 2008, 99, 221–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, K.E.; Hennings, L.; Sharma, N.; Lai, K.; Cleves, M.A.; Wynne, R.A.; Badger, T.M.; Ronis, M.J.J. Alcohol Consumption Promotes Diethylnitrosamine-Induced Hepatocarcinogenesis in Male Mice through Activation of the Wnt/β-Catenin Signaling Pathway. Cancer Prev. Res. 2014, 7, 675–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon-Warner, E.; Walling, T.L.; Schrum, L.W.; McKillop, I.H. Chronic Ethanol Feeding Accelerates Hepatocellular Carcinoma Progression in a Sex-Dependent Manner in a Mouse Model of Hepatocarcinogenesis. Alcohol. Clin. Exp. Res. 2012, 36, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Karim, M.R.; Wanibuchi, H.; Wei, M.; Morimura, K.; Salim, E.I.; Fukushima, S. Enhancing risk of ethanol on MeIQx-induced rat hepatocarcinogenesis is accompanied with increased levels of cellular proliferation and oxidative stress. Cancer Lett. 2003, 192, 37–47. [Google Scholar] [CrossRef]
- Wanibuchi, H.; Wei, M.; Karim, M.R.; Morimura, K.; Doi, K.; Kinoshita, A.; Fukushima, S. Existence of No Hepatocarcinogenic Effect Levels of 2-amino-3,8-dimethylimidazo [4,5-f] quinoxaline with or without Coadministration with Ethanol. Toxicol. Pathol. 2006, 34, 232–236. [Google Scholar] [CrossRef]
- Tatsuta, M.; Iishi, H.; Baba, M.; Yano, H.; Iseki, K.; Uehara, H.; Nakaizumi, A. Enhancement by ethyl alcohol of experimental hepatocarcinogenesis induced by N-nitrosomorpholine. Int. J. Cancer 1997, 71, 1045–1048. [Google Scholar] [CrossRef]
- Yan, G.; Wang, X.; Sun, C.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Chronic Alcohol Consumption Promotes Diethylnitrosamine-Induced Hepatocarcinogenesis via Immune Disturbances. Sci. Rep. 2017, 7, 2567. [Google Scholar] [CrossRef]
- Edamoto, Y.; Hara, A.; Biernat, W.; Terracciano, L.; Cathomas, G.; Riehle, H.-M.; Matsuda, M.; Fujii, H.; Scoazec, J.-Y.; Ohgaki, H. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int. J. Cancer 2003, 106, 334–341. [Google Scholar] [CrossRef]
- Chen, D.; Yan, Y.; Wang, X.; Li, S.; Liu, Y.; Yu, D.; He, Y.; Deng, R.; Liu, Y.; Xu, M.; et al. Chronic alcohol exposure promotes HCC stemness and metastasis through β-catenin/miR-22-3p/TET2 axis. Aging 2021, 13, 14433–14455. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Moon, H.-B.; Son, J.-K.; Jeong, S.; Yu, S.-L.; Yoon, H.; Han, Y.-M.; Lee, C.-S.; Park, J.-S.; Lee, C.-H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, P.Y.; Shin, H.-J.; Kim, D.H.; Kang, S.; Moon, H.-B.; Kang, S.W.; Kim, J.-M.; Park, S.G.; Park, B.C.; et al. Proteomic analysis of liver tissue from HBx -transgenic mice at early stages of hepatocarcinogenesis. Proteomics 2009, 9, 5056–5066. [Google Scholar] [CrossRef]
- Benn, J.; Su, F.; Doria, M.; Schneider, R.J. Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J. Virol. 1996, 70, 4978–4985. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The Transcriptional Transactivation Function of HBx Protein Is Important for Its Augmentation Role in Hepatitis B Virus Replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunsford, H.A.; Sell, S.; Chisari, F. V Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res. 1990, 50, 3400–3407. [Google Scholar]
- Toshkov, I.; Chisari, F.V.; Bannasch, P. Hepatic preneoplasia in hepatitis B virus transgenic mice. Hepatology 1994, 20, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-W.; Liang, K.-H.; Lin, W.-R.; Huang, Y.-H.; Huang, S.-F.; Chen, T.-C.; Yeh, C.-T. Hepatocarcinogenesis in transgenic mice carrying hepatitis B virus pre-S/S gene with the sW172* mutation. Oncogenesis 2016, 5, e273. [Google Scholar] [CrossRef] [Green Version]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuuras, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Santoni-Rugiu, E.; Nagy, P.; Jensen, M.R.; Factor, V.M.; Thorgeirsson, S.S. Evolution of neoplastic development in the liver of transgenic mice co-expressing c-myc and transforming growth factor-alpha. Am. J. Pathol. 1996, 149, 407–428. [Google Scholar] [PubMed]
- Thorgeirsson, S.S.; Santoni-Rugiu, E. Transgenic mouse models in carcinogenesis: Interaction of c- myc with transforming growth factor α and hepatocyte growth factor in hepatocarcinogenesis. Br. J. Clin. Pharmacol. 1996, 42, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvisi, D.F.; Factor, V.M.; Ladu, S.; Conner, E.A.; Thorgeirsson, S.S. Disruption of β-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 2004, 126, 1374–1386. [Google Scholar] [CrossRef]
- Conner, E.A.; Lemmer, E.R.; Omori, M.; Wirth, P.J.; Factor, V.M.; Thorgeirsson, S.S. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene 2000, 19, 5054–5062. [Google Scholar] [CrossRef]
- Colnot, S.; Decaens, T.; Niwa-Kawakita, M.; Godard, C.; Hamard, G.; Kahn, A.; Giovannini, M.; Perret, C. Liver-targeted disruption of Apc in mice activates -catenin signaling and leads to hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 2004, 101, 17216–17221. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in Mice with β-Catenin and Ha-Ras Gene Mutations. Cancer Res. 2004, 64, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.I.; Moon, H.; Kim, D.Y.; Cho, K.J.; Ju, H.; Kim, D.Y.; Ahn, S.H.; Han, K.; Ro, S.W. Development of a transgenic mouse model of hepatocellular carcinoma with a liver fibrosis background. BMC Gastroenterol. 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chisari, F.V.; Filippi, P.; Buras, J.; McLachlan, A.; Popper, H.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc. Natl. Acad. Sci. USA 1987, 84, 6909–6913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, M.; Spano, D.; D’Apolito, M.; Centra, M.; Lasalandra, C.; Capasso, M.; Di Leo, A.; Volinia, S.; Arcelli, D.; Rosso, N.; et al. Gene Expression Analysis in HBV Transgenic Mouse Liver: A Model to Study Early Events Related to Hepatocarcinogenesis. Mol. Med. 2006, 12, 115–123. [Google Scholar] [CrossRef]
- Sell, S.; Hunt, J.M.; Dunsford, H.A.; Chisari, F. V Synergy between hepatitis B virus expression and chemical hepatocarcinogens in transgenic mice. Cancer Res. 1991, 51, 1278–1285. [Google Scholar]
- Lai, M.; Huang, S.; Hsu, C.-W.; Chang, M.-H.; Liaw, Y.-F.; Yeh, C.-T. Identification of nonsense mutations in hepatitis B virus S gene in patients with hepatocellular carcinoma developed after lamivudine therapy. Antivir. Ther. 2009, 14, 249–261. [Google Scholar] [PubMed]
- Ichibangase, T.; Moriya, K.; Koike, K.; Imai, K. A Proteomics Method Revealing Disease-Related Proteins in Livers of Hepatitis-Infected Mouse Model. J. Proteome Res. 2007, 6, 2841–2849. [Google Scholar] [CrossRef]
- Kamegaya, Y.; Hiasa, Y.; Zukerberg, L.; Fowler, N.; Blackard, J.T.; Lin, W.; Choe, W.H.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 2005, 41, 660–667. [Google Scholar] [CrossRef]
- Sandgren, E.P.; Quaife, C.J.; Pinkert, C.A.; Palmiter, R.D.; Brinster, R.L. Oncogene-induced liver neoplasia in transgenic mice. Oncogene 1989, 4, 715–724. [Google Scholar] [PubMed]
- Etiemble, J.; Degott, C.; Renard, C.A.; Fourel, G.; Shamoon, B.; Vitvitski-Trépo, L.; Hsu, T.Y.; Tiollais, P.; Babinet, C.; Buendia, M.A. Liver-specific expression and high oncogenic efficiency of a c-myc transgene activated by woodchuck hepatitis virus insertion. Oncogene 1994, 9, 727–737. [Google Scholar]
- Liu, P.; Terradillos, O.; Renard, C.A.; Feldmann, G.; Buendia, M.A.; Bernuau, D. Hepatocarcinogenesis in woodchuck hepatitis virus/c-myc mice: Sustained cell proliferation and biphasic activation of insulin-like growth factor II. Hepatology 1997, 25, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Coste, A.d.L.; Romagnolo, B.; Billuart, P.; Renard, C.-A.; Buendia, M.-A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the -catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [Green Version]
- Molina-Sánchez, P.; Ruiz de Galarreta, M.; Yao, M.A.; Lindblad, K.E.; Bresnahan, E.; Bitterman, E.; Martin, T.C.; Rubenstein, T.; Nie, K.; Golas, J.; et al. Cooperation Between Distinct Cancer Driver Genes Underlies Intertumor Heterogeneity in Hepatocellular Carcinoma. Gastroenterology 2020, 159, 2203–2220.e14. [Google Scholar] [CrossRef]
- Méndez-Lucas, A.; Lin, W.; Driscoll, P.C.; Legrave, N.; Novellasdemunt, L.; Xie, C.; Charles, M.; Wilson, Z.; Jones, N.P.; Rayport, S.; et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab. 2020, 2, 335–350. [Google Scholar] [CrossRef]
- Méndez-Lucas, A.; Li, X.; Hu, J.; Che, L.; Song, X.; Jia, J.; Wang, J.; Xie, C.; Driscoll, P.C.; Tschaharganeh, D.F.; et al. Glucose Catabolism in Liver Tumors Induced by c-MYC Can Be Sustained by Various PKM1/PKM2 Ratios and Pyruvate Kinase Activities. Cancer Res. 2017, 77, 4355–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Wang, J.; Karagoz, E.; Liang, B.; Song, X.; Shang, R.; Evert, K.; Xu, M.; Che, L.; Evert, M.; et al. Axis inhibition protein 1 (Axin1) Deletion–Induced Hepatocarcinogenesis Requires Intact β-Catenin but Not Notch Cascade in Mice. Hepatology 2019, 70, 2003–2017. [Google Scholar] [CrossRef]
- Yamamoto, M.; Xin, B.; Watanabe, K.; Ooshio, T.; Fujii, K.; Chen, X.; Okada, Y.; Abe, H.; Taguchi, Y.; Miyokawa, N.; et al. Oncogenic Determination of a Broad Spectrum of Phenotypes of Hepatocyte-Derived Mouse Liver Tumors. Am. J. Pathol. 2017, 187, 2711–2725. [Google Scholar] [CrossRef] [Green Version]
- Xin, B.; Yamamoto, M.; Fujii, K.; Ooshio, T.; Chen, X.; Okada, Y.; Watanabe, K.; Miyokawa, N.; Furukawa, H.; Nishikawa, Y. Critical role of Myc activation in mouse hepatocarcinogenesis induced by the activation of AKT and RAS pathways. Oncogene 2017, 36, 5087–5097. [Google Scholar] [CrossRef] [Green Version]
- Zhan, N.; Michael, A.A.; Wu, K.; Zeng, G.; Bell, A.; Tao, J.; Monga, S.P. The Effect of Selective c-MET Inhibitor on Hepatocellular Carcinoma in the MET-Active, β-Catenin-Mutated Mouse Model. Gene Expr. 2018, 18, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P.S. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef]
- Stauffer, J.K.; Scarzello, A.J.; Andersen, J.B.; De Kluyver, R.L.; Back, T.C.; Weiss, J.M.; Thorgeirsson, S.S.; Wiltrout, R.H. Coactivation of AKT and β-Catenin in Mice Rapidly Induces Formation of Lipogenic Liver Tumors. Cancer Res. 2011, 71, 2718–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Murakami, H.; Sanderson, N.D.; Nagy, P.; Marino, P.A.; Merlino, G.; Thorgeirsson, S.S. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993, 53, 1719–1723. [Google Scholar] [PubMed]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qi, X.; Zeng, Z.; Wang, L.; Wang, J.; Zhang, T.; Xu, Q.; Shen, C.; Zhou, G.; Yang, S.; et al. CRISPR/Cas9-mediated p53 and Pten dual mutation accelerates hepatocarcinogenesis in adult hepatitis B virus transgenic mice. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M. Immunology of hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2080. [Google Scholar] [CrossRef]
- Johnston, M.P.; Khakoo, S.I. Immunotherapy for hepatocellular carcinoma: Current and future. World J. Gastroenterol. 2019, 25, 2977–2989. [Google Scholar] [CrossRef]
- Kole, C.; Charalampakis, N.; Tsakatikas, S.; Vailas, M.; Moris, D.; Gkotsis, E.; Kykalos, S.; Karamouzis, M.V.; Schizas, D. Immunotherapy for Hepatocellular Carcinoma: A 2021 Update. Cancers 2020, 12, 2859. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzer, B.; Stout, M.; Fiebiger, E. Antigen Cross-Presentation of Immune Complexes. Front. Immunol. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers 2019, 11, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Lin, L.; Chen, C.-W.; Ou, D.-L. Mouse Models for Immunotherapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1800. [Google Scholar] [CrossRef] [Green Version]
- Verma, B.; Wesa, A. Establishment of Humanized Mice from Peripheral Blood Mononuclear Cells or Cord Blood CD34+ Hematopoietic Stem Cells for Immune-Oncology Studies Evaluating New Therapeutic Agents. Curr. Protoc. Pharmacol. 2020, 89, 1–19. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “Humanized” Mice to Study Human Immunity. Curr. Protoc. Immunol. 2008, 81, 1–21. [Google Scholar] [CrossRef]
- Morillon, Y.M.; Sabzevari, A.; Schlom, J.; Greiner, J.W. The Development of Next-generation PBMC Humanized Mice for Preclinical Investigation of Cancer Immunotherapeutic Agents. Anticancer Res. 2020, 40, 5329–5341. [Google Scholar] [CrossRef]
- Brehm, M.A.; Kenney, L.L.; Wiles, M.V.; Low, B.E.; Tisch, R.M.; Burzenski, L.; Mueller, C.; Greiner, D.L.; Shultz, L.D. Lack of acute xenogeneic graft- versus -host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019, 33, 3137–3151. [Google Scholar] [CrossRef]
- Yao, L.-C.; Cheng, M.; Shultz, L.D.; Keck, J.G. Abstract 5619: PBMC humanized NSG-(K b D b ) null (IA) null mouse model to evaluate immune-oncology drug efficacy. In Proceedings of the AACR Annual Meeting 2020, American Association for Cancer Research, Philadelphia, PA, USA, 27–28 April 2020. [Google Scholar]
- Su, S.; Zhou, H.; Xue, M.; Liu, J.-Y.; Ding, L.; Cao, M.; Zhou, Z.-X.; Hu, H.-M.; Wang, L.-X. Anti-tumor Efficacy of a Hepatocellular Carcinoma Vaccine Based on Dendritic Cells Combined with Tumor-derived Autophagosomes in Murine Models. Asian Pac. J. Cancer Prev. 2013, 14, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, H.; Wei, M.; Mou, T.; Shi, T.; Ma, Y.; Cai, X.; Li, Y.; Dong, J.; Wei, J. Recombinant Adenovirus Expressing a Soluble Fusion Protein PD-1/CD137L Subverts the Suppression of CD8+ T Cells in HCC. Mol. Ther. 2019, 27, 1906–1918. [Google Scholar] [CrossRef]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Zumwalde, N.A.; Gumperz, J.E. Modeling Human Antitumor Responses In Vivo Using Umbilical Cord Blood-Engrafted Mice. Front. Immunol. 2018, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r null humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Serra-Hassoun, M.; Bourgine, M.; Boniotto, M.; Berges, J.; Langa, F.; Michel, M.-L.; Freitas, A.A.; Garcia, S. Human Hematopoietic Reconstitution and HLA-Restricted Responses in Nonpermissive Alymphoid Mice. J. Immunol. 2014, 193, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkening, S.; Stahl, F.; Bader, A. Comparison of primary human hepatocytes and hepatoma cell line hepg2 with regard to their biotransformation properties. Drug. Metab. DIspos. 2003, 31, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.D.; Yuen, G.; Tu, T.; Budzinska, M.A.; Spring, K.; Bryant, K.; Shackel, N.A. In Vitro Models of the Liver: Disease Modeling, Drug Discovery and Clinical Applications. In Tijdschrift voor Geneeskunde; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019; Volume 38, pp. 505–507. ISBN 9780994438188. [Google Scholar]
- Guo, X.; Seo, J.-E.; Li, X.; Mei, N. Genetic toxicity assessment using liver cell models: Past, present, and future. J. Toxicol. Environ. Health Part B 2020, 23, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, V.; Cooreman, A.; Gijbels, E.; Van Campenhout, R.; Gustafson, E.; Ballet, S.; Annaert, P.; Cogliati, B.; Vinken, M. Primary hepatocytes and their cultures for the testing of drug-induced liver injury. Adv. Pharmacol. 2019, 85, 1–30. [Google Scholar] [CrossRef]
- Arellanes-Robledo, J.; Hernández, C.; Camacho, J.; Pérez-Carreón, J.I. In Vitro Models of HCC. In Liver Pathophysiology; Muriel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 563–579. ISBN 9780128043219. [Google Scholar]
- Brambilla, G.; Martelli, A. Human hepatocytes in genotoxicity assays. Pharmacol. Res. 1990, 22, 381–392. [Google Scholar] [CrossRef]
- Aniagu, S.O.; Williams, T.D.; Chipman, J.K. Changes in gene expression and assessment of DNA methylation in primary human hepatocytes and HepG2 cells exposed to the environmental contaminants—Hexabromocyclododecane and 17-β oestradiol. Toxicology 2009, 256, 143–151. [Google Scholar] [CrossRef]
- Ayed-Boussema, I.; Pascussi, J.-M.; Maurel, P.; Bacha, H.; Hassen, W. Effect of Aflatoxin B1 on Nuclear Receptors PXR, CAR, and AhR and Their Target Cytochromes P450 mRNA Expression in Primary Cultures of Human Hepatocytes. Int. J. Toxicol. 2012, 31, 86–93. [Google Scholar] [CrossRef]
- Ayed-Boussema, I.; Pascussi, J.M.; Rjiba, K.; Maurel, P.; Bacha, H.; Hassen, W. The mycotoxin, patulin, increases the expression of PXR and AhR and their target cytochrome P450s in primary cultured human hepatocytes. Drug Chem. Toxicol. 2012, 35, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ayed-Boussema, I.; Pascussi, J.M.; Zaied, C.; Maurel, P.; Bacha, H.; Hassen, W. Ochratoxin A induces CYP3A4, 2B6, 3A5, 2C9, 1A1, and CYP1A2 gene expression in primary cultured human hepatocytes: A possible activation of nuclear receptors. Drug Chem. Toxicol. 2012, 35, 71–80. [Google Scholar] [CrossRef]
- Rieswijk, L.; Claessen, S.M.H.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.J.; Jennen, D.G.J.; de Kok, T.M.C.M.; Kleinjans, J.C.S.; van Breda, S.G.J. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350–352, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Pez, F.; Gifu, P.; Degli-Esposti, D.; Fares, N.; Lopez, A.; Lefrançois, L.; Michelet, M.; Rivoire, M.; Bancel, B.; Sylla, B.S.; et al. In vitro transformation of primary human hepatocytes: Epigenetic changes and stemness properties. Exp. Cell Res. 2019, 384, 111643. [Google Scholar] [CrossRef] [PubMed]
- Kiamehr, M.; Heiskanen, L.; Laufer, T.; Düsterloh, A.; Kahraman, M.; Käkelä, R.; Laaksonen, R.; Aalto-Setälä, K. Dedifferentiation of Primary Hepatocytes is Accompanied with Reorganization of Lipid Metabolism Indicated by Altered Molecular Lipid and miRNA Profiles. Int. J. Mol. Sci. 2019, 20, 2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular Mechanisms Underlying the Dedifferentiation Process of Isolated Hepatocytes and Their Cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef] [PubMed]
- Gijbels, E.; Vanhaecke, T.; Vinken, M. Establishment of Sandwich Cultures of Primary Human Hepatocytes. In Experimental Cholestasis Research; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1981, pp. 99–115. ISBN 978-1-4939-9419-9. [Google Scholar]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell–Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Gross-Steinmeyer, K.; Stapleton, P.L.; Tracy, J.H.; Bammler, T.K.; Strom, S.C.; Buhler, D.R.; Eaton, D.L. Modulation of Aflatoxin B1–Mediated Genotoxicity in Primary Cultures of Human Hepatocytes by Diindolylmethane, Curcumin, and Xanthohumols. Toxicol. Sci. 2009, 112, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Green, C.J.; Parry, S.A.; Gunn, P.J.; Ceresa, C.D.L.; Rosqvist, F.; Piché, M.-E.; Hodson, L. Studying non-alcoholic fatty liver disease: The ins and outs of in vivo, ex vivo and in vitro human models. Horm. Mol. Biol. Clin. Investig. 2018, 41, 1–22. [Google Scholar] [CrossRef]
- Saraswati, S.; Alhaider, A.; Abdelgadir, A.M.; Tanwer, P.; Korashy, H.M. Phloretin attenuates STAT-3 activity and overcomes sorafenib resistance targeting SHP-1–mediated inhibition of STAT3 and Akt/VEGFR2 pathway in hepatocellular carcinoma. Cell Commun. Signal. 2019, 17, 127. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, N.; Zhang, Y.-F.; Fu, H.; Feng, M.; Schneider, D.; Su, L.; Wu, X.; Zhou, J.; Mackay, S.; et al. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology 2020, 158, 2250–2265.e20. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef] [Green Version]
- Czauderna, C.; Palestino-Dominguez, M.; Castven, D.; Becker, D.; Zanon-Rodriguez, L.; Hajduk, J.; Mahn, F.L.; Herr, M.; Strand, D.; Strand, S.; et al. Ginkgo biloba induces different gene expression signatures and oncogenic pathways in malignant and non-malignant cells of the liver. PLoS ONE 2018, 13, e0209067. [Google Scholar] [CrossRef]
- Chen, Z.; Zhuang, W.; Wang, Z.; Xiao, W.; Don, W.; Li, X.; Chen, X. MicroRNA-450b-3p inhibits cell growth by targeting phosphoglycerate kinase 1 in hepatocellular carcinoma. J. Cell. Biochem. 2019, 120, 18805–18815. [Google Scholar] [CrossRef]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef] [PubMed]
- Ran, L.-K.; Chen, Y.; Zhang, Z.-Z.; Tao, N.-N.; Ren, J.-H.; Zhou, L.; Tang, H.; Chen, X.; Chen, K.; Li, W.-Y.; et al. SIRT6 Overexpression Potentiates Apoptosis Evasion in Hepatocellular Carcinoma via BCL2-Associated X Protein–Dependent Apoptotic Pathway. Clin. Cancer Res. 2016, 22, 3372–3382. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.-P.; Varma, S.; Gottesman, M.M. The Clinical Relevance of Cancer Cell Lines. JNCI J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoni, M.; Cortesi, M.; Zamagni, A.; Arienti, C.; Pignatta, S.; Tesei, A. Modeling neoplastic disease with spheroids and organoids. J. Hematol. Oncol. 2020, 13, 97. [Google Scholar] [CrossRef]
- Feng, P.-C.; Ke, X.-F.; Kuang, H.-L.; Pan, L.-L.; Ye, Q.; Wu, J.-B. BMP2 secretion from hepatocellular carcinoma cell HepG2 enhances angiogenesis and tumor growth in endothelial cells via activation of the MAPK/p38 signaling pathway. Stem Cell Res. Ther. 2019, 10, 237. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Gao, X.; Zuo, J.; Hu, B.; Yang, J.; Zhao, J.; Chen, J. STMN1 upregulation mediates hepatocellular carcinoma and hepatic stellate cell crosstalk to aggravate cancer by triggering the MET pathway. Cancer Sci. 2020, 111, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, F.; Ai, J.; Ebrahimi-Barough, S.; Seyhoun, I.; Hajifathali, A.; Muhammadnejad, S.; Hosseinzadeh, F.; Shadnoush, M.; Dabiri Oskouei, F.; Verdi, J. Natural Killer Cell Expansion with Autologous Feeder Layer and Anti-CD3 Antibody for Immune Cell Therapy of Hepatocellular Carcinoma. Asian Pac. J. Cancer Prev. 2019, 20, 3797–3803. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, Y.; Wang, L.; Yao, B.; Chen, T.; Li, Q.; Liu, Z.; Liu, R.; Niu, Y.; Song, T.; et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J. Exp. Clin. Cancer Res. 2019, 38, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yin, D.; Hu, Z.; Luo, C.; Zhou, Z.; Xin, H.; Yang, X.; Shi, Y.; Wang, Z.; Huang, X.; et al. A Positive Feedback Loop Between Cancer Stem-Like Cells and Tumor-Associated Neutrophils Controls Hepatocellular Carcinoma Progression. Hepatology 2019, 70, 1214–1230. [Google Scholar] [CrossRef] [PubMed]
- Wuputra, K.; Lin, C.-S.; Tsai, M.-H.; Ku, C.-C.; Lin, W.-H.; Yang, Y.-H.; Kuo, K.-K.; Yokoyama, K.K. Cancer cell reprogramming to identify the genes competent for generating liver cancer stem cells. Inflamm. Regen. 2017, 37, 15. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Factor, V.M.; Thorgeirsson, S.S. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. J. Hepatol. 2010, 53, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Gehart, H.; Clevers, H. Stem Cell-Derived Liver Cells. In The Liver; Wiley: Hoboken, NJ, USA, 2020; pp. 1015–1021. [Google Scholar]
- Wang, Y.; Takeishi, K.; Li, Z.; Cervantes-Alvarez, E.; Collin de l’Hortet, A.; Guzman-Lepe, J.; Cui, X.; Zhu, J. Microenvironment of a tumor-organoid system enhances hepatocellular carcinoma malignancy-related hallmarks. Organogenesis 2017, 13, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Štampar, M.; Tomc, J.; Filipič, M.; Žegura, B. Development of in vitro 3D cell model from hepatocellular carcinoma (HepG2) cell line and its application for genotoxicity testing. Arch. Toxicol. 2019, 93, 3321–3333. [Google Scholar] [CrossRef] [PubMed]
- Mišík, M.; Nersesyan, A.; Ropek, N.; Huber, W.W.; Haslinger, E.; Knasmueller, S. Use of human derived liver cells for the detection of genotoxins in comet assays. Mutat. Res. Toxicol. Environ. Mutagen. 2019, 845, 402995. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Xu, X.; Guo, X.; Sun, G.; Ma, X. Mesenchymal stem cells enhance the metastasis of 3D-cultured hepatocellular carcinoma cells. BMC Cancer 2016, 16, 566. [Google Scholar] [CrossRef] [Green Version]
- Shah, U.-K.; de Oliveira Mallia, J.; Singh, N.; Chapman, K.E.; Doak, S.H.; Jenkins, G.J.S. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutat. Res. Toxicol. Environ. Mutagen. 2018, 825, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Eilenberger, C.; Rothbauer, M.; Ehmoser, E.-K.; Ertl, P.; Küpcü, S. Effect of Spheroidal Age on Sorafenib Diffusivity and Toxicity in a 3D HepG2 Spheroid Model. Sci. Rep. 2019, 9, 4863. [Google Scholar] [CrossRef] [Green Version]
- Chiew, G.G.Y.; Wei, N.; Sultania, S.; Lim, S.; Luo, K.Q. Bioengineered three-dimensional co-culture of cancer cells and endothelial cells: A model system for dual analysis of tumor growth and angiogenesis. Biotechnol. Bioeng. 2017, 114, 1865–1877. [Google Scholar] [CrossRef]
- Yang, T.; Zhang, W.; Wang, L.; Xiao, C.; Wang, L.; Gong, Y.; Huang, D.; Guo, B.; Li, Q.; Xiang, Y.; et al. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC Cancer 2018, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Van Tienderen, G.S.; Groot Koerkamp, B.; IJzermans, J.N.M.; van der Laan, L.J.W.; Verstegen an Tienderen, M.M.A. Recreating Tumour Complexity in a Dish: Organoid Models to Study Liver Cancer Cells and their Extracellular Environment. Cancers 2019, 11, 1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tharehalli, U.; Svinarenko, M.; Lechel, A. Remodelling and Improvements in Organoid Technology to Study Liver Carcinogenesis in a Dish. Stem Cells Int. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef] [Green Version]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Knutsdottir, H.; Hui, K.; Weiss, M.J.; He, J.; Philosophe, B.; Cameron, A.M.; Wolfgang, C.L.; Pawlik, T.M.; Ghiaur, G.; et al. Human primary liver cancer organoids reveal intratumor and interpatient drug response heterogeneity. JCI Insight 2019, 4, 1–16. [Google Scholar] [CrossRef]
- Renwick, A.B.; Watts, P.S.; Edwards, R.J.; Barton, P.T.; Guyonnet, I.; Price, R.J.; Tredger, J.M.; Pelkonen, O.; Boobis, A.R.; Lake, B. Differential maintenance of cytochrome P450 enzymes in cultured precision-cut human liver slices. Drug Metab. Dispos. 2020, 28, 1202–1209. [Google Scholar]
- Wu, X.; Roberto, J.B.; Knupp, A.; Kenerson, H.L.; Truong, C.D.; Yuen, S.Y.; Brempelis, K.J.; Tuefferd, M.; Chen, A.; Horton, H.; et al. Precision-cut human liver slice cultures as an immunological platform. J. Immunol. Methods 2018, 455, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.W.F.; Betzel, B.; Stoopen, G.; Berends, F.J.; Janssen, I.M.; Peijnenburg, A.A.; Kersten, S. The impact of PPARα activation on whole genome gene expression in human precision cut liver slices. BMC Genom. 2015, 16, 760. [Google Scholar] [CrossRef] [Green Version]
- Plazar, J.; Filipič, M.; Groothuis, G.M.M. Antigenotoxic effect of Xanthohumol in rat liver slices. Toxicol. Vitr. 2008, 22, 318–327. [Google Scholar] [CrossRef]
- Boess, F.; Kamber, M.; Romer, S.; Gasser, R.; Muller, D.; Albertini, S.; Suter, L. Gene expression in two hepatic cell lines, cultured primary hepatocytes, and liver slices compared to the in vivo liver gene expression in rats: Possible implications for toxicogenomics use of in vitro systems. Toxicol. Sci. 2003, 73, 386–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Grunsven, L.A. 3D in vitro models of liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef]
- Prins, G.; Luangmonkong, T.; Oosterhuis, D.; Mutsaers, H.; Dekker, F.; Olinga, P. A Pathophysiological Model of Non-Alcoholic Fatty Liver Disease Using Precision-Cut Liver Slices. Nutrients 2019, 11, 507. [Google Scholar] [CrossRef] [Green Version]
- Gore, E.; Bigaeva, E.; Oldenburger, A.; Kim, Y.O.; Rippmann, J.F.; Schuppan, D.; Boersema, M.; Olinga, P. PI3K inhibition reduces murine and human liver fibrogenesis in precision-cut liver slices. Biochem. Pharmacol. 2019, 169, 113633. [Google Scholar] [CrossRef] [PubMed]
- Crespo Yanguas, S.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Armeanu, S.; Smirnow, I.; Kupka, S.; Wagner, S.; Wehrmann, M.; Rots, M.G.; Groothuis, G.M.M.; Weiss, T.S.; Königsrainer, A.; et al. Human precision-cut liver tumor slices as a tumor patient-individual predictive test system for oncolytic measles vaccine viruses. Int. J. Oncol. 2009, 34, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Granitzny, A.; Knebel, J.; Schaudien, D.; Braun, A.; Steinberg, P.; Dasenbrock, C.; Hansen, T. Maintenance of high quality rat precision cut liver slices during culture to study hepatotoxic responses: Acetaminophen as a model compound. Toxicol. Vitr. 2017, 42, 200–213. [Google Scholar] [CrossRef]
- Paish, H.L.; Reed, L.H.; Brown, H.; Bryan, M.C.; Govaere, O.; Leslie, J.; Barksby, B.S.; Garcia Macia, M.; Watson, A.; Xu, X.; et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 2019, 70, 1377–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramboer, E.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Immortalized Human Hepatic Cell Lines for In Vitro Testing and Research Purposes. In Protocols in In Vitro Hepatocyte Research; Vinken, M., Rogiers, V., Eds.; Springer: New York, NY, USA, 2015; pp. 53–76. ISBN 978-1-4939-2074-7. [Google Scholar]
- Zheng, Y.-H.; Yin, L.-H.; Grahn, T.H.M.; Ye, A.-F.; Zhao, Y.-R.; Zhang, Q.-Y. Anticancer Effects of Baicalein on Hepatocellular Carcinoma Cells. Phyther. Res. 2014, 28, 1342–1348. [Google Scholar] [CrossRef]
- Zhou, T.; Ye, L.; Bai, Y.; Sun, A.; Cox, B.; Liu, D.; Li, Y.; Liotta, D.; Snyder, J.P.; Fu, H.; et al. Autophagy and Apoptosis in Hepatocellular Carcinoma Induced by EF25-(GSH)2: A Novel Curcumin Analog. PLoS ONE 2014, 9, e107876. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, K.P.; Granata, I.; Guarracino, M.R. A computational integrative approach based on alternative splicing analysis to compare immortalized and primary cancer cells. Int. J. Biochem. Cell Biol. 2017, 91, 116–123. [Google Scholar] [CrossRef]
- Du, X.; Wu, L.; Ur Rahman, M.S.; Teng, X.; Teng, L.; Ye, J.; Cao, J. Promoter Hypomethylation Is Responsible for Upregulated Expression of HAI-1 in Hepatocellular Carcinoma. Dis. Markers 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- He, M.; Qin, H.; Poon, T.C.W.; Sze, S.-C.; Ding, X.; Co, N.N.; Ngai, S.-M.; Chan, T.-F.; Wong, N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Kongkavitoon, P.; Butta, P.; Sanpavat, A.; Bhattarakosol, P.; Tangtanatakul, P.; Wongprom, B.; Tangkijvanich, P.; Hirankarn, N.; Palaga, T. Regulation of periostin expression by Notch signaling in hepatocytes and liver cancer cell lines. Biochem. Biophys. Res. Commun. 2018, 506, 739–745. [Google Scholar] [CrossRef]
- Caruso, S.; Calatayud, A.-L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouzé, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, S.; Mihara, Y.; Kondo, R.; Kusano, H.; Akiba, J.; Yano, H. Antiproliferative Effect of Lenvatinib on Human Liver Cancer Cell Lines In Vitro and In Vivo. Anticancer Res. 2019, 39, 5973–5982. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Lieftink, C.; du Chatinier, A.; Gao, D.; Jin, G.; Jin, H.; Beijersbergen, R.L.; Qin, W.; Bernards, R. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut 2020, 69, 727–736. [Google Scholar] [CrossRef]
- Wu, X.-S.; Bao, T.-H.; Ke, Y.; Sun, D.-Y.; Shi, Z.-T.; Tang, H.-R.; Wang, L. Hint1 suppresses migration and invasion of hepatocellular carcinoma cells in vitro by modulating girdin activity. Tumor Biol. 2016, 37, 14711–14719. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Ding, Q.; Li, X.; Ji, X.; Li, L.; Fan, Y.; Xia, Y.; Tian, D.; Liu, M. SWELL1 promotes cell growth and metastasis of hepatocellular carcinoma in vitro and in vivo. EBioMedicine 2019, 48, 100–116. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, H.; Cao, G.; Kou, C.; Liu, T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial-mesenchymal transition in�vitro through activation of the Hedgehog pathway. Oncol. Rep. 2017, 39, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, W.; Chen, D.-T.; Pan, J.-H.; Chen, Y.-H.; Yan, Y.; Li, Q.; Xue, R.-F.; Yuan, Y.-F.; Zeng, W.-A. Lidocaine Induces Apoptosis and Suppresses Tumor Growth in Human Hepatocellular Carcinoma Cells In Vitro and in a Xenograft Model In Vivo. Anesthesiology 2017, 126, 868–881. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F.; Roustazadeh, A.; Shahsavani, H. In vitro effect of the Histone deacetylase inhibitor valproic acid on viability and apoptosis of the PLC/PRF5 human hepatocellular carcinoma cell line. Asian Pac. J. Cancer Prev. 2018, 19, 2507–2510. [Google Scholar] [CrossRef]
- Fowler, P.; Smith, K.; Young, J.; Jeffrey, L.; Kirkland, D.; Pfuhler, S.; Carmichael, P. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. I. Choice of cell type. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 742, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sasaki, Y.; Terasaki, N.; Kawataki, T.; Takekawa, K.; Iwase, Y.; Shimizu, T.; Sanoh, S.; Ohta, S. Comparison of Drug Metabolism and Its Related Hepatotoxic Effects in HepaRG, Cryopreserved Human Hepatocytes, and HepG2 Cell Cultures. Biol. Pharm. Bull. 2018, 41, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.-E.; Tryndyak, V.; Wu, Q.; Dreval, K.; Pogribny, I.; Bryant, M.; Zhou, T.; Robison, T.W.; Mei, N.; Guo, X. Quantitative comparison of in vitro genotoxicity between metabolically competent HepaRG cells and HepG2 cells using the high-throughput high-content CometChip assay. Arch. Toxicol. 2019, 93, 1433–1448. [Google Scholar] [CrossRef]
- Doktorova, T.Y.; Yildirimman, R.; Vinken, M.; Vilardell, M.; Vanhaecke, T.; Gmuender, H.; Bort, R.; Brolen, G.; Holmgren, G.; Li, R.; et al. Transcriptomic responses generated by hepatocarcinogens in a battery of liver-based in vitro models. Carcinogenesis 2013, 34, 1393–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018, 29, 25–38. [Google Scholar] [CrossRef] [PubMed]
- López-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef]
- Tian, J.; Tang, Z.; Xue, Q. [Expressions of the metastasis-associated factors of a new human hepatocellular carcinoma cell line with highly metastatic potential]. Zhonghua Yi Xue Za Zhi 1999, 79, 470–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffelfinger, S.C.; Hawkins, H.H.; Barrish, J.; Taylor, L.; Darlington, G.J. SK HEP-1: A human cell line of endothelial origin. Vitr. Cell. Dev. Biol.-Anim. 1992, 28, 136–142. [Google Scholar] [CrossRef]
- Mills, J.B.; Rose, K.A.; Sadagopan, N.; Sahi, J.; de Morais, S.M.F. Induction of Drug Metabolism Enzymes and MDR1 Using a Novel Human Hepatocyte Cell Line. J. Pharmacol. Exp. Ther. 2004, 309, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Reid, Y.; Gaddipati, J.P.; Yadav, D.; Kantor, J. Establishment of a human neonatal hepatocyte cell line. Vitr. Cell. Dev. Biol.-Anim. 2009, 45, 535–542. [Google Scholar] [CrossRef]
- Pfeifer, A.M.A.; Cole, K.E.; Smoot, D.T.; Weston, A.; Groopman, J.D.; Shields, P.G.; Vignaud, J.M.; Juillerat, M.; Lipsky, M.M.; Trump, B.F. Simian virus 40 large tumor antigen-immortalized normal human liver epithelial cells express hepatocyte characteristics and metabolize chemical carcinogens. Proc. Natl. Acad. Sci. USA 1993, 90, 5123–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, M.; Hirohashi, S. Cell lines from non-neoplastic liver and hepatocellular carcinoma tissue from a single patient. Vitr. Cell. Dev. Biol.-Anim. 1996, 32, 135–137. [Google Scholar] [CrossRef]
- Bogdanos, D.P.; Gao, B.; Gershwin, M.E. Liver Immunology. Compr Physiol. 2013, 2, 567–598. [Google Scholar] [CrossRef]
- Ding, C.; Li, Y.; Guo, F.; Jiang, Y.; Ying, W.; Li, D.; Yang, D.; Xia, X.; Liu, W.; Zhao, Y.; et al. A Cell-type-resolved Liver Proteome. Mol. Cell. Proteom. 2016, 15, 3190–3202. [Google Scholar] [CrossRef] [Green Version]
- Doumba, P.P.; Nikolopoulou, M.; Gomatos, I.P.; Konstadoulakis, M.M.; Koskinas, J. Co-culture of primary human tumor hepatocytes from patients with hepatocellular carcinoma with autologous peripheral blood mononuclear cells: Study of their in vitro immunological interactions. BMC Gastroenterol. 2013, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Baze, A.; Parmentier, C.; Hendriks, D.F.G.; Hurrell, T.; Heyd, B.; Bachellier, P.; Schuster, C.; Ingelman-Sundberg, M.; Richert, L. Three-Dimensional Spheroid Primary Human Hepatocytes in Monoculture and Coculture with Nonparenchymal Cells. Tissue Eng. Part C Methods 2018, 24, 534–545. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. Cell death in a co-culture of hepatocellular carcinoma cells and human umbilical vascular endothelial cells in a medium lacking glucose and arginine. Oncol. Lett. 2017, 13, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Cao, S.; Wang, Y.; Hu, Y.; Liu, H.; Li, J.; Chen, J.; Li, P.; Liu, J.; Wang, Q.; et al. Long non-coding RNA UBE2CP3 enhances HCC cell secretion of VEGFA and promotes angiogenesis by activating ERK1/2/HIF-1α/VEGFA signalling in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 113. [Google Scholar] [CrossRef] [PubMed]
- Serhal, R.; Saliba, N.; Hilal, G.; Moussa, M.; Hassan, G.; El Atat, O.; Alaaeddine, N. Effect of adipose-derived mesenchymal stem cells on hepatocellular carcinoma: In vitro inhibition of carcinogenesis. World J. Gastroenterol. 2019, 25, 567–583. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Lin, Y.; Lu, X.; Zhang, C.; Chen, S.; Jian, Z. IL-6/STAT3 pathway intermediates M1/M2 macrophage polarization during the development of hepatocellular carcinoma. J. Cell. Biochem. 2018, 119, 9419–9432. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Schulte, L.-A.; López-Gil, J.C.; Sainz, B.; Hermann, P.C. The Cancer Stem Cell in Hepatocellular Carcinoma. Cancers 2020, 12, 684. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.-W. Cellular reprogramming and hepatocellular carcinoma development. World J. Gastroenterol. 2013, 19, 8850. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeong, J.; Park, S.; Jin, Y.-W.; Lee, S.-S.; Lee, S.B.; Choi, D. Establishment of Hepatocellular Cancer Induced Pluripotent Stem Cells Using a Reprogramming Technique. Gut Liver 2017, 11, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Sheng, Y.; Yang, J.; Wang, C.; Zhang, R.; Zhu, Y.; Zhang, Z.; Zhang, K.; Yan, S.; Sun, H.; et al. Osteopontin alters DNA methylation through up-regulating DNMT1 and sensitizes CD133+/CD44+ cancer stem cells to 5 azacytidine in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 179. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, H.; Chung, R.T.; Sato, C. An Identification of Novel Therapy for Human Hepatocellular Carcinoma by Using Human Induced Pluripotent Stem Cells. Hepatology 2010, 51, 1089–1090. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, H.; Madson, J. The reprogramming therapy for a patient with advanced hepatocellular carcinoma by using human-induced pluripotent stem (iPS) cells technology. Case Reports 2013, 2013, bcr2013008950. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yan, Q.; Sun, Y.; Nam, Y.; Hu, L.; Loong, J.H.C.; Ouyang, Q.; Zhang, Y.; Li, H.-L.; Kong, F.-E.; et al. A hepatocyte differentiation model reveals two subtypes of liver cancer with different oncofetal properties and therapeutic targets. Proc. Natl. Acad. Sci. USA 2020, 117, 6103–6113. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Dydowiczová, A.; Trosko, J.E.; Bláha, L.; Babica, P. Ready to go 3D? A semi-automated protocol for microwell spheroid arrays to increase scalability and throughput of 3D cell culture testing. Toxicol. Mech. Methods 2020, 30, 590–604. [Google Scholar] [CrossRef]
- Kim, S.M.; Han, J.M.; Le, T.T.; Sohng, J.K.; Jung, H.J. Anticancer and Antiangiogenic Activities of Novel α-Mangostin Glycosides in Human Hepatocellular Carcinoma Cells via Downregulation of c-Met and HIF-1α. Int. J. Mol. Sci. 2020, 21, 4043. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, M.P.; Angel, A.; Delprato, C.B.; Livore, V.I.; Ferretti, A.C.; Lucci, A.; Comanzo, C.G.; de Lujan Alvarez, M.; Quiroga, A.D.; Mottino, A.D.; et al. Sirtuin 1 and 2 inhibitors enhance the inhibitory effect of sorafenib in hepatocellular carcinoma cells. Eur. J. Pharmacol. 2021, 892. [Google Scholar] [CrossRef]
- Varan, G.; Akkın, S.; Demirtürk, N.; Benito, J.M.; Bilensoy, E. Erlotinib entrapped in cholesterol-depleting cyclodextrin nanoparticles shows improved antitumoral efficacy in 3D spheroid tumors of the lung and the liver. J. Drug Target. 2021, 29, 439–453. [Google Scholar] [CrossRef]
- Song, Y.; Lee, S.-Y.; Kim, S.; Choi, I.; Kim, S.-H.; Shum, D.; Heo, J.; Kim, A.-R.; Kim, K.M.; Seo, H.R. Inhibitors of Na+/K+ ATPase exhibit antitumor effects on multicellular tumor spheroids of hepatocellular carcinoma. Sci. Rep. 2020, 10, 5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Kim, J.-S.; Kim, S.-H.; Park, Y.K.; Yu, E.; Kim, K.-H.; Seo, E.-J.; Oh, H.-B.; Lee, H.C.; Kim, K.M.; et al. Patient-derived multicellular tumor spheroids towards optimized treatment for patients with hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 109. [Google Scholar] [CrossRef]
- Ranga, A.; Gjorevski, N.; Lutolf, M.P. Drug discovery through stem cell-based organoid models. Adv. Drug Deliv. Rev. 2014, 69–70, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Prior, N.; Inacio, P.; Huch, M. Liver organoids: From basic research to therapeutic applications. Gut 2019, 68, 2228–2237. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Mead, B.E.; Safaee, H.; Langer, R.; Karp, J.M.; Levy, O. Engineering Stem Cell Organoids. Cell Stem Cell 2016, 18, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Jee, J.H.; Lee, D.H.; Ko, J.; Hahn, S.; Jeong, S.Y.; Kim, H.K.; Park, E.; Choi, S.Y.; Jeong, S.; Lee, J.W.; et al. Development of Collagen-Based 3D Matrix for Gastrointestinal Tract-Derived Organoid Culture. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Cen, J.; Ma, X.; Cui, L.; Qiu, Z.; Zhang, Z.; Li, H.; Yang, R.-Z.; Wang, C.; et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 2019, 21, 1015–1026. [Google Scholar] [CrossRef]
- Kern, M.A.; Haugg, A.M.; Eiteneuer, E.; Konze, E.; Drebber, U.; Dienes, H.P.; Breuhahn, K.; Schirmacher, P.; Kasper, H.U. Ex vivo analysis of antineoplastic agents in precision-cut tissue slices of human origin: Effects of cyclooxygenase-2 inhibition in hepatocellular carcinoma. Liver Int. 2006, 26, 604–612. [Google Scholar] [CrossRef]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef] [Green Version]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Thorgeirsson, S.S. SnapShot: Hepatocellular Carcinoma. Cancer Cell 2014, 25, 550.e1. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, R.; Llovet, J.M.; Han, G.; Tak, W.Y.; Yang, J.; Guglielmi, A.; Paik, S.W.; Reig, M.; Kim, D.Y.; Chau, G.-Y.; et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J. Hepatol. 2016, 64, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Han, G.; Finn, R.S.; Poon, R.T.P.; Blanc, J.-F.; Yan, L.; Yang, J.; Lu, L.; Tak, W.-Y.; Yu, X.; et al. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: A randomized phase III trial. Hepatology 2014, 60, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Labeur, T.A.; Ten Cate, D.W.G.; Bart Takkenberg, R.; Azahaf, H.; van Oijen, M.G.H.; van Delden, O.M.; de Man, R.A.; van Vugt, J.L.A.; IJzermans, J.N.M.; Eskens, F.A.L.M.; et al. Are we SHARP enough? The importance of adequate patient selection in sorafenib treatment for hepatocellular carcinoma. Acta Oncol. 2018, 57, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Groenendijk, F.H.; Mellema, W.W.; van der Burg, E.; Schut, E.; Hauptmann, M.; Horlings, H.M.; Willems, S.M.; van den Heuvel, M.M.; Jonkers, J.; Smit, E.F.; et al. Sorafenib synergizes with metformin in NSCLC through AMPK pathway activation. Int. J. Cancer 2015, 136, 1434–1444. [Google Scholar] [CrossRef]
- Huang, D.Q.; Muthiah, M.D.; Zhou, L.; Jumat, H.; Tan, W.X.; Lee, G.H.; Lim, S.G.; Kow, A.; Bonney, G.; Shridhar, I.; et al. Predicting HCC Response to Multikinase Inhibitors With In Vivo Cirrhotic Mouse Model for Personalized Therapy. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1313–1325. [Google Scholar] [CrossRef]
- Shen, J.; Cai, W.; Ma, Y.; Xu, R.; Huo, Z.; Song, L.; Qiu, X.; Zhang, Y.; Li, A.; Cao, W.; et al. hGC33-Modified and Sorafenib-Loaded Nanoparticles have a Synergistic Anti-Hepatoma Effect by Inhibiting Wnt Signaling Pathway. Nanoscale Res. Lett. 2020, 15, 220. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.M.; Onorato, A.; Cantero, M.J.; Domínguez, L.; Bayo, J.; Fiore, E.; García, M.; Atorrasagasti, C.; Canbay, A.; Malvicini, M.; et al. 4-methylumbelliferone-mediated polarization of M1 macrophages correlate with decreased hepatocellular carcinoma aggressiveness in mice. Sci. Rep. 2021, 11, 6310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-F.; Peng, F.; Li, J.-Y.; Ye, Y.-B. Intratumoral IL-12 Gene Therapy Inhibits Tumor Growth In A HCC-Hu-PBL-NOD/SCID Murine Model. Onco. Targets. Ther. 2019, 12, 7773–7784. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Jiang, H.; Wang, P.; Song, B.; Wang, H.; Kong, X.; Li, Z. Treatment of hepatocellular carcinoma with a GPC3-targeted bispecific T cell engager. Oncotarget 2017, 8, 52866–52876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fu, C.; Kong, Y.; Pan, D.; Wang, Y.; Huang, S.; Li, Z.; Ning, Z.; Lu, X.; Shan, S.; et al. Antitumor and immunomodulatory effects of a novel multitarget inhibitor, CS2164, in mouse hepatocellular carcinoma models. Anticancer. Drugs 2019, 30, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Jilkova, Z.M.; Kuyucu, A.Z.; Kurma, K.; Ahmad Pour, S.T.; Roth, G.S.; Abbadessa, G.; Yu, Y.; Schwartz, B.; Sturm, N.; Marche, P.N.; et al. Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget 2018, 9, 11145–11158. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.; Zheng, H.; Wang, B.; Liu, Y.; Liu, D.; Chen, J.; Xu, W.; Li, A.; Zhu, Y. Synergistic anti-tumor efficacy of sorafenib and fluvastatin in hepatocellular carcinoma. Oncotarget 2017, 8, 23265–23276. [Google Scholar] [CrossRef] [Green Version]
- Afaloniati, H.; Angelopoulou, K.; Giakoustidis, A.; Hardas, A.; Pseftogas, A.; Makedou, K.; Gargavanis, A.; Goulopoulos, T.; Iliadis, S.; Papadopoulos, V.; et al. HDAC1/2 Inhibitor Romidepsin Suppresses DEN-Induced Hepatocellular Carcinogenesis in Mice. Onco. Targets. Ther. 2020, 13, 5575–5588. [Google Scholar] [CrossRef]
- Sung, Y.-C.; Liu, Y.-C.; Chao, P.-H.; Chang, C.-C.; Jin, P.-R.; Lin, T.-T.; Lin, J.-A.; Cheng, H.-T.; Wang, J.; Lai, C.P.; et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics 2018, 8, 894–905. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Chung, A.S.; Mettlen, M.; Ganguly, D.; Lu, T.; Wang, T.; Brekken, R.A.; Hsiehchen, D.; Zhu, H. Immune Checkpoint Inhibition is Safe and Effective for Liver Cancer Prevention in a Mouse Model of Hepatocellular Carcinoma. Cancer Prev. Res. 2020, 13, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Reszegi, A.; Horváth, Z.; Fehér, H.; Wichmann, B.; Tátrai, P.; Kovalszky, I.; Baghy, K. Protective Role of Decorin in Primary Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Takahashi, H.; Kuwashiro, T.; Tanaka, K.; Mori, H.; Ozaki, I.; Kitajima, Y.; Matsuda, Y.; Ashida, K.; Eguchi, Y.; et al. Glucagon-Like Peptide-1 Receptor Agonist Prevented the Progression of Hepatocellular Carcinoma in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 5722. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tian, G.; Zhuang, Z.; Chen, J.; You, N.; Zhuo, L.; Liang, B.; Song, Y.; Zang, S.; Liu, J.; et al. Berberine prevents non-alcoholic steatohepatitis-derived hepatocellular carcinoma by inhibiting inflammation and angiogenesis in mice. Am. J. Transl. Res. 2019, 11, 2668–2682. [Google Scholar]
- Chow, A.K.-M.; Yau, S.W.-L.; Ng, L. Novel molecular targets in hepatocellular carcinoma. World J. Clin. Oncol. 2020, 11, 589–605. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, H.; Hu, M.; Huang, T.; Hu, Y.; Sang, N.; Zhao, Y. Recent progress in treatment of hepatocellular carcinoma. Am. J. Cancer Res. 2020, 10, 2993–3036. [Google Scholar] [CrossRef]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L. Drug Development for Hepatocellular Carcinoma: Knowing the Past Helps to Understand the Future. Oncologist 2014, 19, 1115–1117. [Google Scholar] [CrossRef] [Green Version]






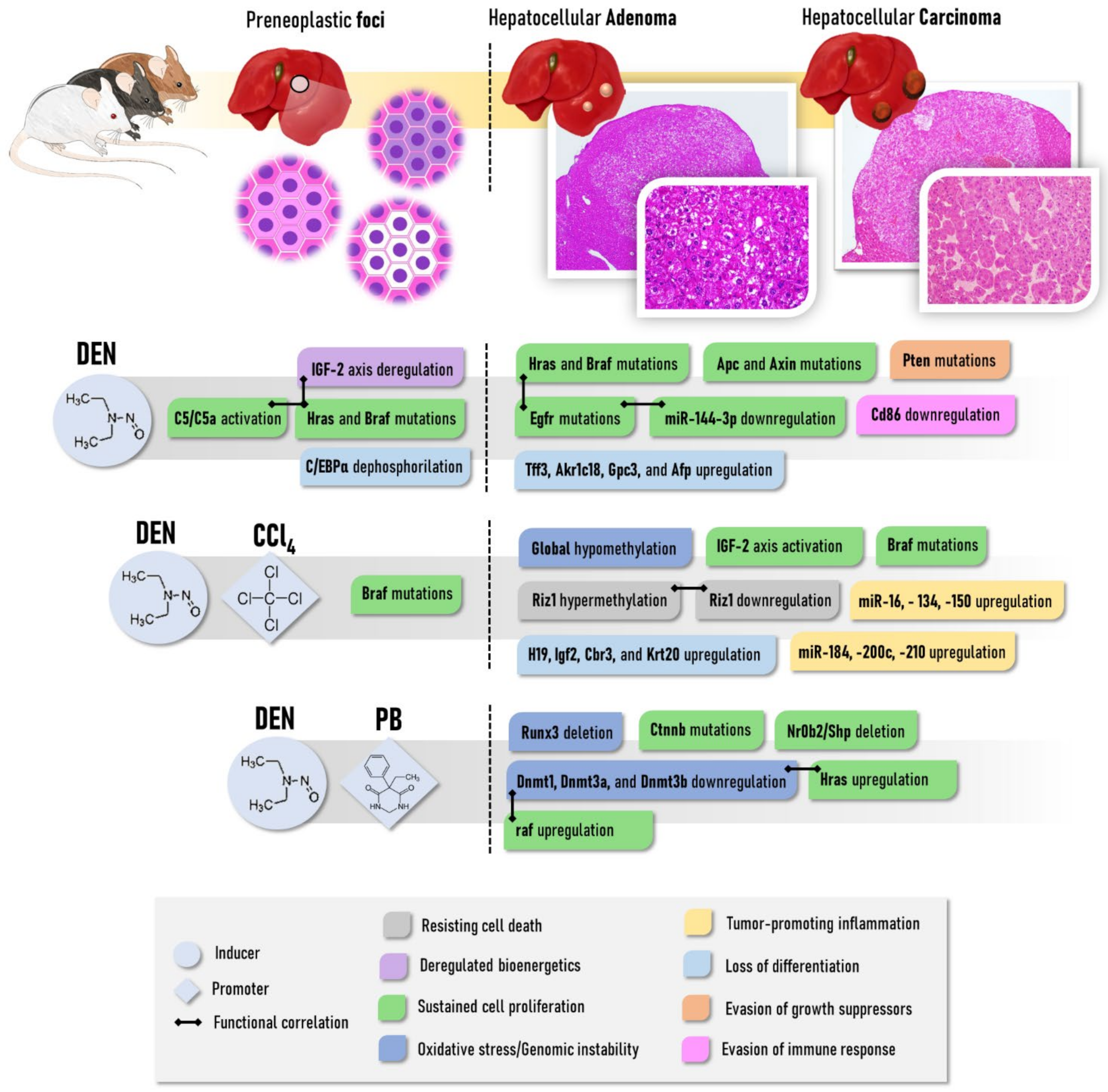{kind=link}
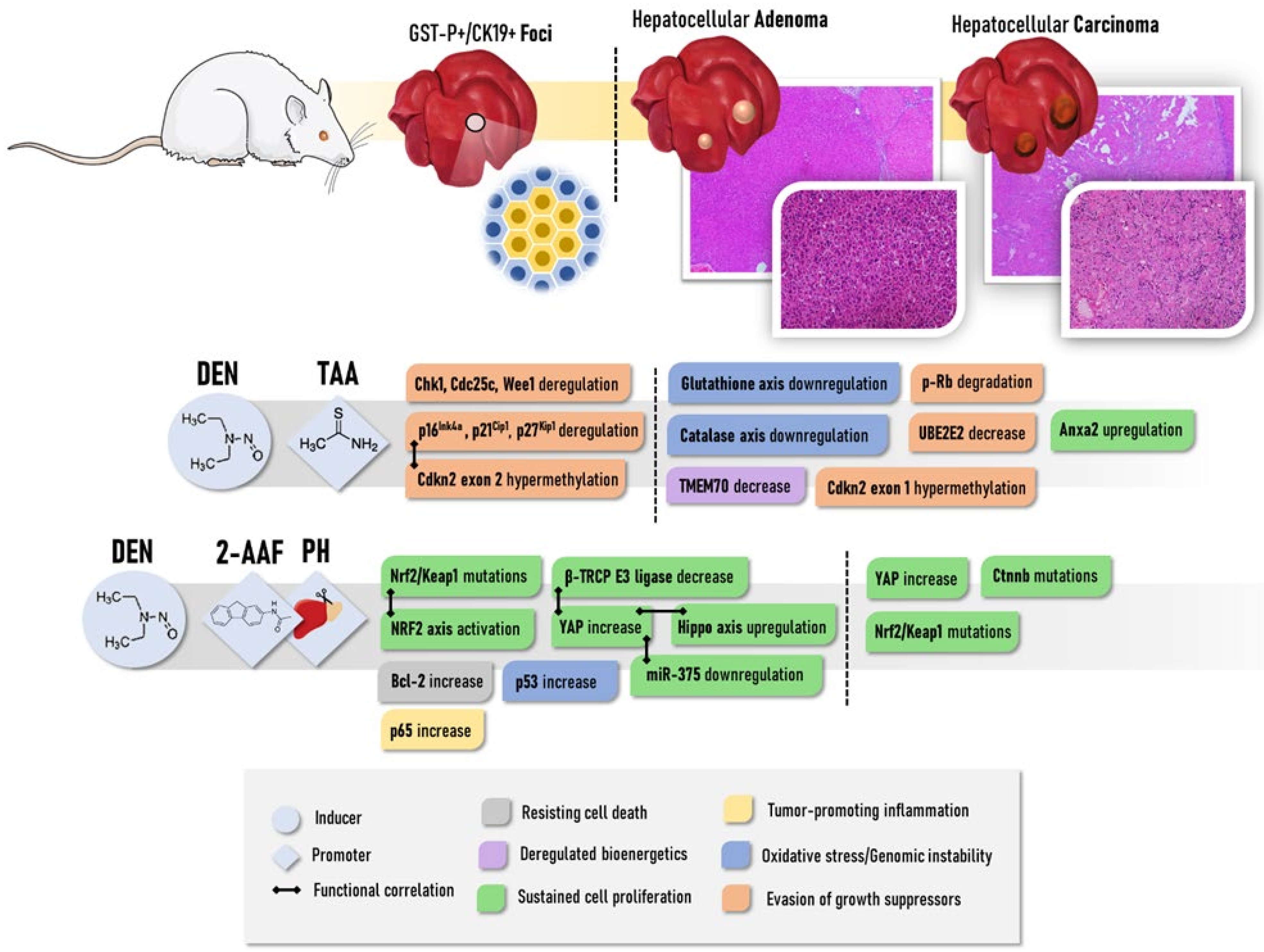{kind=link}
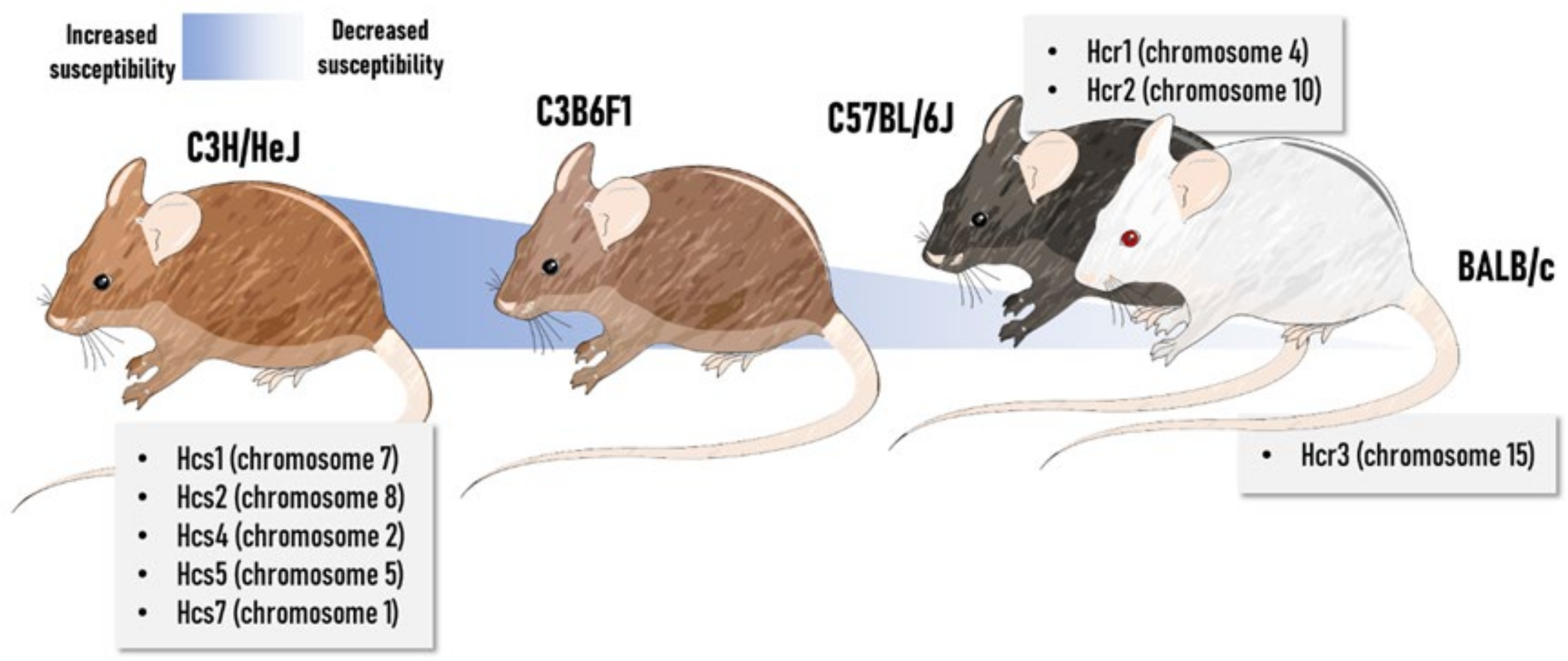{kind=link}
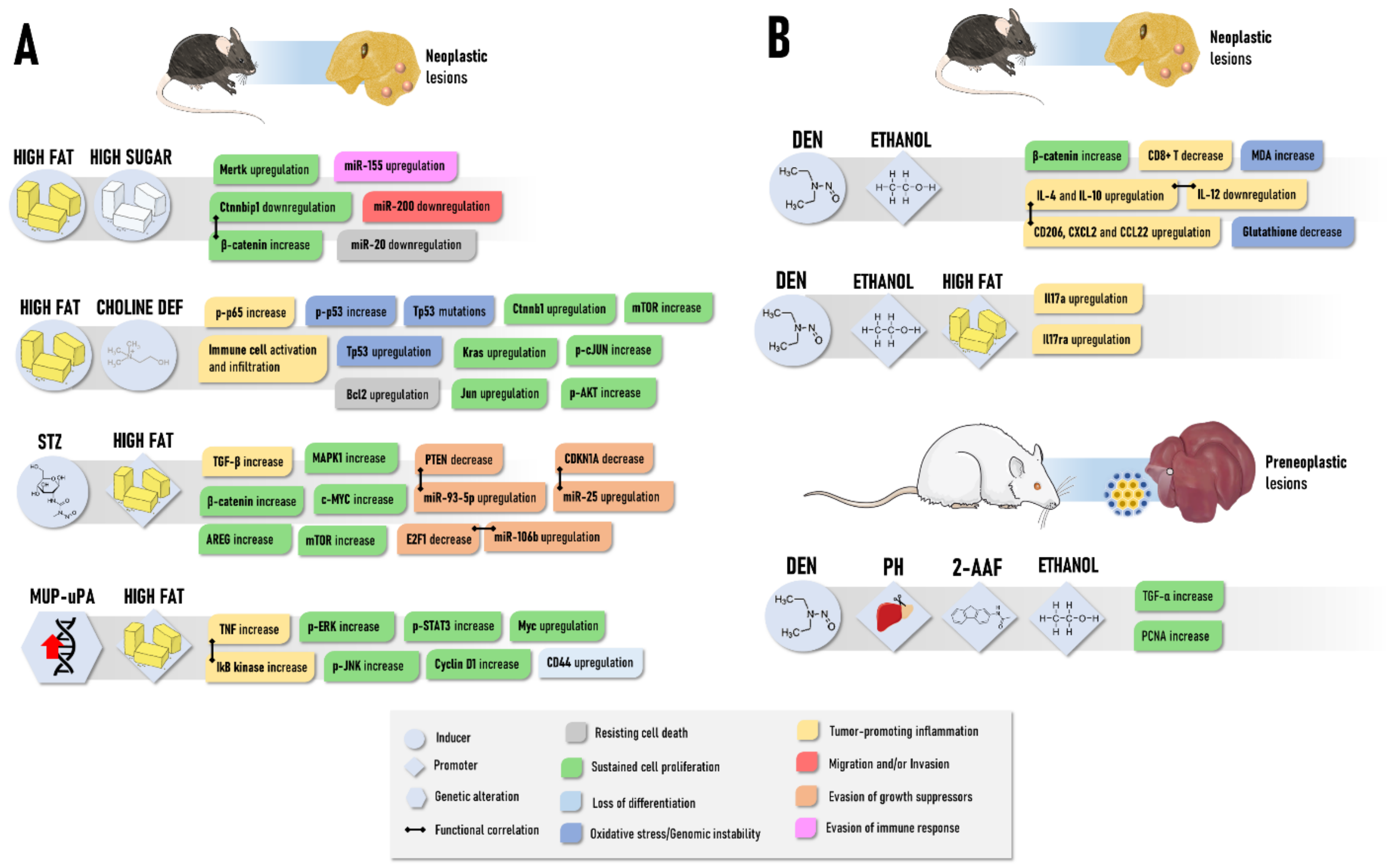{kind=link}
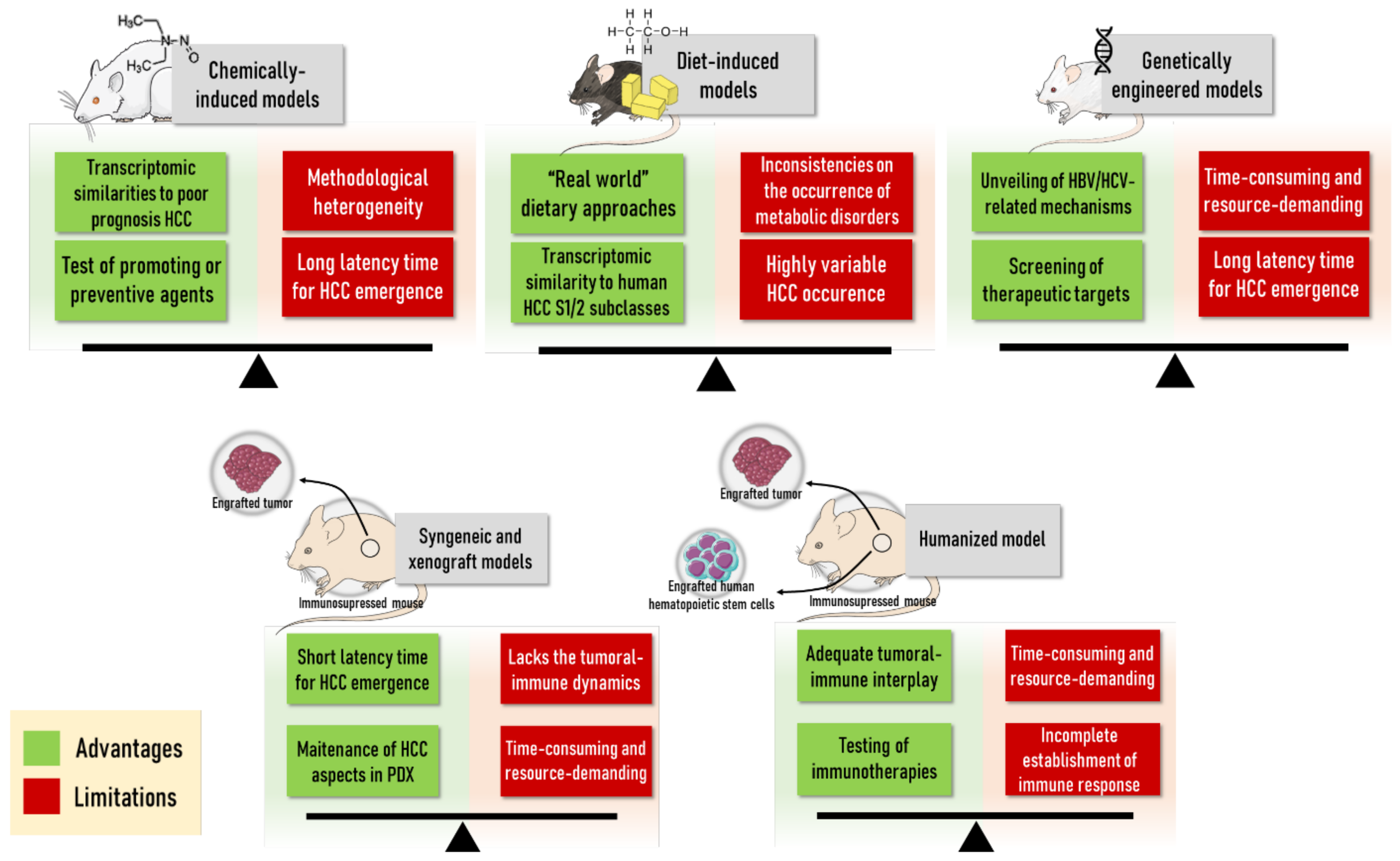{kind=link}
| Model | Procedure | Animal (Species, Strain, Age) | Timepoints and Incidence of Lesions | References |
|---|---|---|---|---|
| Ectopic implantation of human HCC in mouse; |
| Female Balb/c athymic nude mice at 4-week-old; |
| [26] |
| LCI-D20: orthotopic Implantation of human HCC |
| Male BALB/cA nude mice at 4 to 6-week-old |
| [22] |
| HCC cells |
| Male SCID at 6-week-old; |
| [23] |
| HCC-LY5 and HCC-LY10: Ectopic and orthotopic Implantation of human HCC in mice; |
| NOD/SCID male and female mice and T cell-immunodeficient BALB/c-nu/nu mice at 6 to 8-week-old |
| [27] |
| Ectopic implantation from human HCC needle biopsies in mice |
| Nonobese, diabetic/severe combined immunodeficiency gamma-c mice at 10-week-old; |
| [24] |
| Model | Procedure | Animal (Species, Strain, Age) | Timepoints and Incidence of Lesions | References |
|---|---|---|---|---|
| DEN | Single i.p., 90 mg/kg b.w. | Juvenile (5 weeks) C3H/He, DBA/2 and C57BL/6 mice (male) |
| [41] |
| Multiple i.p. 1.5 or 3 mg/kg b.w. for 1 week (4×/week) | Juvenile (6 weeks) B6C3F1 and C3AF1 mice (male) |
| [42] | |
| Drinking water 15 mg/L for 3 weeks | Juvenile (4 weeks) B6C3F1 mice (male) |
| [43] | |
| Multiple i.p. 25, 50, or 75 mg/kg b.w., for 4 or 8 weeks (1×/week) | Juvenile (4 weeks) C57BL/6 mice (male) |
| [44] | |
| Single i.p. 2.5, 10, 25 or 50 mg/kg b.w. | Infant (2 weeks) BALB/c mice (male) |
|
| |
| Single i.p. 5 mg/kg b.w. | Infant (2 weeks) C3H/HeJ, B6C3F1 and C57BL mice (male) |
| [46] | |
| Single i.p. 1 mg/kg b.w. | Infant (2 weeks) C3H/HeJ, B6C3F1 and C57BL mice (male) |
| [47] | |
| Single i.p. 1 mg/kg b.w. | Infant (2 weeks) B6C3F1 mice (male) |
| [48] | |
| Multiple i.p. 20 (1 dose), 30 (1 dose) and 50 mg/kg (6 doses) b.w., for 8 weeks (1×/week) | Infant (2 weeks) C57BL/6 mice (male and female) |
| [49] | |
| Gavage 80 mg/kg b.w. (weeks ~6–7) | Adult (~6–7 weeks) Sprague-Dawley rats (male) |
| [50] | |
| Single i.p., 200 mg/kg b.w. (week 6) | Juvenile (4 weeks) F344 rats (male) |
| [51] | |
| Multiple i.p. 70 mg/kg b.w., for 10 weeks (1×/week) | Adult (6 weeks) Sprague-Dawley rats (male) |
| [52] | |
| Multiple i.p. 30 mg/kg b.w. for 11 weeks (2×/week) | Juvenile (4–5 weeks) Sprague-Dawley rats (male) | [53] | ||
| Multiple gavage 70 mg/kg b.w. for 14 weeks (1×/week) | Juvenile (4–5 weeks) Wistar rats (male) |
| [54] | |
| DEN and CCl4 |
| Infant (2 weeks) C3H/HeJ mice (male) |
| [55] |
| Infant (2 weeks) B6C3F1 mice (male) |
| [48] | |
| Juvenile (4–5 weeks) F344 rats (male) |
| [56] | |
| CCl4 |
| Infant (2 weeks) B6C3F1 mice (male) |
| [48] |
| DEN and TAA |
| Infant (2 weeks) C57BL/6 mice (male and female) |
| [49] |
| Adult (6 weeks) Wistar rats (male) |
| [57] | |
| DEN, TAA and PB |
| Adult (6 weeks) F344 rats (male) |
| [58] |
| DEN and PB |
| Juvenile (5 weeks) C3H/He, DBA/2 and C57BL/6 mice (male) |
| [41] |
| Adult (~6–7 weeks) Sprague-Dawley rats (male) |
| [50] | |
| Juvenile (4 weeks) F344 rats (male) |
| [51] | |
| DEN, 2-AAF and PH |
| Adult Fischer 344 rats (male) |
| [59] |
| Model | Procedure | Animal (Species, Strain, Age) | Timepoints and Incidence of Lesions | References |
|---|---|---|---|---|
| HF/HS diet | High-fat and high sugar diet (~35% hydrogenated coconut oil and soybean oil, ~19% carbohydrate, w/w) for 12, 24 or 48 weeks | Juvenile (4 weeks) C57BL/6J mice (male) | Neoplastic lesions: 0% at weeks 12 and 24 and 20% at week 48 | [165] |
| High-fat diet (~21% fat and 0.1% cholesterol, w/w) and high sugar solution (23.1/18.9 g/L of fructose/glucose) for 56 weeks | Juvenile to adult (6–8 weeks) B6/129 mice (male) | Adenomas: 25% at week 56 Well-differentiated Carcinomas: 100% at week 56 Poorly-differentiated Carcinomas: 37.5% at week 56 | [162] | |
| High-fat (23% w/w, of which 23% saturated, 34% trans, 31% monounsaturated (cis), 12% polyunsaturated) and high sugar solution (23.1/18.9 g/L of fructose/glucose) for 24 or 48 weeks | Juvenile to adult (6–8 weeks) B6/129 mice (male) | Neoplastic lesions: 0 at week 24 and 40% at week 48 | [164] | |
| High-fat and high sugar diet (21.1% fat, 41% sucrose, and 1.25% cholesterol, w/w) and high sugar solution (23.1/18.9 g/L of fructose/glucose) for 12 or 24 weeks | Adult (9 weeks) C57BL/6J mice (male) | Neoplastic lesions: 0% at week 12 and 30% at week 24 | [166] | |
| HF/HS diet and CCl4 | High-fat and high sugar diet (21.1% fat, 41% sucrose, and 1.25% cholesterol, w/w) and high sugar solution for 12 or 24 weeks CCl4: Multiple i.p. 0.2 mL/kg b.w. for 24 or 36 weeks (1×/week) | Neoplastic lesions: 0% at week 12 and 100% at week 24 | ||
| HF diet | High-fat (~24% fat, lard and soybean oil, w/w) for 48 weeks | Juvenile (4–5 weeks) C57BL/6J mice (male and female) | Carcinomas: 2.5% at week 48 | [167] |
| CDHF diet | High-fat (~24% fat, lard and soybean oil, w/w) and choline-deficient diet for 48 weeks | Carcinomas: 25% at week 48 | ||
| CDAHF diet | High-fat (~35% fat, w/w), choline-deficient, L-amino acid-defined, 0.1% methionine diet for 12, 24, 36, 48 or 60 weeks | Juvenile (5 weeks) C57BL/6J mice (male) | Adenomas: 0% at week 12 and 24; 67% at week 36; 100% at weeks 48 and 60 Carcinomas: 0% at week 12 and 24; 17% at week 36; 9% at week 48; 26% at week 60 | [168] |
| CDA diet | Choline-deficient L-amino acid-defined diet for 24 or 36 weeks | Juvenile to adult (6–8 weeks) C57BL/6 mice | Carcinomas: 30–40% at weeks 24 and 36 | [169] |
| CDA diet and CCl4 | Choline-deficient L-amino acid-defined diet for 24 or 36 weeks CCl4: Multiple i.p. 0.2 mL/kg b.w. for 24 or 36 weeks (1×/week) | Carcinomas: 40% at weeks 24; 100% at week 36 | ||
| STAM | STZ: Single s.c. 200 µg (day 2) High fat (32%) diet for 20 weeks | Infant (2 days) C57BL/6J mice (male) | Carcinomas: 100% at weeks 14 to 20 | [170] |
| Pten null | - | Pten null mice (male and female) | Adenomas: 47% at week 44 and 100% at weeks 74–78; Carcinomas: 66% at weeks 74–78 | [171] |
| MUP-uPA transgenic | High-fat (59% kcal from fat) for 32 or 40 weeks | MUP-uPA mice | Adenomas: 50% at week 32 and 71.4% at week 40; Carcinomas: 16.6% at week 32 and 50% at week 40 | [172] |
| Model | Procedure | Animal (Species, Strain, Age) | Timepoints and Incidence of Lesions | References |
|---|---|---|---|---|
| Ethanol | Liquid low (1%, w/w) or high (3%, w/w) ethanol diet for ~100–110 weeks | Juvenile to adult (6–7 weeks) Sprague-Dawley rats (male and female) | Neoplastic lesions: 2% at weeks ~100–110 (for both doses and sexes) | [178] |
| Ethanol in drinking water 2.5% or 5% (v/v) for 104 weeks | Juvenile (4 weeks) B6C3F1 mice (male) | Neoplastic lesions: 34% (2.5%) and 52% (5%) at week 108 Adenomas: 25.5% (2.5%) and 39.6% (5%) at week 108 | [179] | |
| MeIQx and Ethanol | MeIQx: 200 mg/kg diet for 8 weeks Ethanol in drinking water 10 or 20% (v/v) for 16 weeks | Juvenile (3 weeks) F344/DuCrj rats (male) | Adenomas: ~80% (10%) or ~100% (20%) at week 27 Carcinomas: ~20% (10%) or ~50% (20%) at week 27 | [183] |
| Resistant hepatocyte and Ethanol | DEN: Single i.p. 200 mg/kg b.w.,at week 6 2-AAF: 200 mg/kg in diet for 3 weeks PH: at week 9 Ethanol in drinking water 5% (v/v) for 5 or 15 weeks | Juvenile (4 weeks) Wistar rats (male) | Preneoplastic foci: 63%–100% at week, 18 44%–100% at week 28 * Adenomas: 75% at week 18, 94% at week 28 * Carcinomas: 0% at week 18, 0% at week 28 * | [184] |
| DEN and Ethanol | DEN: Single i.p. 200 mg/kg b.w., at week 9 Ethanol in drinking water 5% (v/v) for 16 weeks | Adult (9 weeks) WT rats | Adenomas: 8% at week 25 Carcinomas: 0% at week 25 | [182] |
| Adult (9 weeks) Cx32 dominant-negative transgenic rats | Adenomas: 25% at week 25 Carcinomas: 25% at week 25 | |||
| DEN: Single i.p.10 mg/kg b.w., at week 2 Lieber-Decarli diet (4.9% of ethanol, v/v) for 16 weeks | Infant (2 weeks) C57BL/6 mice (male and female) | Eosinophilic foci: 53% at week 23 Adenomas: 60% at week 23 * Carcinomas: 13% at week 23 * | [185] | |
| DEN: Single i.p. 1 mg/kg b.w. at weeks 3–4 Ethanol in drinking water 5% (v/v) for 3 days, followed by 10% (v/v) for 3 days and 10/20% (v/v) (alternate days) for 8 weeks, during weeks 16 to 24 or 40 to 48 | Juvenile (3–4 weeks) B6C3 mice (male) | Neoplastic lesions: 97.5% at week 48 * | [186] | |
| DEN, Ethanol and HF | DEN: Single i.p. 25 mg/kg b.w. at weeks 2 Liquid ethanol diet (gradually increased from 1% to 2% and 3% (v/v, throughout 3 weeks), and maintained at 3.5% (v/v)) for 18 or 24 weeks. | Infant (2 weeks) C57BL/6 mice (male) | Carcinomas: ~20% at week 18; ~70% at week 24 | [187] |
| Model | Genetic Modification | Timepoints and Incidence of Lesions | References |
|---|---|---|---|
| HBV-transgenic | HBx gene |
| [195] |
| [196] | ||
| HBx, HBsAg, and pre-S gene |
| [199] | |
| [200] | ||
| pre-S/S gene (rtA181T/sW172mutation) |
| [201] | |
| HCV-transgenic | Core gene |
| [202] |
| c-myc transgenic | cnsfo-myc overexpression |
| [203] |
| [204] | ||
| [205] | ||
| c-myc/TGF-α transgenic | Double c-myc/TGF-α overexpression |
| |
| [203] | ||
| E2F-1 transgenic | E2F-1 overexpression |
| [206] |
| c-myc/E2F-1 transgenic | Double c-myc/E2F-1 overexpression |
| [205] |
| Apc knockout | Apc deletion |
| [207] |
| β-catenin/H-ras mutant | Double Catnb/Hras overexpression |
| [208] |
| cMyc + shp53 mice | c-myc overexpression, p53 downregulation |
| [209] |
| Genes | Plasmids | Strain | Timepoints and/or Lesions | References |
|---|---|---|---|---|
| NRasV12 and myr-AKT | 7.5 μg of myr-AKT1; 7.5 μg N-RasV12; SB transposase (25:1) | C57BL/6 | HCC formation and progression after 2 to 4 weeks post-injection | [220] |
| c-Myc and β-catenin | 10 μg pT3-EF1a-MYC; 10 μg pT3-N90-CTNNB1; 2.5 μg SB13-Luc transposase-encoding vector | C57BL/6 | Poorly to moderately differentiated HCC with solid/trabecular pattern, and immunoexpression of CK19 and nuclear β-catenin | [221] |
| β-catenin and tert or pten | 10 μg pT3-N90-CTNNB1; 10 μg pT3-EF1a-Tert or 10 μg pX330-Pten; 2.5 μg SB13-Luc transposase-encoding vector | C57BL/6 | Well to moderately differentiated HCC with trabecular pattern, abundant clear cells, and immuno-expression of glutamine synthetase | [221] |
| c-Myc and axin1 | 10 μg pT3-EF1a-MYC; 10 μg pX330-Axin1; 2.5 μg SB13-Luc transposase-encoding vector | C57BL/6 | Well to moderately differentiated HCCs with trabecular pattern | [221] |
| c-Myc and MCL1 | 10 μg pT3-EF1α-c-MYC-shLuc; 5 μg pT3-EF1α-Mcl1; SB transposase (25:1) | C57BL/6 FVB/N Balb/C | Liver tumor formation after 5 to 8 weeks post-injection | [222,223] |
| c-met and axin1 | 20 μg pT3-EF1α-c-Met; 40 μg pX330-Axin1.1; 0.8 μg pCVM/SB | FVB/N | HCC burden at 9 to 12 weeks post-injection showing membranous immunoexpression of E-Cadherin and absence of glutamine synthetase | [224] |
| c-Myc and myr-AKT | 16–36 µg of mixed plasmids: pT3-EF1a-myrAKT-HA; pT3-EF1α-c-MYC; SB13 transposase-expression plasmid | C57BL/6J | Well to moderately differentiated HCC at 8 to 10 weeks post-injection showing trabecular or nest-like patterns | [225] |
| myr-AKT and/or Hras | 16–43 µg of mixed plasmids: pT3-EF1a-myrAKT-HA; cDNA fragments of FLAG-HRASV12; SB13 transposase-expression plasmid | C57BL/6J | Akt or Hras: multiple HCC associated with lipid accumulation after 20–28 weeks post-injection Akt and Hras: HCC after 8 weeks post-injection with a higher proliferation rate | [226] |
| c-met and β-catenin | 5 μg pT3-EF5a-hMet-V5; 5 μg pT3-EF5α-S33Y-β-catenin-Myc or 5 μg pT3-EF5α-S45Y-β-catenin-Myc; SB transposase (25:1) | FVB/N | Well-differentiated HCC by 6 to 9.5 weeks post-injection | [227,228,229] |
| myr-AKT and c-met | pT3-EF1α -HA-myr-AKT1; pT3-EF1α-V5-c-Met; SB transposase (25:1) | FVB/N | Lethal burden of HCC within 6 to 8 weeks post-injection showing admixture of clear, lipid-rich and lipid-poor, basophilic cells | [229] |
| myr-AKT and β-catenin | pT3-EF5-AKT; pT3-EF1α-ΔN90-β-catenin. | FVB/N C57BL/6 | Progression to HCC only in vivo passage of steatotic tumor cells from hepatocellular adenomas | [228] |
| In Vivo Model | Advantages | Disadvantages | Applications | References |
|---|---|---|---|---|
| PHH monolayer | - Similar to in vivo phenotype - High biotransformation capacity | - High donor-to-donor variability - Progressive dedifferentiation - Fail to represent complex in vivo environment | - Biotransformation studies - Toxicity studies - Drug-induced liver injury studies - Studies related to initiation and promotion of HCC - Liver disease studies | [252,253,254,255,256,257,264,266,271] |
| PHH sandwich cultures | - Prolonged viability - Retained morphology | - Altered protein expression over time - High donor-to-donor variability | - Studies of HCC initiation by carcinogens - Drug-induced liver injury studies | [252,261,268,272,273] |
| Liver cell lines monolayer | - Easy to use - Stable phenotype - Reproducible - Fit for high-throughput and high-content analyses - Allow genetic manipulation - Low cost | - Fail to represent intertumor and intratumor diversity - Less differentiated than PHH - Reduced or absent biotransformation capacity - Fail to represent complex in vivo environment | - Drug-screening - Safety testing - Genotoxicity studies - HCC biology studies - Studies assessing molecular and (epi)genetic modifications in HCC - Overexpression/silencing studies | [253,254,256,274,275,276,277,278,279,280] |
| Co-culture models | - Closer resemblance to in vivo cell heterogeneity and tumor-specific micro-environment - More pathologically relevant model by allowing cell-cell interactions | - Lack of standard protocols - High interlaboratory variability | - Studying influences of non-parenchymal cells and micro-environments on HCC | [253,280,281,282,283,284,285] |
| Stem cell-derived model | - High culture stability - Expandable - Reproducible - Metabolism that resembles PHH | - Lack of standard protocols for isolation of primary CSC | - HCC therapy studies - Studies related to initiation, promotion and drug resistance of HCC | [286,287,288] |
| Spheroid models | - Retained morphology and phenotypic functions - Higher biotransformation capacity compared 2D culture - Display oxygen and nutrient gradients - Closer resemblance to in vivo tumor-specific micro-environment - Closer resemblance to in vivo cell heterogeneity when combining co-culture techniques with spheroid models | - High donor-to-donor variability of PHH - More time-consuming and expensive than 2D culture - Lacks uniformity depending on spheroid-forming method - Low throughput depending on spheroid-forming method - Long-term culture is difficult | - Liver function studies - (Geno)toxicity studies - Liver disease studies - Drug delivery and efficacy studies - Studies investigating role of stem cells - Tumor-growth studies - Angiogenesis studies - Immunotherapy studies | [226,253,254,267,268,289,290,291,292,293,294,295,296] |
| Organoid model | - Mimic functionality and architecture of native tissue - Fit for high-throughput analyses - Expandable - Cryopreservation is possible - Allow genetic manipulation - Retention of tumor heterogeneity - Limited starting material is required - Based on healthy or tumorigenic material | - High cost - Time-consuming - Average success rate | - Regenerative medicine - Personalized drug discovery - Toxicity studies - Gene therapy studies - HBV-related carcinogenesis - Model liver cancer initiation - Molecular and cellular characterization of HCC - Study of inter- and intratumor diversity in HCC | [220,280,284,297,298,299,300,301,302,303] |
| Precision-cut-liver slices | - Capture complex in vivo micro-environment - Retained polarized morphology - Low cost - Automation possible - Closely resemble in vivo gene expression - Based on healthy or tumorigenic material | - Less suitable for high-throughput analyses - Labor-intensive preparation and incubation - Variable culture conditions between studies jeopardizing reproducibility - Reduced albumin and cytochrome P450 expression over time - Technically difficult - Limited availability | - Immunological studies - Toxicity studies - Genotoxicity assessment - Drug-screening - Liver disease studies | [266,288,304,305,306,307,308,309,310,311,312,313,314,315,316] |
| Cell Line | Cancer Type | HBV/HCV | Gender | Age | Race | References |
|---|---|---|---|---|---|---|
| HepG2 | Hepatoblastoma | -/- | Male | 15 | African | [324,337,338] |
| Huh-7 | HCC | -/- | Male | 57 | Asian | [324,337] |
| Hep3B | HCC | +/- | Male | 8 | African-American | [324,337] |
| HepaRG | HCC | -/+ | Female | / | European | [324,333,337] |
| MHCC97 | HCC | +/unknown | Male | 39 | Asian | [324,337,339] |
| PLC/PRF/5 | HCC | +/- | Male | 24 | African | [324] |
| SK-HEP-1 | Adenocarcinoma | -/unknown | male | 52 | European | [324,340] |
| SNU-475 | HCC | +/- | Male | 43 | Asian | [324] |
| SNU-423 | HCC | +/- | Male | 40 | Asian | [324] |
| SNU-449 | HCC | +/- | Male | 52 | Asian | [324] |
| C3A | Hepatoblastoma | -/- | Male | 15 | European | [277] |
| SNU-387 | HCC | +/- | Female | 41 | Asian | [324] |
| Cell Line | Immortalization Method | Gender | Age | Race | References |
|---|---|---|---|---|---|
| Fa2N-4 | SV40 large T antigen Transfection | Female | 12 | Unknown | [317,341] |
| NeHepLxHT | hTERT Retroviral vector | Male | <1 month | Unknown | [337,342] |
| THLE-2 | SV40 large T antigen Retroviral vector | Male | Adult | Unknown | [343] |
| PH5CH | SV40 large T antigen Lipid mediated gene transfer | Male | 58 | Unknown | [317,344] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romualdo, G.R.; Leroy, K.; Costa, C.J.S.; Prata, G.B.; Vanderborght, B.; da Silva, T.C.; Barbisan, L.F.; Andraus, W.; Devisscher, L.; Câmara, N.O.S.; et al. In Vivo and In Vitro Models of Hepatocellular Carcinoma: Current Strategies for Translational Modeling. Cancers 2021, 13, 5583. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215583
Romualdo GR, Leroy K, Costa CJS, Prata GB, Vanderborght B, da Silva TC, Barbisan LF, Andraus W, Devisscher L, Câmara NOS, et al. In Vivo and In Vitro Models of Hepatocellular Carcinoma: Current Strategies for Translational Modeling. Cancers. 2021; 13(21):5583. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215583
Chicago/Turabian StyleRomualdo, Guilherme Ribeiro, Kaat Leroy, Cícero Júlio Silva Costa, Gabriel Bacil Prata, Bart Vanderborght, Tereza Cristina da Silva, Luís Fernando Barbisan, Wellington Andraus, Lindsey Devisscher, Niels Olsen Saraiva Câmara, and et al. 2021. "In Vivo and In Vitro Models of Hepatocellular Carcinoma: Current Strategies for Translational Modeling" Cancers 13, no. 21: 5583. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13215583